
Abstract
The 72-kD inducible heat shock protein (HSP72) can attenuate cerebral ischemic injury when overexpressed before ischemia onset. Whether HSP72 overexpression is protective when applied after ischemia onset is not known, but would have important clinical implications. Fifty-seven rats underwent middle cerebral artery occlusion for 1 hour. Defective herpes simplex viral (HSV) vectors expressing hsp72 with lacZ as a reporter were delivered 0.5, 2, and 5 hours after ischemia onset into each striatum. Control animals received an identical vector containing only lacZ. Striatal neuron survival at 2 days was improved by 23% and 15% when HSP72 vectors were delayed 0.5 and 2 hours after ischemic onset, respectively (P < 0.05). However, when delayed by 5 hours, HSP72 overexpression was no longer protective. This is the first demonstration that HSP72 gene transfer even after ischemia onset is neuroprotective. Because expression from these HSV vectors begins 4 to 6 hours after injection, this suggests that the temporal therapeutic window for HSP72 is at least 6 hours after ischemia onset. Future strategies aimed at enhancing HSP72 expression after clinical stroke may be worth pursuing. The authors suggest that in the future HSP72 may be an effective treatment for stroke.
The stress response has been identified as a highly conserved mechanism of coordinated reprogramming of gene expression in response to environmental challenges. Although the synthesis of most proteins is decreased, the transcription of certain stress response genes is up-regulated. Among these, heat shock proteins (HSPs) are induced by a variety of central nervous system insults, such as cerebral ischemia, seizures, neurotoxin exposure, and other metabolic stress (Massa et al., 1996; Sharp et al., 1999). The 72-kD HSP (HSP72, also known as the inducible form of HSP70) has been studied extensively and is thought to function as a molecular chaperone, thus preventing premature protein folding and facilitating protein translocation to target areas. Heat shock proteins also may remove denatured proteins and assist in new protein synthesis (Welch, 1993; Kiang and Tsokos, 1998). It has been debated extensively whether cells expressing HSP72 after cerebral ischemia will die or survive. Therefore, the neuroprotective role of HSP72 has been controversial. With the availability of transgenic animals or viral vector mediated gene transfer, it is now possible to investigate the specific role of different proteins. The authors previously showed that overexpressing HSP72 in cultured neurons protected them from heat shock (Fink et al., 1997) and that gene therapy with HSP72 is neuroprotective in rat models of stroke and epilepsy when delivered before the insult (Yenari et al., 1998a). Others (Plumier et al., 1997; Rajdev et al., 1999) have similarly demonstrated that transgenic mice overexpressing HSP70 are protected against stroke.
For gene therapy to have more clinical relevance, the authors investigated here whether delayed, postischemic application of the viral vector protects striatal neurons against transient middle cerebral artery occlusion (MCAO) in rats.
MATERIALS AND METHODS
Amplicon plasmids
The construction of amplicon plasmids pα22βgalα4hsp72 and pα22βgalα4s has been described in detail elsewhere (Lawrence et al. 1995; Fink et al., 1998). Briefly, the rat inducible HSP72 was isolated from a pHSP-8 clone (provided by Drs. S. M. Massa and F. R. Sharp, University of California, San Francisco, CA), ligated with a polyadenylation (polyA) signal of the human cytomegalovirus immediate early gene and inserted into the pα22βgal downstream of the HSV α4 promoter. The pα22βgal contained the E. coli lacZ gene, a simian virus 40 polyA signal and α22 of HSV as promoter (Fink et al., 1998). Because both promoters are immediate early genes of HSV with similar kinetics, the authors have previously shown coexpression of the two genes in this bicistronic system (Ho 1994; Fink et al., 1997; Yenari et al., 1998a). The HSV oriS and the “a” sequence also were included to provide the necessary cis-signals for replication and packaging of the amplicon DNA. pα22βgalα4s, which lacks the HSP72 gene and contains a stop codon after the promoter (“s”), was used to produce control vector.
