
Abstract
The current study was designed to determine the effect of recombinant heme oxygenase-1 (HO-1) gene expression on endothelial function in cerebral arteries. Isolated canine basilar arteries were exposed ex vivo (30 minutes at 37°C) to an adenoviral vector (1010 PFU/mL, total volume 300 μL) encoding either the HO-1 gene (AdCMVHO-1) or the β-galactosidase (β-Gal) reporter gene (AdCMVβ-Gal). Twenty-four hours after transduction, arterial rings were suspended in organ chamber for isometric force recording. Endothelium-dependent relaxations were obtained in response to bradykinin (10−10 to 10−6 mol/L) during contraction to uridine-5′-triphosphate (UTP; 3 × 10−6 to 3 × 10−5 mol/L). Certain rings were incubated with oxyhemoglobin (OxyHb; 10−5 mol/L) overnight (16 to 18 hours of 24 hours). Expression and localization of recombinant protein were shown by Western blot analysis and immunohistochemistry. Endothelium-dependent relaxation to bradykinin and endothelium-independent relaxation to forskolin (10−9 to 10−5 mol/L) and DEA-NONOate (10−10 to 10−5 mol/L) were identical in β-Gal– and HO-1–transduced arteries. Exposure to OxyHb caused impairment of endothelium-dependent relaxation to bradykinin (P < 0.01). In contrast, OxyHb did not affect endothelium-dependent relaxation in arteries expressing recombinant HO-1 (P > 0.05). This protective effect of HO-1 was reversed by coincubation with tin protoporphyrin (SnPP9; 10−5 mol/L), a selective inhibitor of HO-1 (P < 0.01). Basal levels of 3′,5′-cyclic monophosphate (cGMP) in HO-1–transduced vessels were not significantly different from those in β-Gal–transduced vessels. Pretreatment with OxyHb significantly reduced cGMP level in β-Gal–transduced rings (P < 0.01), whereas it had no effect in HO-1–transduced rings. These results demonstrate that HO-1 gene transfer does not affect endothelial and smooth muscle function of normal arteries, and that expression of recombinant HO-1 in cerebral arteries protects vasomotor function against OxyHb-induced injury.
Cerebral vasospasm is a frequent and potentially life-threatening complication in survivors of subarachnoid hemorrhage. Oxyhemoglobin (OxyHb) plays an important role in pathogenesis of vasospasm. It produces contraction of cerebral arteries either by direct contractile effect on smooth muscle cells or by scavenging endothelial nitric oxide (NO) (Cook and Vollrath, 1995). Furthermore, autooxidation of OxyHb releases free radicals, and degradation of OxyHb increases free iron and heme that, in turn, may cause oxidative injury (Misra and Fridovich, 1972).
Heme oxygenase (HO) is a rate-limiting enzyme in heme degradation (Maines, 1997). Accumulating evidence suggests that HO-1 is a stress-response protein induced by a variety of stimuli including heat shock, cytokines, hypoxia, and hemolysis (Otterbein et al., 1999; Amersi et al., 1999; Abraham et al., 1995; Soares et al., 1998; Hancock et al., 1998). Moreover, enzymatic activity of the inducible isoform of HO-1 appears to play an important role in hemoglobin clearance and the pathogenesis of cerebral vasospasm (Suzuki et al., 1999).
Previous studies demonstrated that adenovirus-mediated delivery of HO-1 protects lungs and liver from oxidative injury, supporting the idea that HO-1 activity is an essential mechanism responsible for maintenance of cellular homeostasis (Otterbein et al., 1999; Amersi et al., 1999). The effect of recombinant HO-1 expression on vasomotor function of intact cerebral arteries has not been studied. The authors hypothesized that expression of recombinant HO-1 may protect the cerebral arterial wall against OxyHb-induced injury. Adenovirus-mediated gene transfer was used to characterize the effect of recombinant HO-1 on the vasomotor reactivity of isolated cerebral arteries and to determine whether overexpression of HO-1 can protect the vascular wall against OxyHb-induced injury.
MATERIALS AND METHODS
Preparation of oxyhemoglobin
Oxyhemoglobin was prepared according to the method of Martin et al. (1985) with some modification. Bovine hemoglobin (Sigma Chemical, St. Louis, MO, U.S.A.), 330 mg in 5 mL perfusion solution (10−3 mol/L of hemoglobin solution), was reduced to OxyHb by addition of 35 mg sodium hydrosulfite (Na2 S2 O4; Sigma) and was separated by passage through a 250-mL Sephadex G50 column (Sigma). The concentration of the OxyHb was determined by a co-oximeter (Instrumentation Laboratory, Lexington, MA, U.S.A.). Commercially available OxyHb may be contaminated by phospholipids, pyrogens, and free iron (D'Agnillo and Alayash, 2000). The possibility exists that these contaminants may in part contribute to the effect of OxyHb.
