
Abstract
Although the neuroprotective effects of hypothermia have been known for a long time, the molecular correlates of this neuroprotection are poorly understood. In this study, the authors investigated how hypothermia affects inflammatory responses in the brain elicited by systemic injection of IL-1β. Leukocyte rolling and adhesion were quantified in pial venules (20 to 50 μm) of C57/Bl6 mice 4 hours after intraperitoneal injection of IL-1β (5 μg/kg) using an open cranial window and intravital microscopy. Animals were subjected to moderate hypothermia (32°C) or normothermia (37°C) for 1 or 4 hours after IL-1β injection. Significant increases in leukocyte rolling and adhesion were observed in IL-1β–injected animals as compared with sham controls. Whereas 1-hour hypothermia did not affect IL-1β–induced leukocyte rolling and adhesion, 4-hour hypothermia caused a reduction in both rolling and adhesion. Molecular mechanisms of hypothermic effects were investigated in cultured human cerebral endothelial cells exposed to IL-1β (50 U/mL) for 4 hours at 37°C or 32°C followed by 18 hours at 37°C. Human cerebral endothelial cells exposed to IL-1β at 32°C showed attenuated NF-κB activation determined by the Luciferase yellow reporter gene assay and reduced expression of IL-8 and IL-1β measured by reverse transcriptase–polymerase chain reaction and enzyme-linked immunosorbent assay. Intracellular adhesion molecule-1 was induced to similar levels (threefold over control) at both temperatures. The expression of CD18 on neutrophils in vitro was not affected by either IL-1β or hypothermia. These findings suggest that mechanisms by which hypothermia reduces leukocyte rolling and adhesion include suppression of inflammatory gene transcription in brain endothelial cells.
Inflammatory brain injury is an important pathophysiologic mechanism associated with ischemia–reperfusion and traumatic brain injury (TBI). The hallmark of postischemic, posttraumatic brain inflammation, or both is neutrophil infiltration into brain parenchyma (Barone et al. 1991; Hallenbeck, 1996). Migration of neutrophils from the microvascular lumen into the brain parenchyma involves a cascade of cellular responses mediated by specific adhesion pathways, which have been typified into a rolling/activation/adhesion/transmigration paradigm (Carlos and Harlan, 1994; Hallenbeck, 1996). Circulating leukocytes tether to selectins expressed on activated endothelial cells, decelerate, and sense the local environment and endothelial surface for chemotactic signals. After activation by chemokines, the strong adhesion of leukocytes is mediated by the leukocyte β2 integrins and endothelial adhesion molecules including the immunoglobulin family (Carlos and Harlan, 1994). Subsequent transendothelial migration of leukocytes is accompanied by functional blood–brain barrier (BBB) disruption mediated in part by enzymes released from activated leukocytes (del Zoppo, 1997). Work in both animal and cell culture models have shown that intracellular adhesion molecule-1 (ICAM-1) is a principal mediator of neutrophil adhesion to cerebral endothelial cells after ischemia (Wang and Feuerstein, 1995; Stanimirovic et al., 1997) and in response to cytokines (Carlos and Harlan, 1994). Furthermore, strong experimental evidence suggests that depleting leukocytes, administering anti-CD18 antibodies, or blocking adhesion molecules (including ICAM-1) provides protection after cerebral ischemia (Chopp et al., 1996; Kogure et al., 1996).
Inflammatory activation of endothelial cells and leukocytes is coordinated by the activation and secretion of proinflammatory cytokines from parenchymal and vascular cells. A number of transcription factors, most notably nuclear factor-κB (NF-κB), have been shown to regulate cytokine and adhesion molecule gene expression. In response to oxidants, endotoxins, or cytokines (for example, IL-1β), NF-κB translocates to the nucleus where it stimulates the transcription of proinflammatory genes such as ICAM-1, cytokines (for example, IL-1β), and chemokines (for example, IL-8, macrophage chemo-attractant protein-1 [MCP-1]) (Manning et al., 1995) in endothelial cells. In contrast, the activity of leukocyte integrins is regulated predominantly by increasing the affinity for the ligand or avidity of adhesion (Gahmberg, 1997).
Hypothermia has been shown both clinically (Marion et al., 1997) and experimentally (Clark et al., 1996; Colbourne et al., 1997) to be neuroprotective. Moderate hypothermia is most beneficial when applied immediately after the insult and for longer duration (Colbourne et al., 2000). Proposed mechanisms of action of hypothermia include decreased BBB permeability (Smith and Hall, 1996; Dietrich et al., 1990), decreased glutamate concentrations (Busto et al., 1989), decreased generation of free radicals (Thoresen and Wyatt, 1997), and antiinflammatory effects (Maier et al., 1998; Chatzipanteli et al., 2000; Toyoda et al., 1995). Previous investigations have shown that hypothermia may decrease neutrophil migration in vitro and in vivo (Biggar et al., 1984), suggesting that reducing secondary inflammation in the central nervous system is a possible mechanism of hypothermic neuroprotection.
In this study, the authors demonstrate that moderate hypothermia results in reduction of leukocyte rolling and adhesion in pial microvasculature of mice induced by the systemic injection of IL-1β. The authors further show that hypothermia significantly reduces IL-1β–stimulated NF-κB activation and the expression of inflammatory cytokine IL-1β and chemokine IL-8 in human brain endothelial cells in vitro, whereas it has no effect on CD18 expression in isolated human neutrophils. These results suggest a novel role for hypothermia in the modulation of inflammatory gene transcription in brain endothelium. The study proposes that mechanisms by which hypothermia provides neuroprotection include a reduction in leukocyte trafficking across the blood–brain barrier caused by attenuated expression of inflammatory cytokines in vascular cells.
MATERIALS AND METHODS
Animals
All animals in this study were male C57/Bl6 mice (Charles River, Montreal, Canada) weighing 20 to 24 g. Animals were housed in groups of two with food and water available ad libitum in pathogen-free facilities. Animal procedures were performed in accordance with the University of Ottawa Animal Care Committee and Canadian Council of Animal Care.
Temperature modulation
Mice were randomized to 1 of 4 groups: sham-operated (n = 7) animals received 0.9% saline intraperitoneally (IP) and were maintained at 37°C for 4 hours; control (n = 6) animals were injected with IL-1β (5 μg/kg) IP and maintained at 37°C for 4 hours; hypothermic animals were injected with IL-1β (5 μg/kg) IP and maintained at 32°C for either 1 hour (n = 7) just before video recordings or for 4 hours (n = 7) commencing immediately after IL-1β injection. Mice were anesthetized by IP injection of 10 mg/kg xylazine and 150 mg/kg ketamine. Under a dissecting microscope, a thermistor was placed intracranially through a burr hole in the left parietal bone for brain temperature monitoring. Both rectal and cerebral temperatures were recorded, and rectal temperature was kept within 0.5°C of the target temperature throughout the experimental period using a heated water mat and an overhead heating lamp. Rectal and cerebral temperatures were continuously monitored and recorded every 10 minutes.
Intravital microscopy and open cranial window
At 3.5 hours after IL-1β injection, an open cranial window was made. Briefly, a hole was made in the right parietal bone as described by Rosenblum et al. (1994), and the pia was superfused with warmed artificial cerebrospinal fluid bubbled with a gas mixture (6%CO2, 10%O2, balance N2). Animals then were placed under the intravital microscope (Olympus BHMJ modular focusing mount and BH2-RFCA illuminator on a custom made stand, Hitachi video camera, Sony monitor, and Panasonic WJ-810 time code generator). Acridine orange (17 μg/g) was infused through a tail vein and the microscope was focused on 20- to 50-μm diameter venules using a 20× long working distance objective. The microcirculation was illuminated with a mercury vapor lamp through a 495-nm excitation filter (BH2-DMV; Olympus) for fluorescence or with a fiberoptic epi-illuminator (Olympus highlight 3000) for bright field illumination. At 4 hours after IL-1β injection, videocassette recordings were made using a Mitsubishi (model HS-U69) sVHS video recorder for 30 seconds each at 2 to 3 venules per animal. Mocha (Jandel Scientific, San Rafael, CA, U.S.A.) image analysis software was used for measuring microvessel diameter and distances in the microvessels after capture of an image to a personal computer using a Targa+ (Truevision, Indianapolis, IN, U.S.A.) framegrabber.