HSV vector
Protocols for generating viral vectors for this study using the amplicon system have been previously described in detail (Ho, 1994, Yenari et al., 1998a). Briefly, pα22βgalα4hsp72 and pα22βgal were transfected into E5 cells using Liptofectamine according to the manufacturer's protocol. Twenty-four hours after transfection with the plasmid, cultures were superinfected with helper virus d120 (multiplicity of infection 0.1). These cells were harvested when 100% cytopathic effect was developed. Stocks were further purified by centrifugation and resuspension in phosphate-buffered saline (PBS). The titers of helper virus were determined on E5 cells by using a standard plaque assay. The titers of amplicon vectors were determined on Vero cells by quantifying the number of β-galactosidase (β-gal is the gene product of lacZ) expressing cells. Assays revealed plasmid titers of 5.2 to 9.1 × 106, 1.3 to 1.91 × 106, 4.0 × 107 (vectors/mL) and d120 helper virus titers of 1.84 to 2.75 × 107, 4.25 to 9.13 × 106, 6.25 × 107 (PFU/mL) of experiments at 30 minutes, 2, and 5 hours respectively. Plasmid helper virus ratios were 1:3 (0.5 hour), 1:3.5 (2 hours), 1:1.6 (5 hours).
In vitro ischemia.
Pure neuronal cell cultures were prepared from fetal mice (15-day gestation). Tissue was minced, suspended in Eagle's minimal essential medium (MEM), supplemented with 21 mmol/L glucose, trypsinized, tritunated, and plated in 24-well plates coated with poly-d-lysine and laminin at a density of 3.5 hemispheres per plate. Plating media contained 10% fetal bovine serum. Twenty-four to 28 hours after plating, 60% of culture medium was replaced with glial conditioned medium and cytosine arabinoside (Ara-C, 3 mmol/L). Ten to 11 days later, cultures were transferred into an anaerobic chamber (Forma Scientific; O2, tension < 0.02%), washed into deoxygenated, glucose-free balanced salt solution (BSSO), and incubated at 37°C within the anaerobic chamber for 1 hour. Oxygen–glucose deprivation was ended by replacing the medium with MEM supplemented with glucose (20 mmol/L) sodium bicarbonate (26 mmol/L) and returning the cultures to the normoxic incubator. Cells were collected for Western blots 3 hours later.
Western blots.
Cell lysates from the neuronal cultures were separated on 10% sodium dodecyl sulfate polyacrylamide gel electrophoresis. Protein bands were electrotransferred onto polyvinylidene difluoride membranes. The membranes were stained with Ponceau Red to confirm equal protein loading and transfer, then probed for inducible HSP72. Membranes were blocked with 5% nonfat dry milk (in PBS, pH 7.4) for 1 hour, then probed for 2 hours with an antibody to detect inducible HSP72 (1:500 dilution), SPA–810, mouse monoclonal (92). Membranes were washed with PBS containing 0.1% Tween 20 and were incubated with horseradish peroxidase conjugated anti-rabbit IgG for 1 hour. Bound antibody was visualized with the RCL system (Amersham) according to the manufacturer's directions. Optical densities of the autoradiograms were measured using a computer-assisted program (BioRad multianalyst GS 700; Hercules, CA, U.S.A.).