Construction, propagation, and purification of adenoviral vectors
A 1.0-kbp XhoI-Hind III fragment from the rat HO-1 cDNA clone pRHO-1 (Shibahara et al., 1985) containing the entire coding region was cloned into plasmid pAC-CMVpLpA. Recombinant HO-1 adenovirus Ad5-HO-1 was generated by homologous recombination in 911 cells after cotransfection with the pAC-HO-1 plasmid and plasmid pJM17. Isolation, propagation, and titering of recombinant adenovirus were performed as described previously (Kolls et al., 1994). AdCMVβ-Gal was used in all gene transfer experiments as a control; it was propagated, isolated, and quantified as described elsewhere (Chen et al., 1997).
Gene transfer
All procedures were in accordance with Institutional Animal Care and Use Committee guidelines of Mayo Clinic. Rings (4-mm-long) of basilar arteries were taken from mongrel dogs (18 to 27 kg) anesthetized with 30 mg/kg intravenous Pentothal (Abbott Laboratories, Abbott Park, IL, U.S.A.). To remove blood, arterial rings were gently rinsed with cold (4°C) modified Krebs-Ringer bicarbonate solution (control solution; composition in mmol/L: NaCl, 118.3; KCl, 4.7; CaCl2, 2.5; MgSO4, 1.2; KH2 PO4, 1.2; NaHCO3, 25.0; calcium-ethylenediamine-tetraacetic acid, 0.026; and glucose, 11.1). Loose perivascular tissue was removed carefully and rings were cut (4-mm-long). Rings were randomly assigned for gene transfer and were transduced with adenoviral vectors (1010 pfu/mL, total volume = 300 μL) in minimal essential medium (MEM; containing 0.1% BSA, 100 U/mL penicillin, and 100 μg/mL streptomycin) for 30 minutes at 37°C. Rings then were transferred to fresh MEM and were incubated for 24 hours at 37°C in a CO2 incubator (5% CO2; Forma Scientific). Certain rings were incubated with OxyHb (10−5 mol/L) or tin protoporphyrin (SnPP9; 10−5 mol/L) overnight (16 to 18 hours). The next day, rings were suspended for isometric force recording in an organ chamber. The viral titer and incubation time were considered to be optimal for ex vivo gene transfer on the basis of results of the authors' previous studies (Chen et al., 1997; Tsutsui et al., 1998). Nontransduced arteries used as control for certain experiments were incubated in MEM alone for 24 hours.
Western blot analysis of recombinant heme oxygenase-1
Soluble proteins were extracted by mincing and homogenizing tissues in lysis buffer (pH7.5) containing 50 mmol/L Tris-HCl, 0.1 mmol/L EDTA, 0.1 mmol/L EGTA, 0.1% sodium dodecyl sulfate, 0.1% deoxycholate, 1% IGEPAL, and a 1000-fold dilution of a mammalian protease inhibitor cocktail (all from Sigma). The homogenate was rotated for 1 hour at 4°C and then centrifuged (14,000 g) for 5 minutes. After centrifugation, the supernatant was collected and the total protein concentration was measured by DC Protein Assay Kit (Bio-Rad, Hercules, CA, U.S.A.). Prestained protein markers (Bio-Rad), recombinant heat shock protein 32 (HSP32) as a positive control, and 150 μg of sample were loaded into precast 15% sodium dodecyl sulfate–polyacrylamide gel electrophoresis gels (Bio-Rad). The resolved proteins were transferred to 0.2 μm nitrocellulose membrane on a semi-dry electrophoretic transfer cell (Bio-Rad) for Western blot analysis. Blots were blocked overnight (4°C) and incubated with rabbit anti–HO-1 polyclonal antibody (1:500, Stressgen, Victoria, BC, Canada) at room temperature. Bands were visualized by ECL (Amersham, Arlington Heights, IL, U.S.A.).
Immunohistochemical analysis of gene expression
Recombinant HO-1 protein expression was shown by immunohistochemical staining of transduced arterial rings that were frozen in O.C.T. compound (Sakura Torrance, CA, U.S.A.). Five-micrometer cross-sections were cut and dried onto slides for 45 minutes (37°C). Sections then were fixed in cold acetone, and endogenous peroxide was blocked using 0.1% sodium azide/0.3% hydrogen peroxide. Nonspecific protein binding was blocked with a 30-minute incubation in 10% normal rabbit serum/0.5% Tween 20 in phosphate-buffered 0.5 mol/L saline. Sections then were incubated for 1 hour with a monoclonal antibody (2.2 μg/mL) raised against native rat HO-1 (Stressgen). The secondary antibody was biotinylated rabbit anti-mouse IgG, followed by peroxidase-conjugated streptavidin. Color development used 3-amino-9-ethylcarbazole and hematoxylin counterstaining. For control studies, specificity of HO-1 immunolabeling was examined by the following three methods: (1) omission of the primary antibody in the incubation medium, (2) HO-1 immunostaining of AdCMVβ-Gal–transduced vessels, and (3) immunostaining of AdCMVHO-1–transduced vessels with an isotype-matched nonrelevant antibody.