The number of leukocytes rolling and adhering to pial venules was determined during playback of the videotapes. Rolling leukocytes were quantified as the number of rolling cells passing a reference line in the pial venules over 30 seconds and adhering as those stationary on the walls of pial venules (100-μm lengths) for longer than 2 seconds.
Cell culture
Human cerebral endothelial cell (HCEC) cultures are routinely isolated from small samples of human temporal lobe excised surgically from patients treated for idiopathic epilepsy. The isolation and cultivation procedures for HCEC are a modification of those published by Gerhart et al. (1988). Both primary and propagated HCEC cultures were grown in media containing 65% M199, 10% fetal calf serum, 5% human serum, 20% murine melanoma cell conditioned media (mouse melanoma, Cloudman S91, clone M-3, melanin producing cells), 5 μg/mL insulin, 5 μg/mL transferrin, 5 ng/mL selenium, and 10 μg/mL endothelial cell growth supplement (Collaborative Biomedical Products, Bedford, MA, U.S.A.). Cell cultures derived by these procedures exhibit a polygonal, “cobblestone” morphology, immunoreactivity for the endothelial cell markers, Factor VIII-related antigen, and angiotensin-converting enzyme, as well as high levels of the cerebral endothelium-specific enzymes: gamma glutamyl transpeptidase and alkaline phosphatase. The morphologic, phenotypic, biochemical, and functional characteristics of HCEC cultures used in this study have been described in detail previously (Stanimirovic et al., 1997).
IL-1β stimulation
Human cerebral endothelial cells were exposed to the proinflammatory cytokine IL-1β (50 U/mL; Upstate Biotechnology, Lake Placid, NY, U.S.A.) for 4 hours in serum-free M199 medium at 37°C or 32°C in 5%CO2/95% air. The media then were replaced with 10% growth media and cells were maintained at 37°C for an additional 18 hours. Cell media were collected for determination of secreted IL-8, and cell mRNA was extracted for reverse transcription–polymerase chain reaction (RT-PCR). Separate cell cultures exposed to the same conditions were used for measuring NF-κB transcriptional activity and for determining ICAM-1 expression. Exposure of HCEC to 32°C for 4 hours did not affect cell viability as determined by propidium iodide staining.
Reporter-gene assay for NF-κB
Four double-stranded NF-κB-response elements [5′GATCTTGGATAGGGGACTTTCCCGGGTACA3′] with their complementary sequences were premultimerized and inserted into a Bgl-II site in a linearized and dephosphorylated pGL3 promoter vector (Promega, Madison, WI, U.S.A.). The construct was dideoxysequenced to confirm the orientation and the number of the NF-κB response element insertions. Human cerebral endothelial cells grown in 24-well plates to 90% confluence were transfected with either an empty pGL3 promoter vector or the promoter vector containing NF-κB–responsive elements for 2.5 hours using SuperFect (QIAGEN, Mississauga, ON, Canada) transfection kit as per manufacturer's protocol. Cells then were washed and recovered in growth media for 18 hours at 37°C. The media then were removed, cells were washed once in M199 medium, and exposed to IL-1β for 4 hours at 37°C or 32°C. Human cerebral endothelial cells then were washed twice with Ca2+/Mg2+ -free HBSS and then lysed in 50 μL cell lysis reagent (Promega). Luciferase yellow (LY) activity was determined using a kit from Promega. The luciferase assay reagent containing D-luciferin was added to aliquots of cell lysates and chemiluminescence was measured at 25°C using a chemiluminescence counter (MicroBeta TriLux, Wallac Oy, Finland). Efficiency of transfection was controlled by simultaneously transfecting HCEC with β-galactosidase reporter plasmid. Total cell protein was determined in each sample by the method of Lowry et al. (1951). Light units emitted from samples were read against a standard curve (Recombinant Luciferase, Promega) and normalized to β-galactosidase activity and protein levels in cell lysates.
ELISA
An enzyme-linked immunosorbent assay (ELISA) was used to quantify ICAM-1 expression in HCEC and IL-8 secretion into culture media of HCEC. ELISA for ICAM-1 was performed essentially as described by Stanimirovic et al. (1997). Briefly, HCEC cultures grown in 96-well microtiter plates (2 × 104 cells/100 μL per well were sequentially incubated with a primary monoclonal anti-human ICAM-1 antibody (2 μg/mL; clone CD54, Upstate Biotechnology, Lake Placid, NY, U.S.A.) for 1 hour at 37°C, followed by 1:500 diluted peroxidase-conjugated goat anti-mouse IgG in phosphate-buffered saline (PBS) for 45 minutes at 37°C. Nonspecific binding sites were blocked with 2% bovine serum albumin in PBS for 30 minutes at 37°C. After each incubation, the plates were washed three times with PBS. Color was developed by the addition of a 100 μL of 1 mg/mL solution of the horseradish peroxidase substrate, 2,2′-azinobis (3 ethylbenzthiazoline-6-sulfonic acid) (ImmunoPure ABTS). The reaction was stopped after 5 minutes by the addition of an equal volume of 1% sodium dodecyl sulfate to each well. Optical density of the developed color was read at 405 nm using a SpectraMAX (Molecular Devices, Menlo Park, CA, U.S.A.) microplate reader.
ELISA assay for IL-8 was performed as described in Zhang et al. (1999). Human cerebral endothelial cell media (collected from 2 × 105 cells/dish) were centrifuged at 14000 g for 5 minutes at 4°C and an aliquot (100 μL) was used in ELISA. The levels of IL-8 were determined using a commercially obtained ELISA kit (BioSource, Camarillo, CA, U.S.A.). Assays were performed according to manufacturer's instructions in at least three independent experiments.
Reverse transcriptase–polymerase chain reaction
Total RNA was extracted from HCEC grown in 35-mm Petri dishes using Tri reagent (500 μL/dish) and was purified using the protocol provided by the manufacturer (GibcoBRL, Burlington, Ontario). The RNA pellets were resuspended in 25 μL DEPC-treated dH2 O and were incubated at 55°C for 10 minutes. RNA (0.5 μg) then was mixed with 0.5 μg of oligo (dT) primers, heated for 10 minutes at 70°C, and then chilled on ice.
Synthesis of single-stranded cDNA was performed by reverse transcription (42°C, 1 hour) in a reaction mixture (final volume = 20 μL) containing 4 μL of 5X first strand buffer (Gibco BRL), 2 μL of 0.1 mol/L dithiothreitol, 1μL of 10 mmol/L dNTP, 100 U RNase H reverse transcriptase (SuperScript II, Gibco-BRL), and 10 μL DEPC-treated dH2 O. The reverse transcriptase was inactivated by heating the reaction mixture to 70°C for 10 minutes. The 20 μL of the resulting reaction mixture then was diluted with 20 μL dH2 O, and cDNA from this mixture was subsequently used in 25 μL PCR reaction.