Focal cerebral ischemia
The Stanford University Administrative Panel of Laboratory Animal Care approved all experimental protocols. Fifty-seven male Sprague-Dawley rats (Charles River, Wilmington, MA, U.S.A.) weighing 280 to 310 g were initially anesthetized with 3% halothane plus oxygen and air (ratio: 0.2/0.8 L/min) by face mask. When surgical plans of anesthesia were attained, halothane was decreased to 1% to 1.5%. Depth of anesthesia was assessed by hindleg withdrawal every 15 minutes throughout the procedure. After exposing the skull, burr holes were drilled at coordinates AP: 0 mm, ML: 3.5 mm, relative to Bregma. Dura mater was kept intact and the animal was prepared for middle cerebral artery occlusion. The common carotid (CCA), external carotid, and pterygopalatine arteries were exposed and ligated on the left side. The left internal carotid artery (ICA) was transiently occluded with a microsurgical clip, and an arteriotomy was made in the CCA. A 3.0-monofilament suture (Ethicon, Sommerville, NJ, U.S.A.) with a rounded tip was inserted into the CCA and advanced through the ICA to the ostium of the MCA to occlude the MCA (Longa et al., 1989). The suture was left in place for 1 hour and then removed to allow reperfusion for 48 hours. At the appropriate time point after the onset of ischemia, rats were positioned in a stereotactic frame and viral vector was injected 0.5, 1, and 5 hours after MCAO. Using a Hamilton syringe (Hamilton Company, Reno, NV, U.S.A.), 2.5 μL viral vector was injected at coordinates AP: 0 mm, ML: 3.5 mm, relative to Bregma and depths of 4.5 mm and 5.5 mm in both striata. The first injection was on the ischemic side, followed by the nonischemic side 15 minutes later. After the injections were completed, wounds were closed and the animal was allowed to recover. Animals then were taken to the animal intensive care unit for postoperative monitoring. Throughout the procedure temperature, electrocardiogram, and respiration rate were monitored and kept in physiologic ranges.
Histopathology and cell counts
At 2 days after ischemia, animals were killed by an overdose of halothane and were transcardially perfused with 75 mL normal saline followed by 75 mL of 3% paraformaldehyde (PFA)/20% sucrose solution.
After postfixing in 3% PFA/20% sucrose solution for 1 to 2 days, 30-μm frozen sections in the coronal plane were taken at 100-μm increments 1 mm anterior and posterior to the needle track. Slices were stained with X-gal (5′-Bromo-4-chloro-3indoly-β-D-galactopyranoside; Molecular Probes, Eugene, OR, U.S.A.), a chromogenic substrate for β-gal, and counterstained with cresyl violet to allow the identification of healthy, intact, and virally targeted neurons. Brains without infarction were excluded from the analysis. The number of positive-staining, teal-colored neurons from 10 consecutive slices (5 anterior and 5 posterior to the injection site) was counted at 40× magnification by an investigator blinded to treatment groups. The number of surviving neurons was expressed as the ratio of positive blue neurons in the ischemic striatum compared with the contralateral nonischemic striatum. Differences in survival could be inferred by the severity of the ischemia; therefore, the authors examined brains for infarct size. From the cresyl violet (Sigma Chemicals, St. Louis, MO, U.S.A.) stained sections, infarct sizes also were graded using a semiquantitative scale: 0, no stroke; 1, stroke in striatum only; 2, striatum and some cortex; 3, complete distribution of MCA. This scale previously has been shown to reasonably reflect the quantitative measurement of infarct size (Yenari et al., 2001).
TUNEL staining.
To determine whether other forms of cell death may be caused by the vectors themselves, the authors injected uninjured rat brains (3 rats per group) with vectors and normal saline. After X-gal staining, TUNEL stains were performed to determine whether these vectors might cause cell death by DNA damage such as apoptosis. TUNEL staining was performed according to kit (ApopTag kit; Intergen Purchase, NY, U.S.A.). Briefly, PFA-fixed tissue was treated with proteinase K for 15 minutes and then washed. TdT enzyme then was applied in a 30/70 enzyme to buffer ratio and incubated at 37° for 1 hours and then washed. Anti-digoxigenin conjugate was applied for 30 minutes, then washed, followed by diaminobenzidine (Sigma) to visualize nuclei and vector targeted neurons. Positive controls were thymus pretreated with DNAase and then proceeded as above.
Statistical analysis.
Standard statistical methods were used to analyze the data. Differences in survival between groups were determined by Student's t-test. Differences in infarct size were determined using the Mann–Whitney U test. Statistical significance was determined as P < 0.05. All data are presented as mean ± SD.