Analysis of vascular reactivity
Twenty-four hours after gene transfer, rings were connected to an isometric force-displacement transducer (Grass FT03; Grass Instrument, Quincy, MA, U.S.A.) and suspended in an organ chamber filled with 25 mL of control solution (37°C, pH 7.4) aerated with 94% O2/6% CO2. Isometric tension was recorded continuously. Rings were allowed to stabilize at a resting tension of 0.2 to 0.4 g for 1 hour. Each ring then was gradually stretched to the optimal point of its length-tension curve (approximately 3.0 g) as determined by the contraction to 10−5 mol/L uridine 5′-triphosphate (UTP). All experiments were conducted in the presence of 10−5 mol/L indomethacin to eliminate the possible influence of endogenous cyclooxygenase. Incubation time with indomethacin or l-nitroarginine methylester (L-NAME) was 30 or 15 minutes, respectively. To evaluate relaxation responses, the rings were contracted with median effective concentration (EC50) of UTP (3 × 10−6 to 3 × 10−5 mol/L) before the addition of agonists. Concentration-response curves were obtained in a cumulative fashion. Several rings prepared from the same artery were studied in parallel. Relaxations were expressed as a percentage of maximal relaxations induced by 3 × 10−4 mol/L papaverine.
Measurement of 3′,5′-cyclic monophosphate
A radioimmunoassay technique was used to determine the levels of 3′,5′-cyclic monophosphate (cGMP). Rings were initially incubated in MEM, in a CO2 incubator at 37 °C for 30 minutes. Then the rings were incubated another 30 minutes in 3-isobutyl-1-methylxanthine (IBMX; 10−4 mol/L) to inhibit the degradation of cyclic nucleotides by phosphodiesterases. Then the rings were removed from the medium and were frozen quickly in liquid nitrogen. After homogenization, cGMP levels were measured by a cGMP radioimmunoassay kit (Amersham). Protein assay was conducted by DC Protein Assay Kit (Bio-Rad). Rings taken from the same dogs were studied in parallel.
Drugs
The following pharmacologic agents were used: UTP, bradykinin, papaverine hydrochloride, forskolin, L-NAME, indomethacin, IBMX, hemoglobin, sodium hydrosulfite, Sephadex G-50 (Sigma), DEA-NONOate (Cayman Chemical, Ann Arbor, MI, U.S.A.), and SnPP9 (ICN Biomedicals, Costa Mesa, CA, U.S.A.). Drugs were dissolved in distilled water, and volumes of less than 0.15 mL were added to the organ chambers. Solutions of DEA-NONOate and forskolin in the highest concentration were prepared in 1.5 mol/L Tris (pH 8.8) and DMSO (Sigma), respectively. Concentrations of all drugs are expressed as the final mol/liter concentration in the control solution.
Statistical analysis
Results of this study are expressed as mean ± SD. Four to eight rings were cut from one basilar artery. Results were obtained by repeating the given protocol in basilar arteries obtained from 5 to 8 dogs in each experimental group. “n” refers to the number of the dogs. An unpaired Student's t-test was used to detect significant differences between the two groups. Factorial analysis of variance followed by Bonferroni/Dunn's post hoc test was used to detect significant differences in multiple comparisons. Half-maximal effective concentrations (EC50) were calculated by nonlinear regression and were expressed as −log mol/L. Concentration-response curves were analyzed by repeated measured analysis of variance followed by Bonferroni/Dunn's post hoc test. Statistical significance was accepted as P < 0.05.
RESULTS
Expression and localization of recombinant protein
Expression of recombinant HO-1 protein was confirmed in HO-1–transduced basilar arteries by Western blot analysis (n = 5). A representative blot is shown in Fig. 1. Immunohistochemistry showed that recombinant HO-1 was localized predominantly in the adventitial layer (Fig. 2A). Some endothelial cells also were stained positively when arteries were transduced with AdCMVHO-1. However, β-Gal–transduced vessels did not show HO-1 immunoreactivity (Fig. 2B).

Heme oxygenase-1 (HO-1) protein expression in canine basilar arteries after 24 hours transduction with AdCMVHO-1 vector. Western blot analysis revealed the overexpression of HO-1 protein in the HO-1–transduced vessels, but not in β-galactosidase–transduced vessels (n = 5).