Polymerase chain reaction amplifications were performed in a final volume of 25 μL containing 1× reaction buffer (Promega), 1.5 mmol/L MgCl2, 0.2 mmol/L each of 4 dNTPs, 0.4 μmol/L each of 2 pairs of the primers (primers for an internal control gene as well as the primers for either IL-8 or IL-1β; Table 1), 2.5 U Taq DNA polymerase, and 4 μL cDNA. Amplification reactions then were performed in a PTC-200 Peltier Thermal Cycler (MJ Research, Watertown, MA, U.S.A.) in 0.2-mL tubes (Rose Scientific, Alberta, Canada). All amplifications were performed using a denaturation step at 94°C for 30 seconds, an annealing step at 55°C for 45 seconds, and a polymerization step at 72°C for 40 seconds. Polymerase chain reaction amplifications were linear between 20 and 35 cycles for β-actin, from 25 to 45 cycles for IL-1β, and from 25 to 40 cycles for IL-8. All experiments then were performed using conditions optimized for linear amplification.
Sense and antisense sequences of primers used in PCR reactions in this study

Polymerase chain reaction (PCR) using the above primers generated a 289-bp DNA fragment for IL-8, a 275-bp fragment for IL-1β, and a 504-bp fragment for β-actin.
IL-8 and β-actin amplification was performed in the same PCR reaction containing 5 μL diluted cDNA for 35 cycles. Ten microliters of the PCR product then was subjected to gel electrophoresis as described below. IL-1β was amplified using 10 μL diluted cDNA for 45 cycles. Internal control (β-actin) was amplified using 1 μL diluted cDNA in a 50-μL reaction for 35 cycles. Equal amounts of internal control and IL-1β PCR products then were mixed and 10 μL were subjected to electrophoresis on a 1.7% agarose gel in 1× Tris-Borate EDTA buffer containing 0.5 μg/mL ethidium bromide and were photographed.
Flow cytometry
Blood samples were collected from healthy volunteer subjects into heparinized tubes. All analyses were performed within 2 hours of blood collection. Blood was diluted 1:10 in PBS and aliquoted into 24-well culture plates. Control cells and cells exposed to IL-1β (100 U/mL), peptide neutrophil chemoattractant N-formyl-1-methionyl-1-leucyl-1-phenylalanine (fMLP) (5 mmol/L; Sigma Chemicals, St. Louis, MO, U.S.A.), or bacterial lipopolysaccharide (LPS; 1 μg/mL; Sigma Chemicals) were incubated at either 37°C or 32°C for 1 hour at 4% CO2. Cells then were harvested and washed with PBS/0.1% NaN3. Contaminating erythrocytes were removed by 2 cycles of brief hypotonic lysis in ice-cold 0.15% NaCl. Cells then were resuspended in PBS/NaN3 containing 2 mg/mL IgG for 10 minutes to block nonspecific binding.
Surface expression of CD18 was examined using one-color flow cytometric analysis. Stimulated and control cells were incubated with 10 μL (determined by titration) mouse anti-human FITC-CD18 mAb (BD Biosciences, Mississauga, ON, Canada) for 15 minutes at room temperature in the dark, washed, and resuspended in PBS/0.1% NaN3. Autofluorescence and isotype control (FITC-IgG1/2; Sigma Chemicals) fluorescence was determined for each experimental group. Neutrophils were gated based on forward scatter and side scatter characteristics. CD18 expression then was measured in gated populations as mean channel fluorescence intensity (Becton-Dickinson FACScan Flow Cytometer). A total of 50,000 events were recorded for each sample. Data were analyzed using the WinMDI software package (J. Trotter, Scripps Institute, San Diego, CA, U.S.A.).
Statistical analysis
All data are expressed as mean ± SD. Differences between experimental groups were compared using a one-way analysis of variance followed by Bonferroni multiple comparisons or Fisher protected least square difference multiple comparisons. P < 0.01 was considered statistically significant.
RESULTS
Temperature recordings
Rectal (Fig. 1A) and intracerebral (Fig. 1B) temperatures were measured at 10-minute intervals. Both rectal and intracerebral temperatures in the hypothermic groups were significantly reduced by 4.5°C to 5°C (Fig. 1A and 1B) compared with the normothermic groups, indicating effective maintenance of moderate hypothermia in this mouse model. No significant variations in single animals or between animals within any group were found (data not shown). Cerebral temperature was less than rectal temperature in all experimental groups. Similar observations have been reported by other groups (Maier et al., 1998).
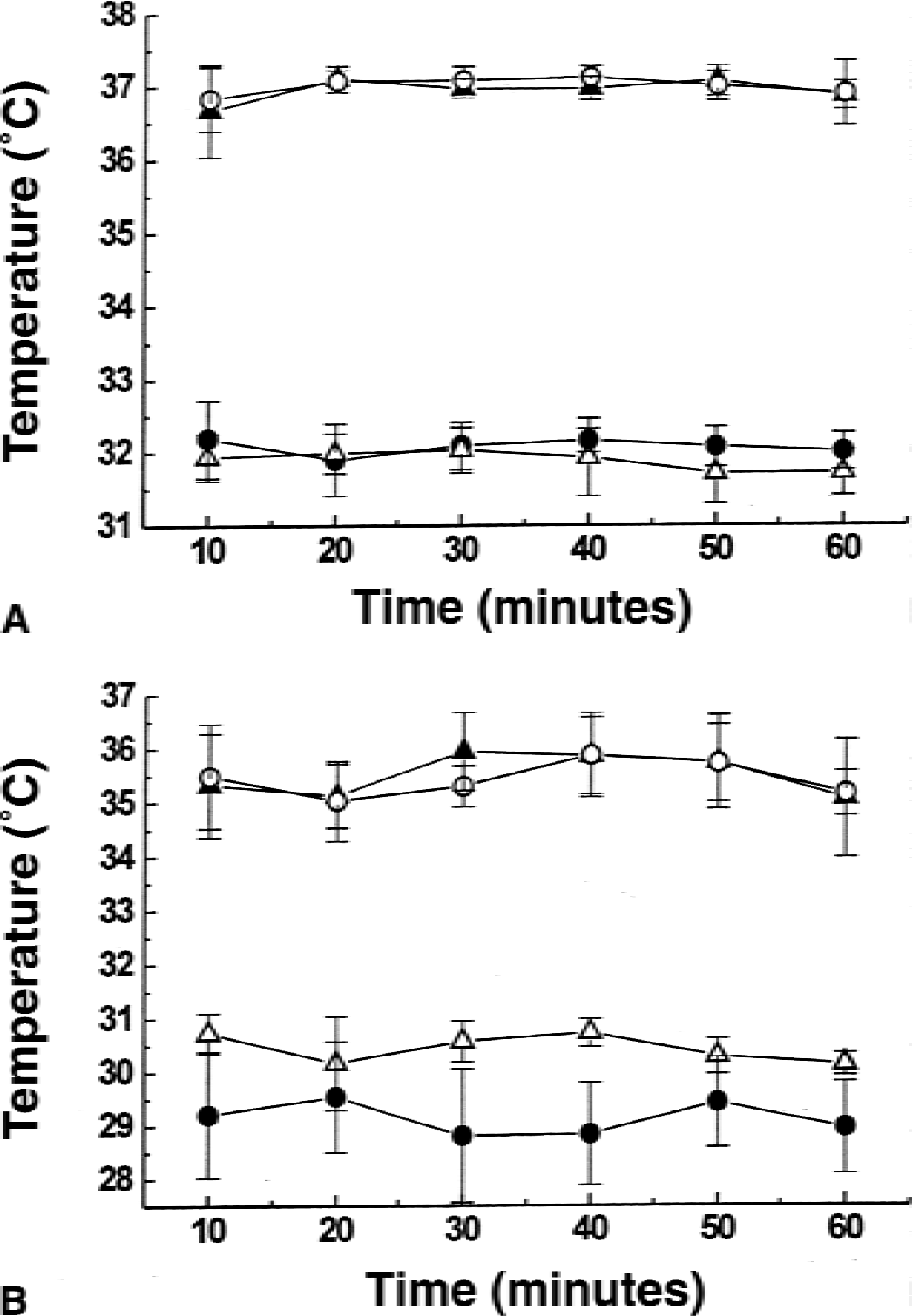
Temperature recordings in experimental groups of animals (Sham, 37°C, open circles; IL-1β, 37°C, closed triangles; IL-1β, 32°C for 1 hour, closed circles; IL-1β, 32°C for 4 hours, open triangles) used in the study. Rectal
Effects of hypothermia on leukocyte rolling and adhesion induced by systemic injection of IL-1β
Leukocyte rolling and adhesion to the endothelium of pial venules were monitored using intravital microscopy. In the sham group there was virtually no rolling or adhering leukocytes observed (Fig. 2A). An increase in numbers of rolling and adhering leukocytes was seen in animals injected with IL-1β as compared with sham-operated controls (Fig. 2A and 2B). Quantitative analysis showed that IL-1β induced significant increases in both rolling (Fig. 3A) and adhering (Fig. 3B) leukocytes. In animals subjected to 1-hour hypothermia 3 hours after IL-1β injection, the numbers of rolling and adhering leukocytes were not different from those in the normothermic, IL-1β–injected group (Fig. 3A and 3B). However, in animals subjected to a 32°C hypothermia for 4 hours after IL-1β injection, a complete inhibition of leukocyte rolling (Figs. 2C and 3A) and attenuation of leukocyte adhesion (Fig. 3B) were detected. All pial venules chosen for measurements of leukocyte rolling and adhesion were 20 to 50 μm and no significant differences in average venule diameter between groups were found (Fig. 3C), suggesting that observed differences in leukocyte rolling and adhesion were not caused by differences in vessel diameter.