RESULTS
No differences were found in infarct scores between the treated and untreated groups, implying that the severity of ischemia was similar between groups (Table 1). The striatum was infarcted in all animals, and a few brains showed infarction in the overlying cortex as well. No significant differences were found in temperature, respiratory rate, or heart rate between the groups. Several hundred neurons could be transfected by the authors' method with approximately 10% overall efficiency of infection.
Infarct score data
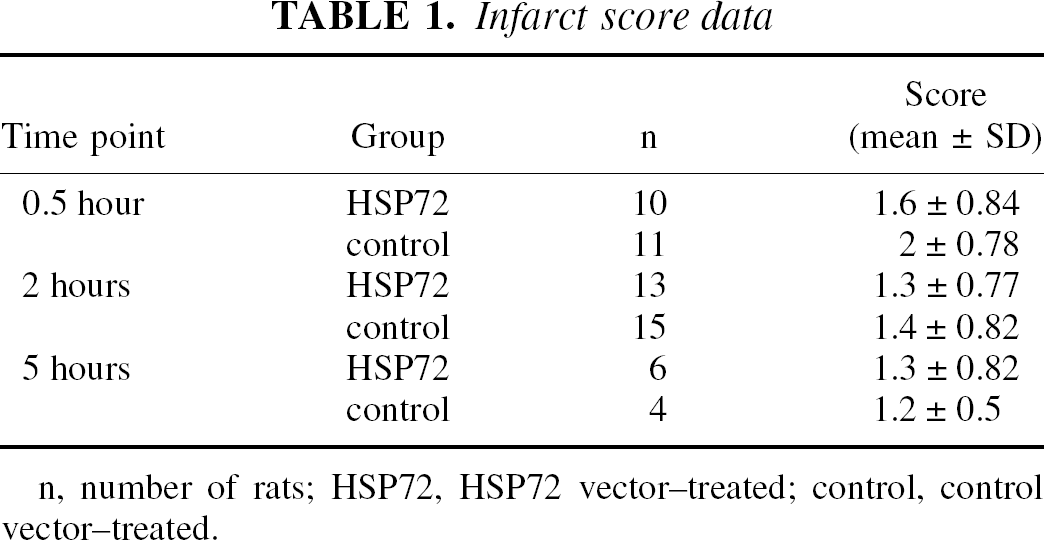
n, number of rats; HSP72, HSP72 vector–treated; control, control vector–treated.
Western blot analysis of cell extracts from neuronal cultures transfected with Hsp72 increased Hsp72 protein expression. Hsp72 showed greater than 10-fold higher Hsp72 vectors compared with control vectors (α4s) and more than 100-fold after mock transfection (PBS) (Fig. 1A) in noninjury paradigm. Similarly, Hsp72 vector transfection increased Hsp72 protein expression by 4-fold in cultured neurons after oxygen–glucose deprivation compared with control vector transfection, and 6-fold higher compared with mock transfection (Fig. 1B).