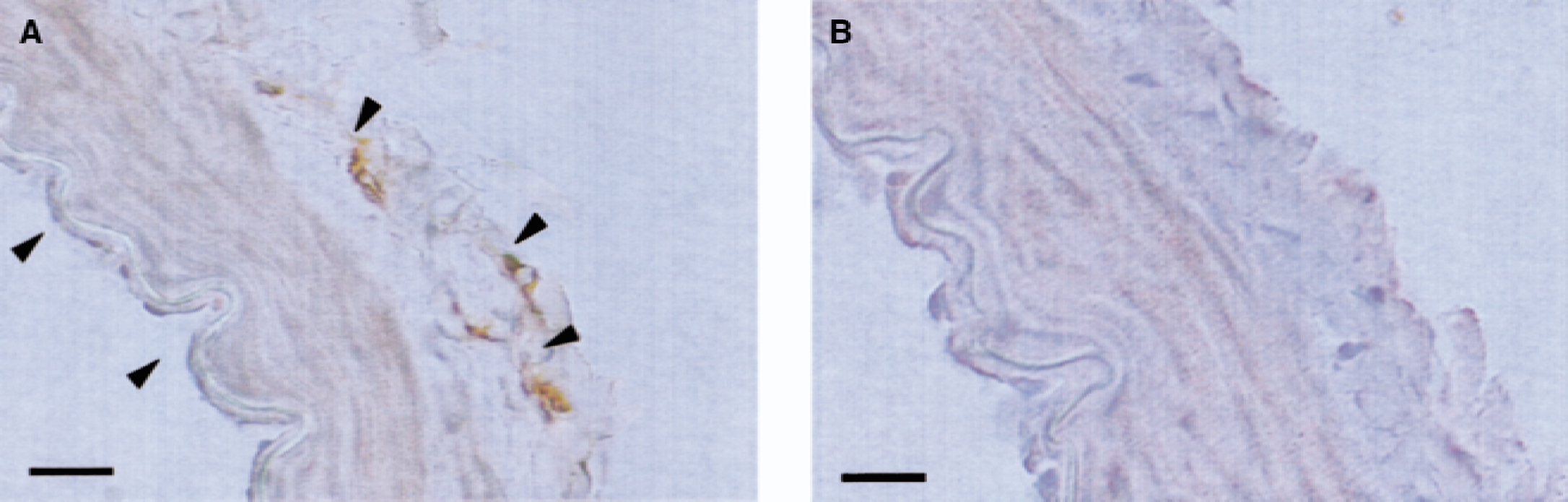
Immunohistochemical staining for recombinant heme oxygenase-1 (HO-1) protein after 24 hours HO-1 transduction.
Endothelium-dependent relaxation to bradykinin
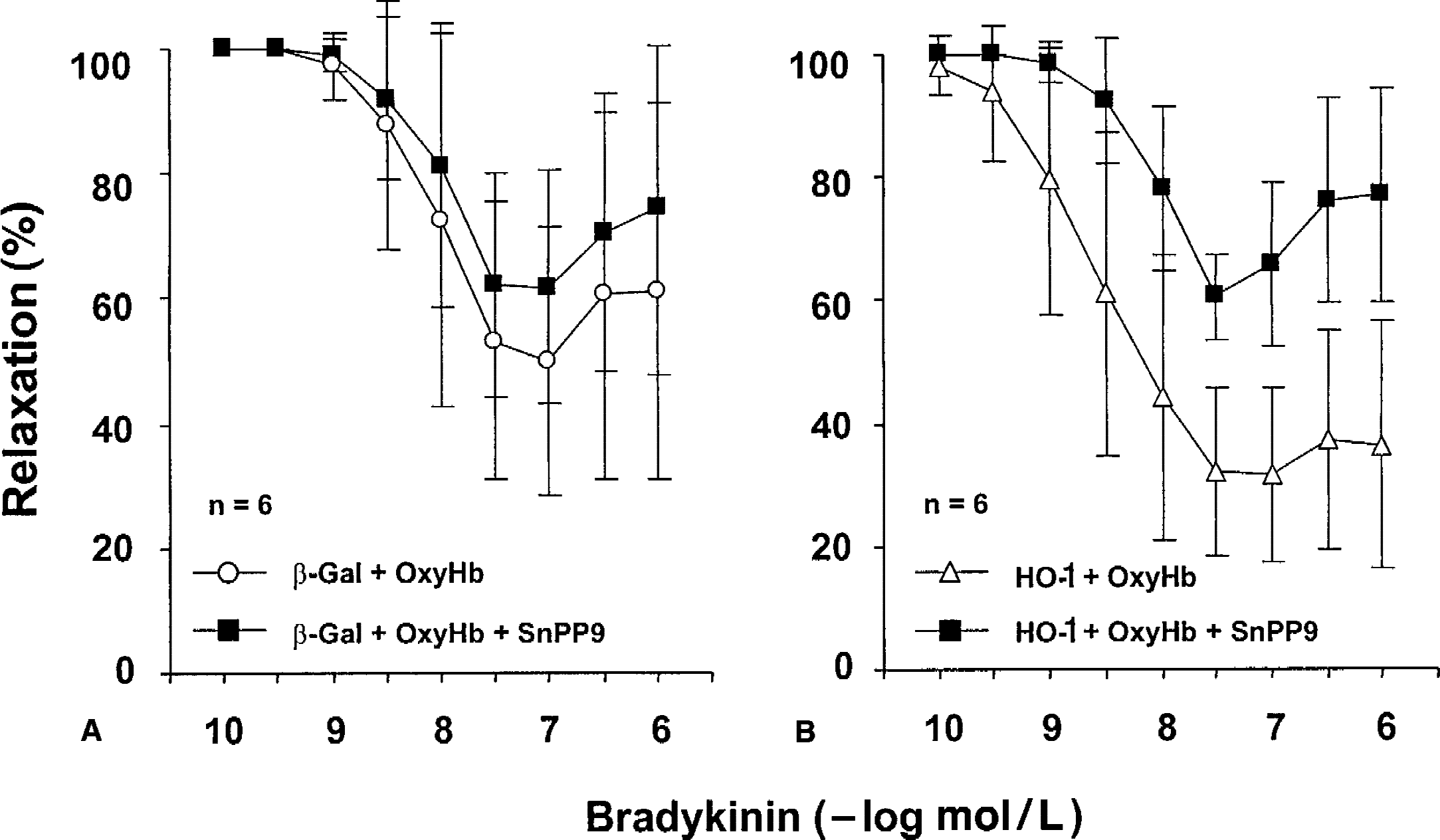
During contraction to UTP (3 × 10−6 to 3 × 10−5 mol/L), bradykinin (10−10 to 10−6 mol/L) caused endothelium-dependent relaxations. L-NAME (3 × 10−4 mol/L) abolished relaxations to bradykinin (data not shown), suggesting that the effect of bradykinin was exclusively mediated by NO. The relaxations to bradykinin in control, β-Gal–transduced, and HO-1–transduced vessels were identical (Fig. 3). In control rings (nontransduced rings), incubation with OxyHb significantly attenuated the relaxation to bradykinin (maximal relaxation = 50.0 ± 10.3%, EC50 = 8.16 ± 0.37, n = 5). Similarly, in β-Gal–transduced rings, incubation with