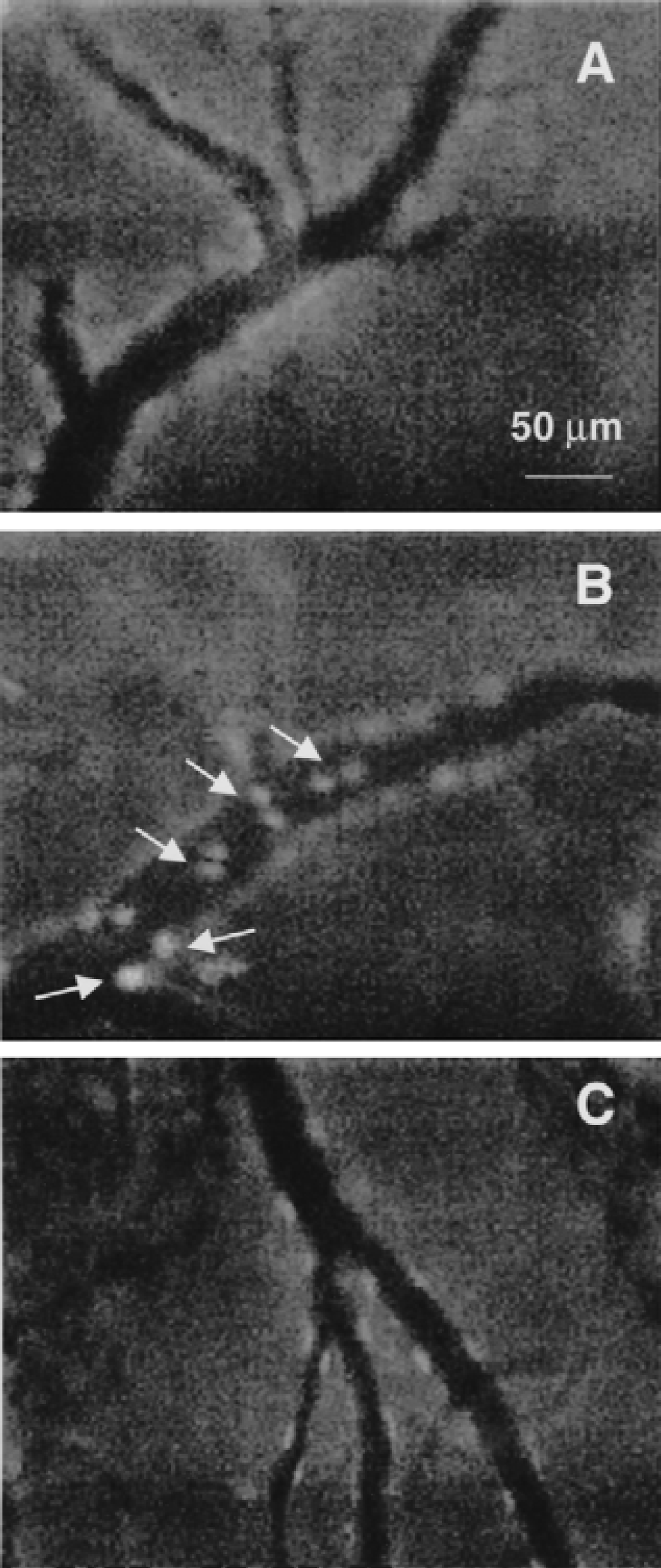
Representative images of pial venules obtained during open cranial window recording of leukocyte rolling and adhesion with a CCD camera.