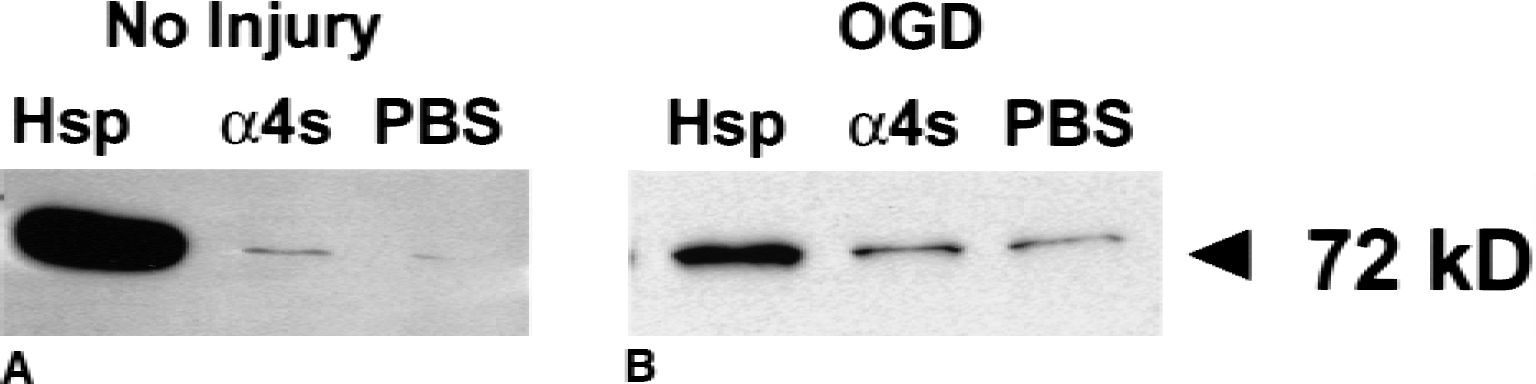
Hsp72 gene transfer increases Hsp72 protein expression. Western blots of cell extracts from cultured neurons show that Hsp72 is increased greater than 10-fold after transfection by Hsp72 vectors (Hsp), compared with control vectors (α4s), and more than 100-fold after mock transfection (phosphate-buffered saline, PBS)
The authors previously showed that helper virus injection did not lead to gross neurotoxicity (Ho, 1994). The authors found here that TUNEL staining was absent in brains of vector (Hsp72 and control vectors) and saline-injected rats as late as 3 days after injection (data not shown).
HSP72 overexpression improved striatal neuron survival when vectors were delivered 0.5 to 2 hours after ischemia onset (Figs. 2 and 3). When HSP72 vectors were delivered 0.5 hour after ischemia onset, striatal neuron survival was 23% greater compared with control vector injected (P < 0.05) (Fig. 3A). When vector was delayed by 2 hours, survival was 15% greater than control (P < 0.05) (Fig. 3B). However, when delayed by 5 hours, HSP72 overexpression was no longer protective (Fig. 3C). Table 2 shows the absolute number of neurons counted for each time point. Analysis of absolute counts revealed significance only at the 30-minute time point. Therefore, by using each animal as its own internal control, the authors were capable of reducing the variability and increasing the statistical power of this model.
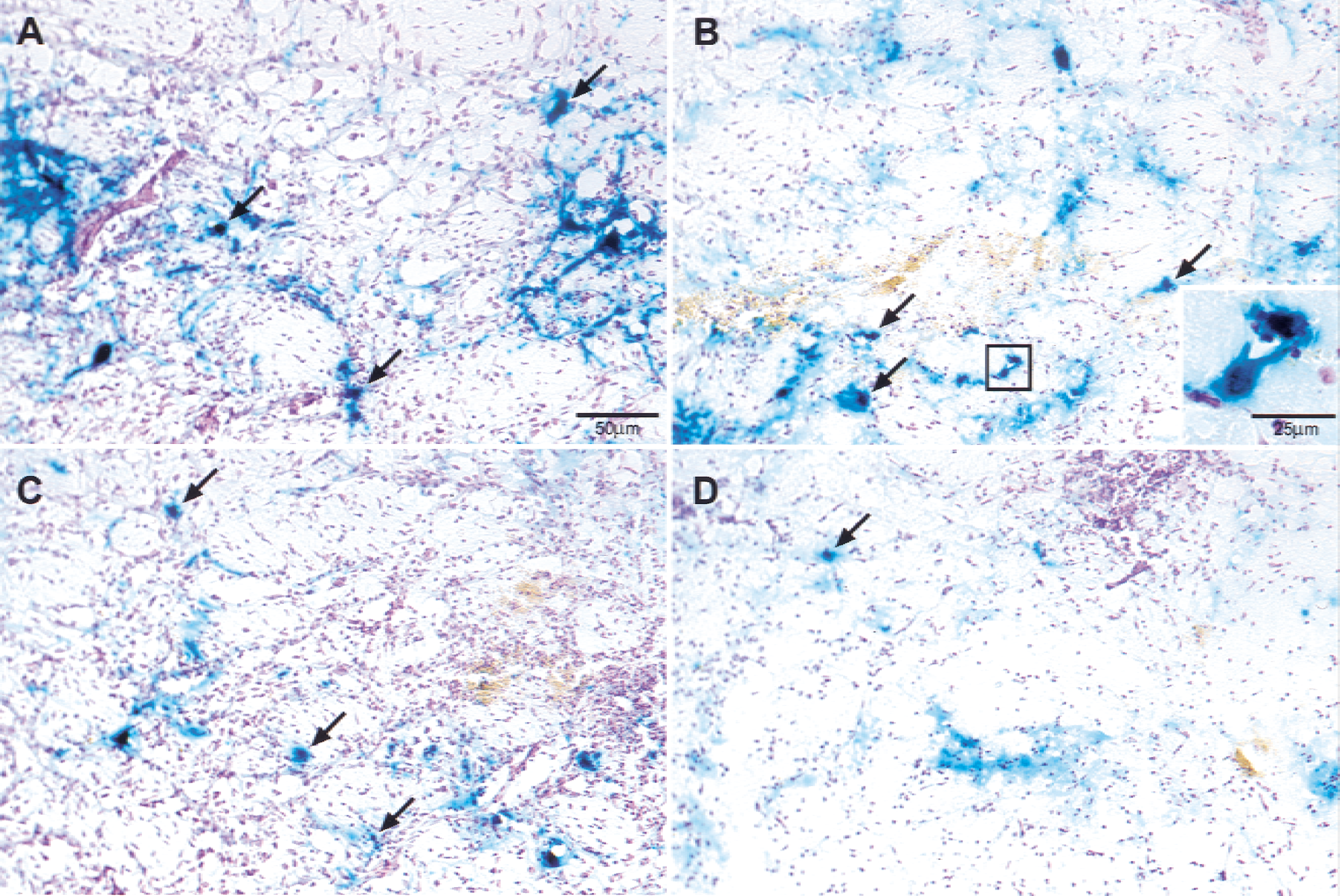
Several surviving vector targeted, X-gal-positive neurons (arrows) are observed in nonischemic
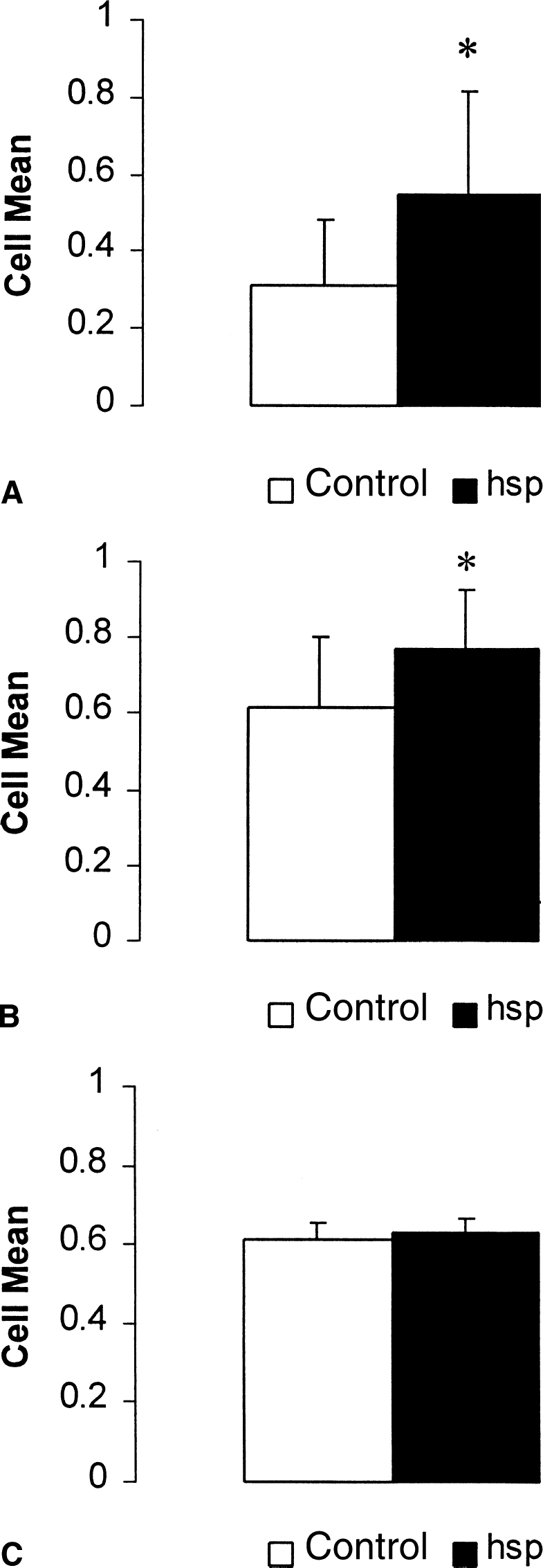
Striatal neuron survival after 1-hour middle cerebral artery occlusion and viral vector injection (HSP72 or control) at 0.5
Absolute counts of surviving vector-targeted neurons, 48 hours after 1-hour MCAO

P < 0.05.
DISCUSSION