OxyHb significantly attenuated the relaxation to bradykinin (Fig. 4A, P < 0.01). In contrast, attenuation of endothelium-dependent relaxations by OxyHb was prevented in arteries expressing recombinant HO-1 (Fig. 4B, P > 0.05).
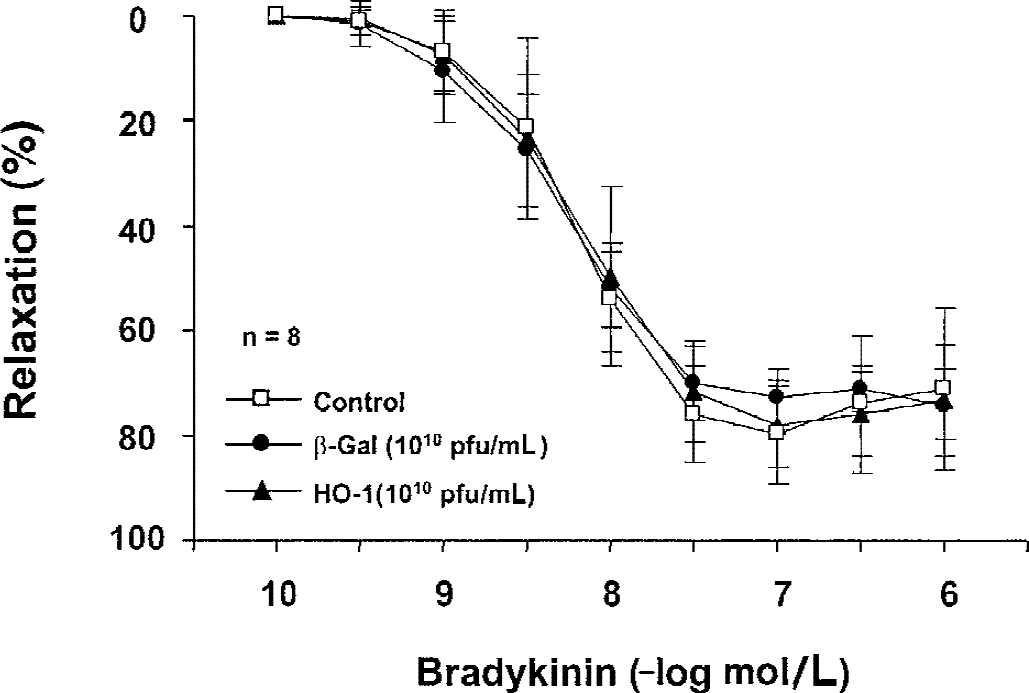
Concentration-response curves to bradykinin in control, β-galactosidase (β-Gal)-transduced, and heme oxygenase-1 (HO-1)-transduced rings. Relaxations were obtained during contractions induced by uridine-5′-triphosphate (UTP; 3 × 10−6 to 3 × 10−5 mol/L). Data are mean ± SD and are expressed as a percentage of maximal relaxation induced by 3 ×10−4 mol/L papaverine; 100% = 4.82 ± 0.195 g, 4.12 ± 0.273 g, and 4.16 ± 0.437 g, in control, β-Gal, and HO-1, respectively (n = 8). Gene transfer with adenoviral vector did not affect UTP-induced contraction and endothelium-dependent relaxation to bradykinin.