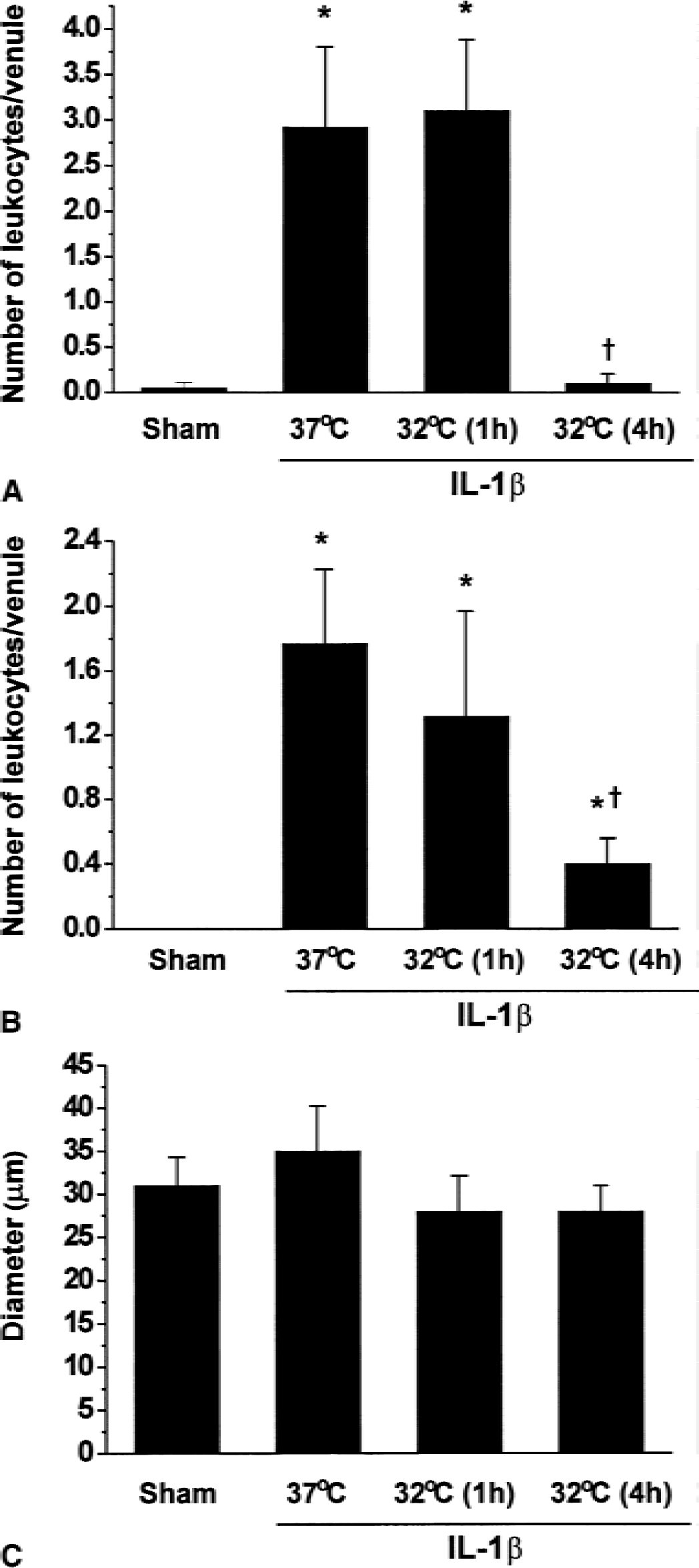
Effects of hypothermia on IL-1β–induced leukocyte rolling and adhesion in the pial microcirculation of mice. Average number of rolling
Effect of hypothermia on IL-1β–induced NF-κB activation in HCEC
Potential mechanisms by which hypothermia attenuates IL-1β–induced rolling and adhesion of leukocytes to pial vessels in vivo were investigated in cultured human cerebromicrovascular endothelial cells exposed to IL-1β. Human cerebral endothelial cells were exposed to IL-1β at either 37°C or 32°C for 4 hours. The NF-κB transcriptional activity was determined in HCEC transfected with the LY reporter gene plasmid at the end of 4-hour exposure to IL-1β. NF-κB–mediated transcription of LY increased 2- to 2.5-fold in HCEC exposed to IL-1β at 37°C as compared with control cells (Fig. 4). Exposure of HCEC to IL-1β at 32°C resulted in reduced NF-κB transcriptional activity as compared with that observed at 37°C (Fig. 4). Human cerebral endothelial cells transfected with control vector plasmid did not respond to any of the above experimental treatments by LY activation (data not shown).
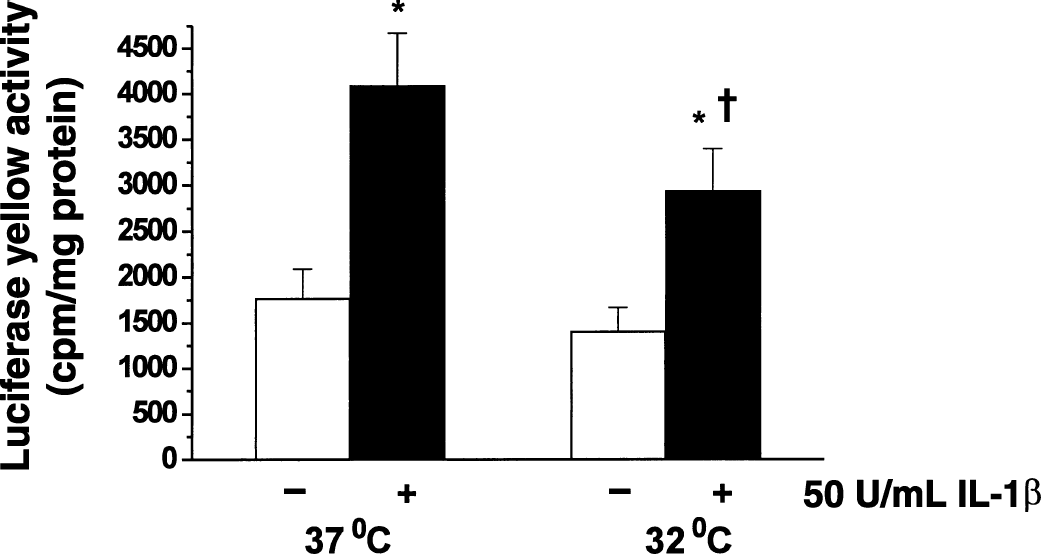
Effects of hypothermia on IL-1β–induced NF-κB activity in human cerebral endothelial cells (HCEC). Cells were incubated in the presence or absence of IL-1β (50 U/mL) at 37°C or 32°C for 4 hours. Transcriptional activation of NF-κB was determined by transfecting HCEC with NF-κB–pGL3 plasmid vector as described in Materials and Methods. Values shown are mean ± SD of triplicate cell cultures in a representative experiment. *Significant difference from corresponding temperature control. †Significant difference (P < 0.01; ANOVA) from IL-1β–treated cells at 37°C.
Effect of hypothermia on IL-1β–induced ICAM-1, IL-8, and IL-1β expression in HCEC
To examine whether reduced NF-κB activation by hypothermia results in attenuated expression of proinflammatory genes in HCEC, the expression of ICAM-1, IL-1β, and IL-8 in HCEC exposed to IL-1β at 37°C or 32°C for 4 hours and then recovered for 18 hours in IL-1β–free media was determined by RT-PCR and ELISA. IL-1β has been previously shown to cause inflammatory activation of HCEC, including the up-regulation of ICAM-1 (Stanimirovic et al, 1997), IL-8, and MCP-1 expression (Zhang et al., 1999).
IL-1β stimulated ICAM-1 expression in HCEC by 3-fold (Fig. 5). There was no significant difference in ICAM-1 expression when HCEC were exposed to IL-1β at 37°C and 32°C (Fig. 5). However, a significant reduction in IL-8 (Fig. 6A) and IL-1β (Fig. 6B) mRNA expression was seen in HCEC exposed to IL-1β at 32°C. The secretion of IL-8 into cell media, as measured by ELISA, also was significantly reduced in HCEC subjected to IL-1β at 32°C as compared with 37°C (Fig. 7).
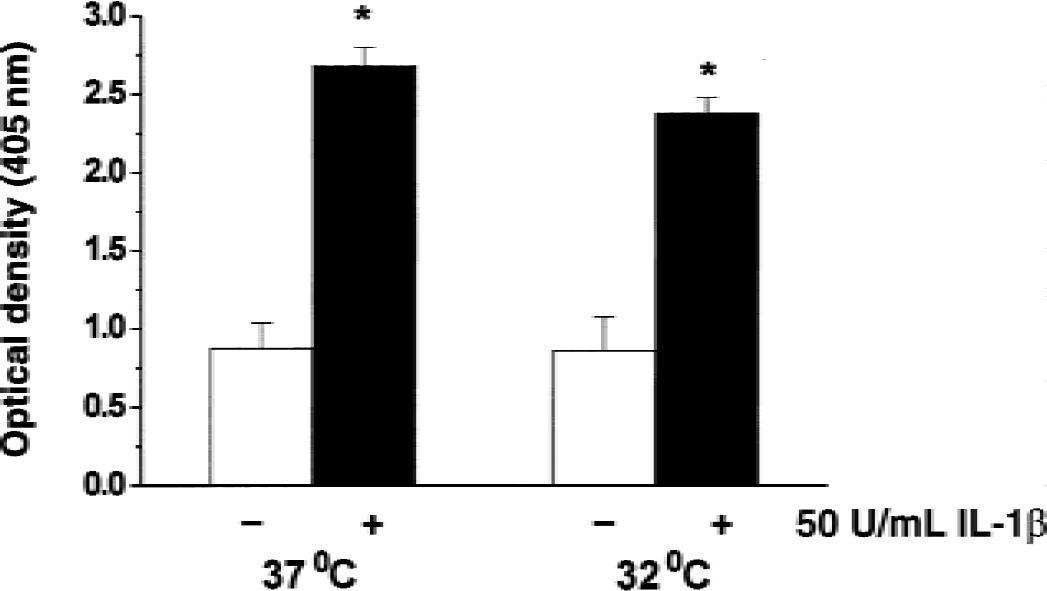
Effects of hypothermia on IL-1β–induced intracellular adhesion molecule-1 (ICAM-1) expression in human cerebral endothelial cells (HCEC). Cells were incubated in the presence or absence of 50 U/mL IL-1β at 37°C or 32°C for 4 hours and then recovered in IL-1β–free media for 18 hours. ICAM-1 expression was determined by ELISA as described in Materials and Methods. Values shown are mean ± SD of six replicates in a representative experiment. Similar results were obtained in at least three separate HCEC isolations.