The authors previously have shown that HSP72 overexpression protected neurons against experimental stroke and epilepsy (Yenari et al., 1998a). However, in that study, viral vector was delivered 12 hours before stroke onset. In this study, the authors show that postinsult HSP72 overexpression protects striatal neurons against stroke as well. To the authors' knowledge, this is the first study demonstrating effective postischemic gene transfer with HSP72.
Studies in which HSP72 was overexpressed have shown that HSP72 protected cultured neurons against heat shock (Uney et al., 1993; Fink et al., 1997) and ischemialike insults (Papadopoulos et al., 1996; Amin et al., 1996). Glial cells also were protected against similar insults (Uney et al., 1993; Xu and Giffard, 1997; Lee, 2001). At the whole animal model level, such mice overexpressing HSP72 were protected against MCAO (Radford et al., 1996; Plumier et al., 1997) and myocardia ischemia (Plumier et al., 1995). Radjdev and colleagues (Rajdev et al., 1999) have shown that HSP72 reduced the overall infarct size in transgenic mice. However, other groups could not demonstrate any differences of infarct size using similar paradigms in other HSP72 transgenic strains (Plumier et al., 1997; Lee, 2001). The reason for these discrepancies is not known, but they could be caused by a lower level of HSP72 overexpression in the latter transgenic strains.
The authors have previously shown using similar bipromoter HSV vectors that transferred genes are capable of altering cell physiology. Ho et al. (1993) demonstrated that transfection with a HSV vector expressing glut-1 gene increased glucose uptake in the brain, and injection of a gene encoding calbindin D28K led to reduced calcium ion mobilization after hypoglycemia (Meier et al., 1997).