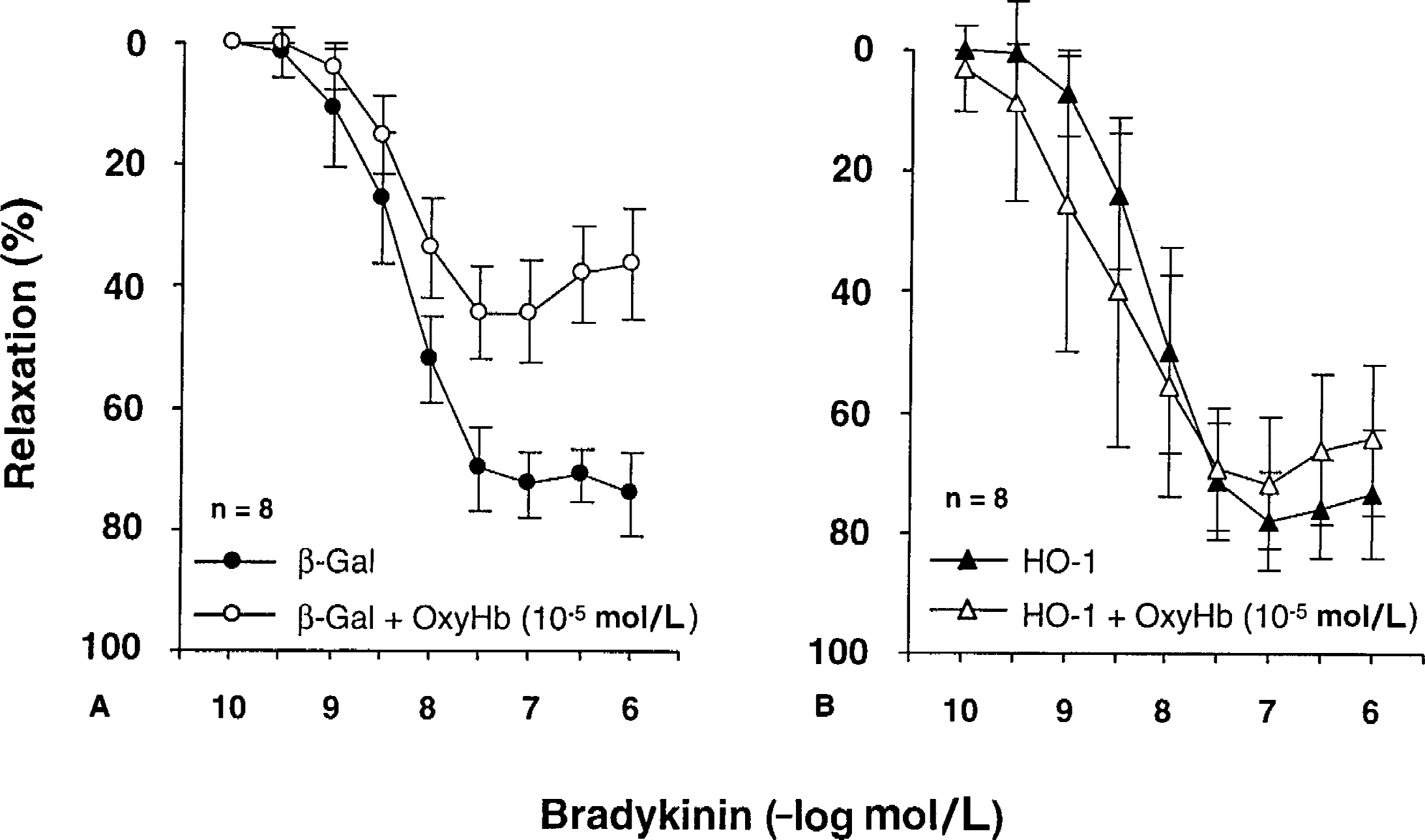
Concentration-response curves to bradykinin in canine basilar artery rings in
Endothelium-independent relaxations to forskolin and DEA-NONOate
During contractions to UTP (3 × 10−6 to 3 × 10−5 mol/L), forskolin (10−8 to 10−5 mol/L) and DEA-NONOate (10−9 to 10−5 mol/L) caused concentration-dependent relaxations. Relaxation to forskolin and DEA-NONOate were identical in β-Gal– and HO-1–transduced rings with and without preincubation with OxyHb (10−5 mol/L;Table 1).