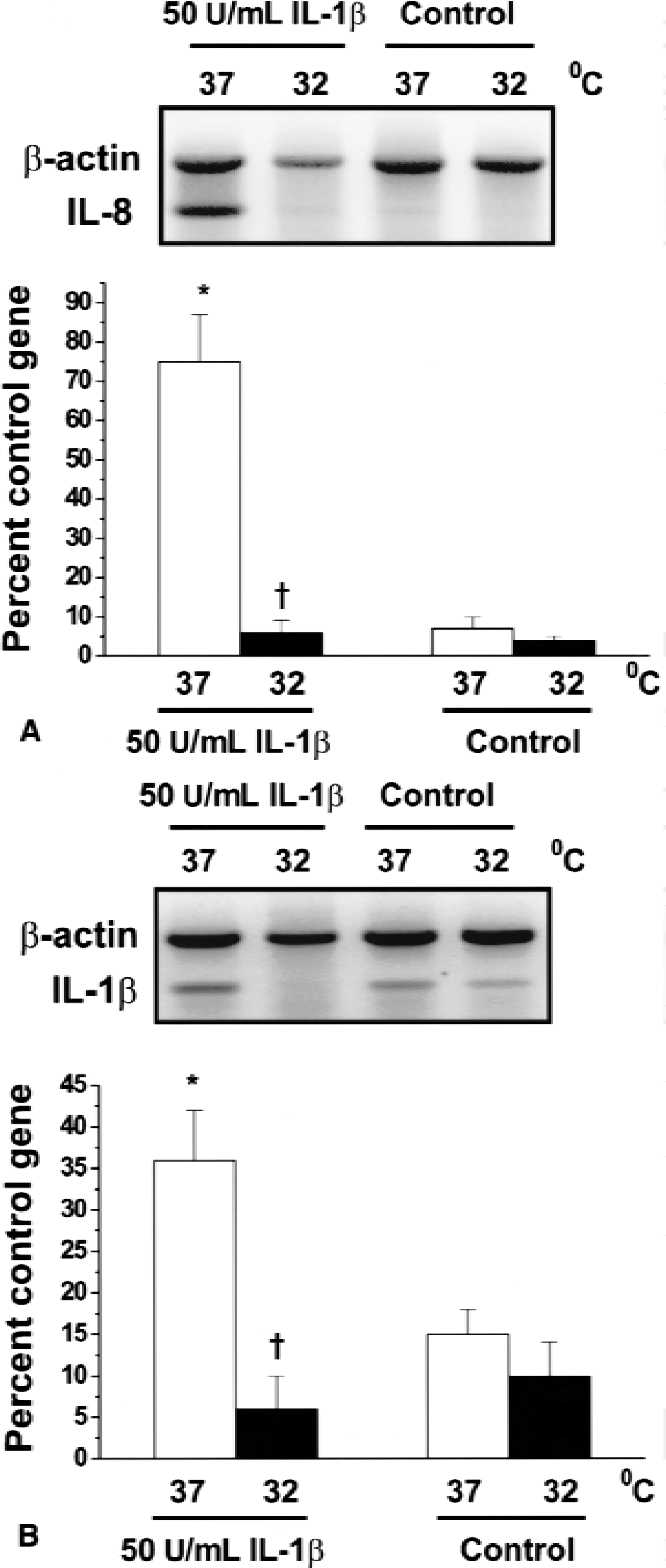
Effects of hypothermia on IL-1β–induced IL-8
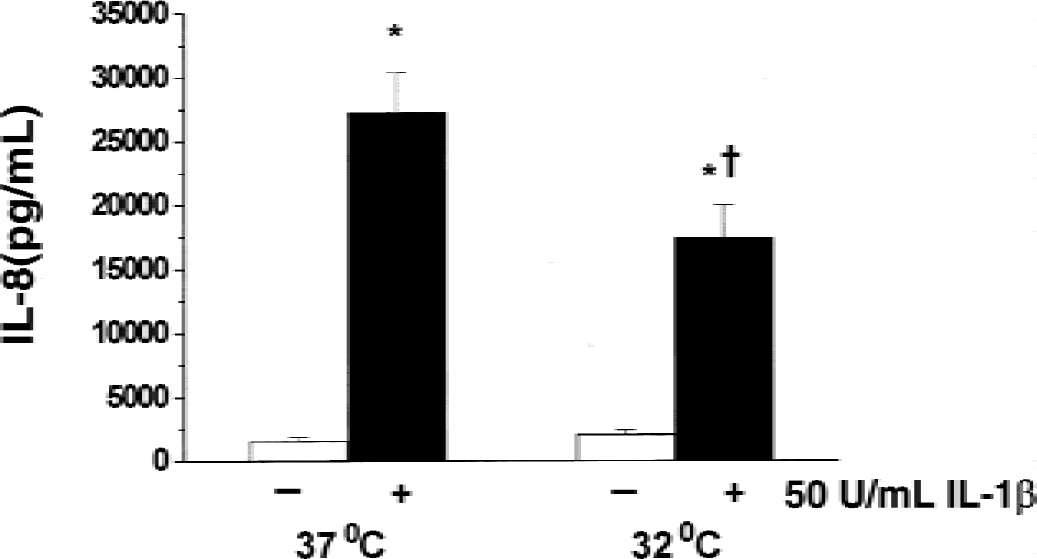
Effects of hypothermia on IL-1β–induced IL-8 secretion in human cerebral endothelial cells (HCEC). Cells were incubated in the presence or absence of 50 U/mL IL-1β at 37°C or 32°C for 4 hours and then recovered in IL-1β–free media for 18 hours. IL-8 secretion was measured in cell media by sandwich ELISA as described in Materials and Methods. Values shown are means ± SD of six replicates in a representative experiment. Similar results were obtained in at least three separate HCEC isolations. *Significant difference from corresponding temperature control. †Significant difference (P < 0.01; ANOVA) from IL-1β-treated cells at 37°C.
Effect of hypothermia on CD18 expression and neutrophil chemotaxis
To investigate the effects of hypothermia on neutrophil activation, neutrophil CD18 expression was determined in whole blood exposed to IL-1βin vitro using flow cytometry. Two known stimulants of neutrophil β2 integrin activity, LPS and fMLP (Gahmberg, 1997), were used as positive controls. Whereas both LPS and fMLP significantly increased neutrophil CD18 expression (Fig. 8; Table 2), IL-1β had no significant effect after either 15 minutes or 1 hour of exposure (Fig. 8; Table 2). Similarly, IL-1β failed to increase neutrophil motility and fMPL-induced chemotaxis (data not shown). Hypothermia applied for either 15 minutes or 1 hour during stimulation of neutrophils with IL-1β, fMLP, or LPS did not have a significant effect on neutrophil CD18 expression (Table 2).