The mechanism of neuroprotection by HSP72 is not yet known; however, it may be related to its chaperone functions leading to the prevention of protein malfolding and aggregation (Kiang and Tsokos, 1998; Yenari et al., 1999). Interestingly, although the protein binding properties of HSP72 are adenosine triphosphate–dependent (Freeman et al., 1995), HSP72 variants with mutations in the adenosine triphosphate binding domain still protect tumor cells from heat shock (Li et al., 1992). Ischemic injury, in particular, is notable for a reduction in metabolic stores. Therefore, an adenosine triphosphate–independent mechanism underlying the observed neuroprotection is plausible, but has yet to be studied in the appropriate models. Others studies have shown that in some systems it can prevent apoptosis (Mosser et al., 2000) and inhibit activation of the transcription factor NF-κB. In nonneuronal cells there is evidence that HSP72 prevents apoptosis. Jaattela and colleges (Jaattela et al., 1998) showed that HSP72 could prevent apoptosis downstream of caspase activation. However, others have demonstrated that HSP72 can prevent the release of cytochrome c from mitochondria, thus preventing the formation a functional apoptosome and subsequent processing of procaspase-9 and −3 (Mosser et al., 2000). Although these studies suggest that HSP72 prevents apoptosis, it has yet to be convincingly shown in the brain. HSP72 also may protect by regulating the transcription of potentially damaging genes. Heneka et al. (2000) reported that HSP72 inhibits the transcription factor NF-κB and NOS-2 activation in an in vivo model of cerebral inflammation. Feinstein et al. (1996) reported that heat shocked glial cells had elevated HSP72 and reduced nuclear NF-κB levels. Selectively overexpressing HSP72 also inhibited NF-κB translocation. Because inflammation and apoptosis may occur late in the ischemic cascade, this could partially explain the observed protection here when HSP72 was overexpressed after insult.