Effect of transduction and OxyHb on endothelium-independent relaxation
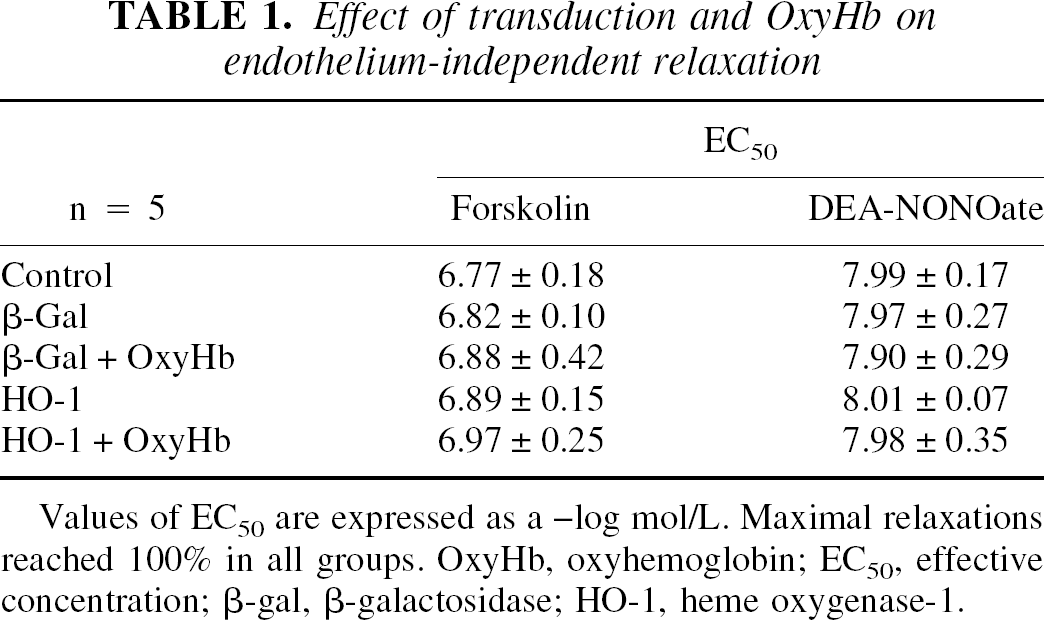
Values of EC50 are expressed as a –log mol/L. Maximal relaxations reached 100% in all groups. OxyHb, oxyhemoglobin; EC50, effective concentration; β-gal, β-galactosidase; HO-1, heme oxygenase-1.
Effects of SnPP9 on bradykinin-induced relaxation
Overnight incubation with SnPP9 (10−5 mol/L) had no effect on endothelium-dependent relaxation to bradykinin in control (nontransduced) arteries (Fig. 5, n = 5). Endothelium-independent relaxation to forskolin and DEA-NONOate in control arteries also was not affected by preincubation with SnPP9 (Table 2). In β-Gal–transduced rings coincubated with OxyHb, SnPP9 had no effect on the relaxation to bradykinin (Fig. 6A, P > 0.05). However, in HO-1–transduced rings coincubated with OxyHb, overnight incubation with SnPP9 significantly reduced relaxation to bradykinin (Fig. 6B, P < 0.01).
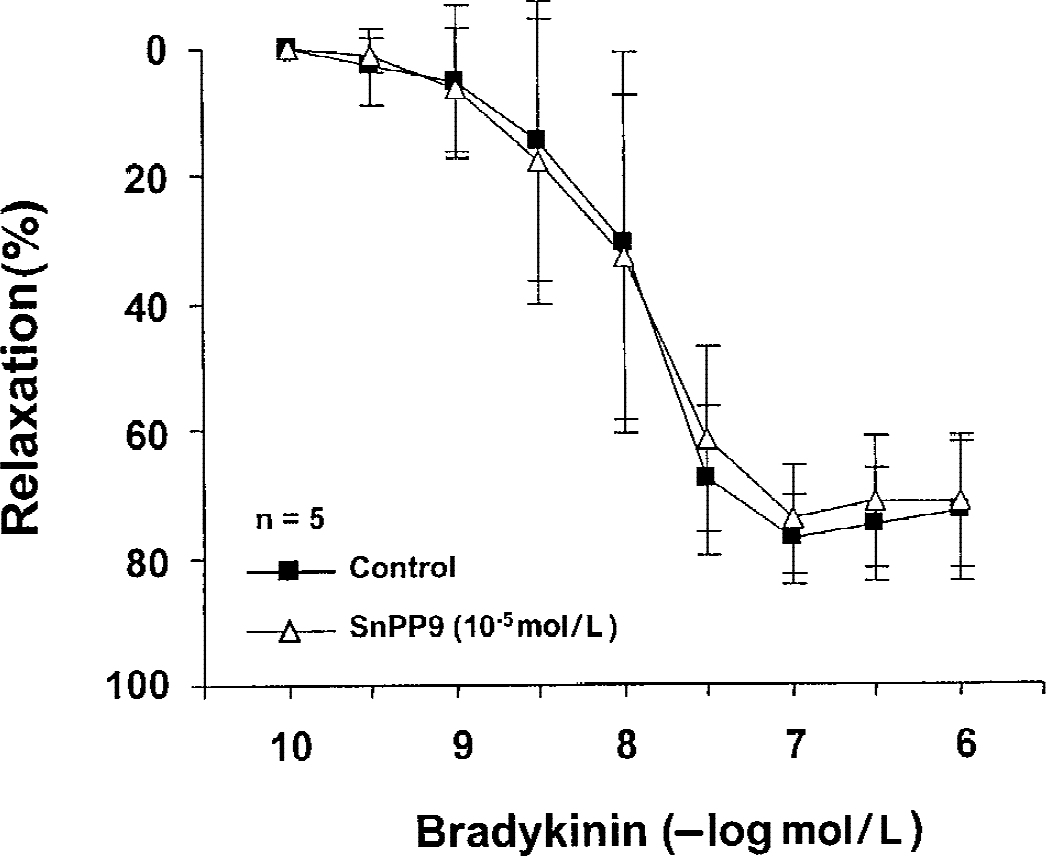
Effects of preincubation with SnPP9 on the endothelium-dependent relaxation to bradykinin in control canine basilar arteries (without virus). Relaxations were obtained during contractions induced by uridine-5′-triphosphate (3 × 10−6 to 3 × 10−5 mol/L). Data are mean ± SD and are expressed as a percentage of maximal relaxation induced by 3 × 10−4 mol/L PPV; 100% = 5.4 ± 0.678 g and 5.2 ± 0.457 g, in control and with SnPP9, respectively (n = 5). SnPP9 had no effect on the relaxation to bradykinin in control vessels.