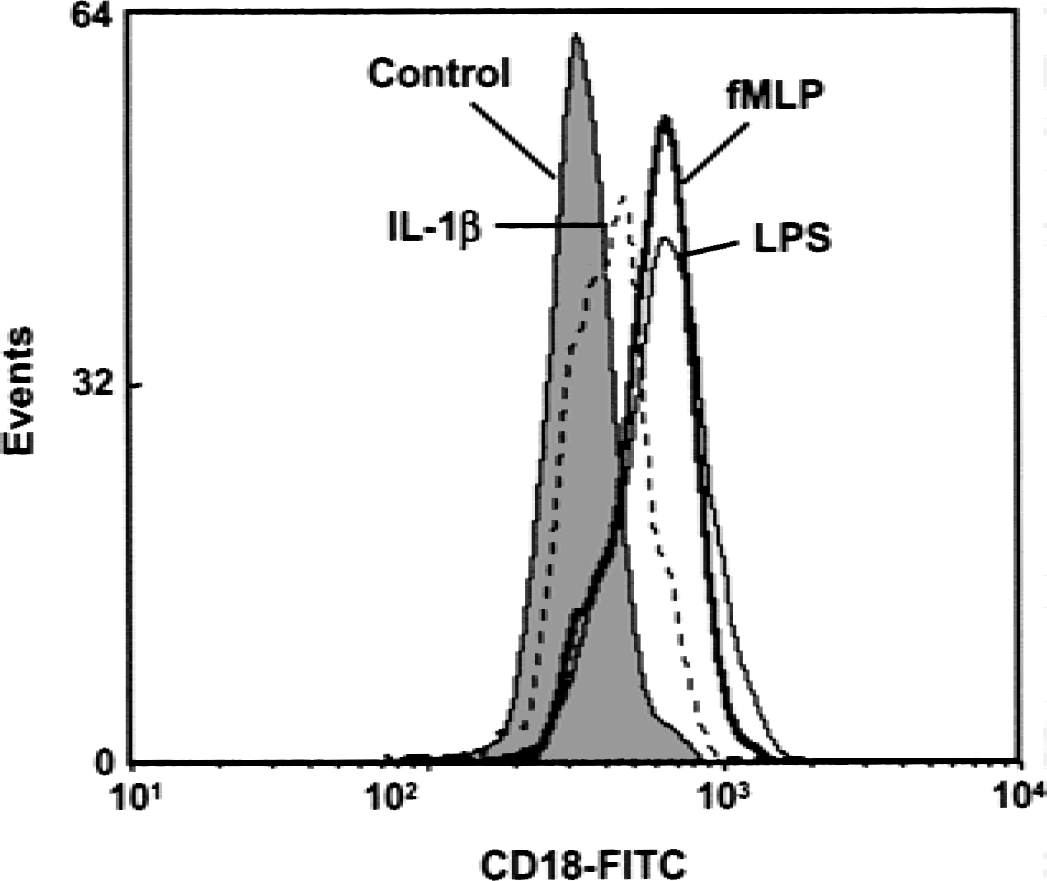
Expression of CD18 in human neutrophils from the whole blood subjected to IL-1β, fMLP, and lipopolysaccharide (LPS) determined by flow cytometry. Control media, 100 U/mL IL-1β, 5 mmol/L fMLP, or 1 μg/mL LPS were added to whole blood for 1 hour at 37°C. Neutrophils then were isolated and labeled with anti–CD18-FITC antibody and subjected to flow cytometry as described in Materials and Methods. The histogram is representative of experiments performed in blood samples from three volunteer subjects.
CD18 expression in neutrophils subjected to IL-1β, LPS, or fMLP at 37°C or 32°C determined by flow cytometry
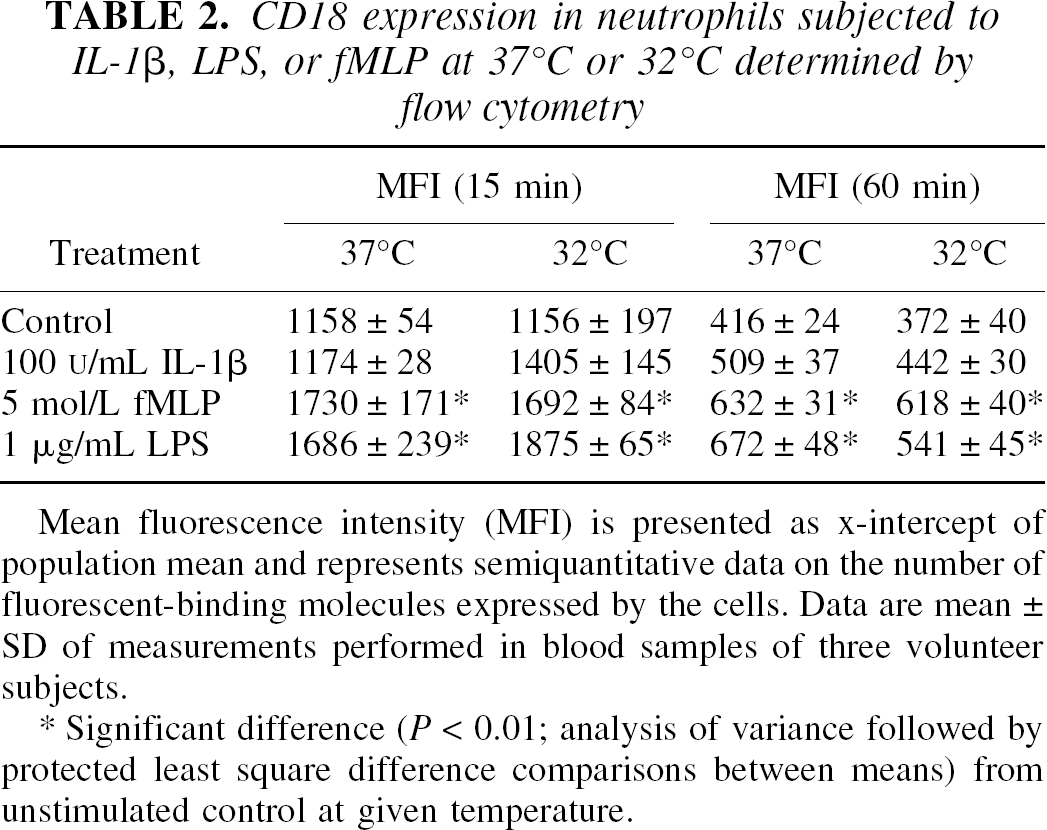
Mean fluorescence intensity (MFI) is presented as x-intercept of population mean and represents semiquantitative data on the number of fluorescent-binding molecules expressed by the cells. Data are mean ± SD of measurements performed in blood samples of three volunteer subjects.
Significant difference (P < 0.01; analysis of variance followed by protected least square difference comparisons between means) from unstimulated control at given temperature.
DISCUSSION
This study provides evidence that moderate hypothermia reduces rolling and adhesion of peripheral leukocytes to pial venules induced by systemic application of proinflammatory cytokine IL-1β in mice. Potential mechanisms of these effects were investigated in cultured human brain microvascular endothelial cells and neutrophils. Hypothermia was shown to attenuate IL-1β–induced inflammatory activation of HCEC by: reducing the activation of NF-κB, a transcription factor known to regulate the expression of proinflammatory genes including adhesion molecules, cytokines, and chemokines; reducing the expression of cytokine IL-1β and chemokine IL-8 mRNA; and inhibiting the secretion of immunoreactive IL-8 into cell media. In contrast, neither IL-1β nor hypothermia changed neutrophil CD18 expression and chemotaxis. Taken together, these observations suggest that moderate hypothermia attenuates IL-1β–induced leukocyte rolling and adhesion in the brain in part by suppressing inflammatory activation of brain endothelial cells. The study implies that suppression of secondary brain inflammation may be an important modality and mechanism of neuroprotective actions of hypothermia.