The HSP72 vector used for this experiment contains hsp72 and lacZ genes under control of two separate promoters. Reporter gene expression began as early as 4 to 6 hours after vector injection, peaked at approximately 12 to 24 hours, and decreased after 24 hours, but counts were still 25% to 30% of peak counts from days 2 through 4 (Fink et al., 1997; Lawrence et al., 1997; Yenari et al., 1998a). The authors have shown previously that vector-infected neurons coexpress HSP72 and β-gal in vivo for at least 18 hours after injection (Yenari et al., 1998b) and in vitro for at least 40 hours (Fink et al., 1997), but no longer than 60 hours. The authors also have studied transfection with α4s control vector alone and observed that it does not induce an endogenous HSP72 (Yenari et al., 1998a). Interestingly, β-gal staining was still observed at 60 hours after injection even though HSP72 expression was absent. This is likely because of the longer tissue half-life of β-gal compared with HSP72. Although the duration of expression of the 2 genes may be different, both the α4 and α22 promoters are immediate early genes of HSV and show similar, though not necessarily identical, kinetics of expression (Ho et al., 1995).
In the current study, viral vector delivery was performed approximately 0.5, 2, and 5 hours after stroke onset. As described above, vector expression began 4 to 6 hours after injection; therefore, vector expression began as early as 4.5 (0.5-hour delay) to 6 hours (2-hour delay) after ischemia onset where protection was observed. No protection was observed when vector was delayed 5 hours (with expression beginning as early as 9 hours). Notably, endogenous HSP72 protein expression begins 4 to 6 hours after insult (Welsh et al., 1992; Kinouchi et al., 1993). Therefore, gene transfer may have bolstered the endogenous HSP72 expression by additional HSP synthesis and persistence of HSP72 for several hours. Gene transfer at these times may act to improve the cell's natural defense mechanisms.
Consequently, the current results imply a potential therapeutic temporal window for HSP72 overexpression up to 6 hours after ischemia onset. This postinsult neuroprotective effect has potential clinical implications for future gene therapy using HSP72 or other strategies that cause HSP72 overexpression (Yenari et al., 1998b; Sapolsky and Steinberg, 1999). Furthermore, the authors believe this is the first study to show that postischemia Hsp72 overexpression is protective. Although it is shown here that gene therapy with HSP72 is feasible, there are still limitations regarding the extent and number of neurons that these vectors can infect, as well as the route of administration. The authors could not alter overall infarct size, which is likely because of the relatively small number of cells infected by the current vector. Therefore, future studies should explore improved vector delivery systems or further purification to higher titers. In addition, the duration of HSP72 overexpression necessary for neuroprotection should be investigated in more detail to evaluate the efficacy of HSP72 as a postinsult therapeutic tool against stroke.
Footnotes
Acknowledgments:
The authors thank Dr. N. A. DeLuca, University of Pittsburgh, Pittsburgh, Pennsylvania, for providing HSV d120 and E5cell lines, and Guo Hua Sun, David Kunis, Danye Cheng, and Beth Houle for preparation of the figures.