Historically, hypothermia has been used to protect the brain after TBI, cardiac arrest, and neonatal asphyxia. However, temperatures used early on were low (28°C–30°C) and led to complications when used for prolonged periods. Recent investigation demonstrating that smaller reductions in brain temperature (32°C to 34°C) provide neuroprotection against global ischemia (Buchan and Pulsinelli, 1990) has led to renewed interest in this field. Although clinical trials currently are being conducted to investigate therapeutic applications of hypothermia in adults after stroke (Schwab et al., 1998) and TBI (Jiang et al., 2000), the mechanisms by which hypothermia provides protection are not fully understood. In experimental models of cerebral injury, hypothermia has been shown to reduce neuronal injury, promote postischemic metabolic recovery, attenuate excitatory amino acid release, reduce BBB disruption, and improve functional outcome (Colbourne et al., 1997). Hypothermia also has been shown to protect murine astrocytes and neuronal cells in culture against simulated ischemia in vitro (Shuaib et al., 1993).
The authors provide the first strong evidence that a moderate 32°C whole body hypothermia strongly reduces manifestations of inflammation in cerebral microvasculature, including leukocyte rolling and adhesion in pial venules. Both cerebral microvasculature (del Zoppo, 1997) and leukocytes (Barone et al. 1991; Matsuo et al., 1994) have been implicated in the pathogenesis of cerebral ischemia and stroke, yet few investigations into endothelial and leukocyte function and brain inflammatory responses during hypothermia have been performed. In a recent study, hypothermia has been shown to attenuate leukocyte rolling after focal cerebral ischemia in rat pial venules (Ishikawa et al., 1999).
The inflammation model used in this study was a systemic injection of IL-1β rather than a specific brain insult. It has been shown that endogenous IL-1β is a key mediator of endothelial activation, inflammation and secondary brain damage in cerebral ischemia and trauma (Rothwell, 1999). After the onset of focal cerebral ischemia, both IL-1β mRNA and protein are up-regulated within hours, and the levels of IL-1β expression correlate well with ischemic brain damage (Rothwell, 1999). Moreover, exogenous administration of IL-1β has been used in various inflammatory models to up-regulate inflammatory mediators, leukocyte rolling, and extravasation (Nourshargh et al., 1995).
The demonstration that 4-hour moderate hypothermia abolishes leukocyte rolling and reduces adhesion to cerebromicrovascular endothelium reveals a novel role for hypothermia in suppressing inflammatory brain responses. It is worth emphasizing that antiinflammatory effects of hypothermia in pial venules were seen only when the duration of hypothermia was protracted over a 4-hour period of IL-1β stimulation, whereas a transient 1-hour hypothermia was ineffective. These findings are in agreement with previous reports that hypothermia is neuroprotective only when maintained for an extended duration (Colbourne et al., 1997).
Molecular mechanisms involved in hypothermic suppression of leukocyte rolling and adhesion in vivo were investigated in human cerebral endothelial cells and neutrophils in vitro. Hypothermia was found to inhibit IL-1β–induced NF-κB activation, expression of IL-1β, and expression and secretion of IL-8 in HCEC, whereas it had no appreciable effect on neutrophil CD18 expression and chemotaxis. It has been reported that in vitro hypothermia of 25°C transiently inhibits transcriptional activation and surface expression of E-selectin and neutrophil adherence to human umbilical vein endothelial cells stimulated with IL-1 or TNFα (Haddix et al., 1996). This study provides additional evidence that antiinflammatory actions of hypothermia are caused by reduced activation of transcription factors that regulate inflammatory genes in endothelial cells, such as NF-κB. NF-κB is an important activator of inflammatory genes in cultured endothelial cells (Chen and Manning, 1995) and is up-regulated after diseases and insults characterized by inflammation (Manning et al., 1995). In addition to NF-κB, IL-1β has been shown to induce other transcription factors such as AP-1, C/EBP, and HIF-1, some of which cooperate with NF-κB in regulating and amplifying inflammatory gene expression (Shi et al., 1999). Both AP-1 and C/EBP binding sites have been mapped in the promoter region of ICAM-1 gene (Hou et al., 1994). Differing effects of hypothermia on NK-κB, AP-1, and C/EBP may explain why hypothermia failed to reduce IL-1β–induced ICAM-1 expression in HCEC despite the observed reduction in NF-κB activity.
The ability of hypothermia to reduce IL-1β–stimulated IL-8 expression and secretion in HCEC provides a tentative explanation for the observed reduction in leukocyte rolling and adhesion observed in pial circulation in vivo. Although various vascular beds may use different regulatory pathways to control leukocyte–endothelial interactions, the observations obtained in this study and in the literature suggest that hypothermia is capable of transiently inhibiting the expression of the activated phenotype of endothelial cells. Hypothermia may disrupt the rolling and adhesion cascade by reducing chemokine expression and elaboration by brain endothelium. Chemokines have been shown to play a key role in leukocyte activation and adhesion to vessel walls (Ransohoff, 1997). IL-8, a prototypic neutrophil chemoattractant shown to play a role in postischemic brain inflammation (Ransohoff, 1997), is produced by HCEC and human astrocytes (Zhang et al., 1999) in response to cytokines and hypoxia. Presentation of IL-8 by endothelial cells has been shown to be a seminal step allowing leukocytes to activate and firmly adhere to vessel walls. Furthermore, anti–IL-8 antibody has been shown to inhibit neutrophil-mediated tissue injury, neutrophil infiltration, and to reduce cerebral infarct (Mukaida et al., 1998). Because hypothermia does not affect ICAM-1 expression by HCEC or CD18 expression by neutrophils, it is possible that under hypothermic conditions leukocytes fail to activate and adhere to endothelium caused by reduced expression and secretion of IL-8 by endothelial cells. However, the possibility cannot be excluded that hypothermia changes the affinity of neutrophil integrins for endothelial ligands or causes neutrophil compartmentalization in the circulation such that it prevents effective neutrophil interactions with endothelial cells.
In vivo inflammatory phenomena are influenced by shear flow rates, vascular tone, and other adjacent cell types that are all absent in HCEC culture model. For example, the authors recently have shown that human astrocytes and HCEC cooperate in regulating endothelial expression of inflammatory cytokines and chemokines (Zhang et al., 2000). In addition to reducing proinflammatory activation of brain endothelial cells, hypothermia likely influences other components of the cerebromicrovascular bed to affect changes in physiologic responses observed in vivo. Regardless, the combined in vitro and in vivo approach used in this study has generated valuable clues into potential molecular equivalents of antiinflammatory actions of hypothermia.
Although this study does not provide a direct experimental link between antiinflammatory actions of moderate hypothermia and neuroprotection, it is tempting to speculate that suppression of leukocyte rolling, adhesion, and ultimately infiltration into the injured brain is an important mechanism by which hypothermia affords neuroprotection. As neuroprotection provided by hypothermia represents one of the most promising therapeutic tools for treating TBI and ischemia–reperfusion, this new understanding of antiinflammatory actions of hypothermia may assist in the development of new therapeutic strategies for treating brain diseases characterized by inflammation, such as a combination of transient hypothermia and antiinflammatory drugs.
Footnotes
Acknowledgments:
The authors thank Dr. Pierre Lemieux for providing an NF-κB reporter plasmid vector, Drs. Charlie Thompson and Wandong Zhang for advice and guidance, and Cathie Smith and Bill Weiss for expert technical assistance.