Effect of preincubation with SnPP9 on the endothelium-dependent relaxation to bradykinin in
Effect of SnPP9 and OxyHb on endothelium-independent relaxation

Values of EC50 are expressed as a –log mol/L. Maximal relaxations reached 100% in all groups. OxyHb, oxyhemoglobin; EC50, effective concentration.
Basal cGMP production
Basal production of cGMP was not different between β-Gal– and HO-1–transduced vessels (Fig. 7). Incubation with OxyHb significantly decreased the level of cGMP in β-Gal–transduced rings. However, transduction with HO-1 preserved basal production of cGMP in arteries exposed to OxyHb (P < 0.01, Student t-test).
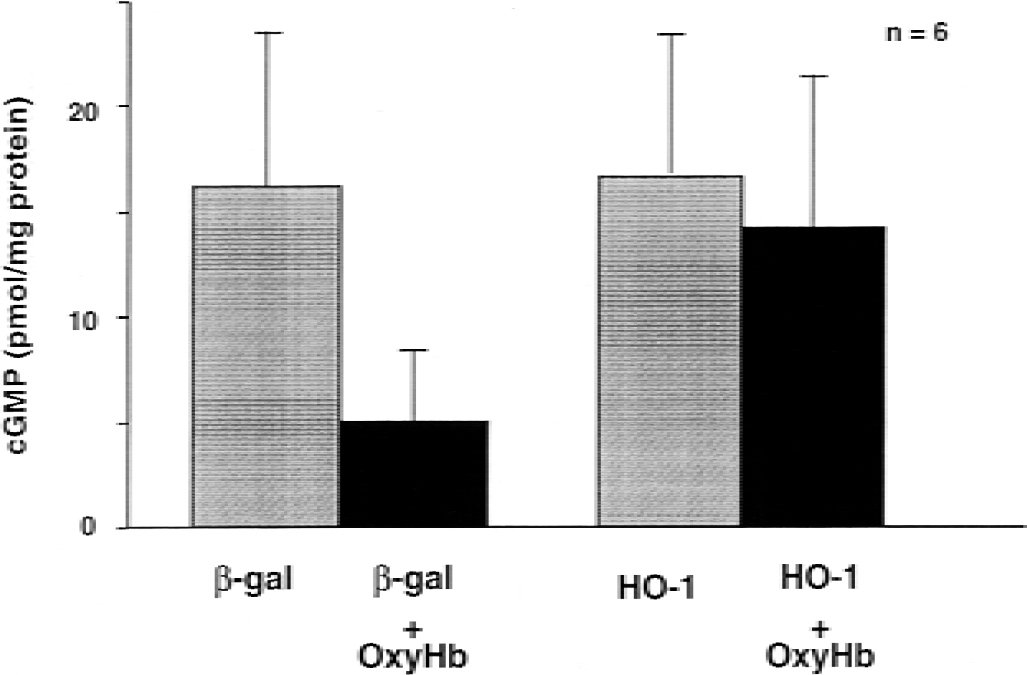
Basal levels of 3′,5′-cyclic monophosphate (cGMP) production. Columns and bars represent mean ± SD. Transduction with heme oxygenase-1 (HO-1) did not increase basal level of cGMP compared with β-galactosidase (β-Gal)-transduced vessels. Preincubation with oxyhemoglobin significantly decreased the cGMP level in β-Gal–transduced vessels, but did not affect in HO-1– transduced vessels.
DISCUSSION
This is the first study to examine the effect of recombinant HO-1 on vasomotor function of cerebral arteries. The current findings demonstrate that: (1) ex vivo adenovirus-mediated transfer of recombinant HO-1 to cerebral arteries transduces adventitia and endothelium, 2) OxyHb impairs endothelium-dependent relaxation, 3) expression of recombinant HO-1 protects vessels against OxyHb-induced endothelial dysfunction, and 4) the protective effect of HO-1 is reversed by SnPP9, an HO inhibitor.
OxyHb is the most likely mediator responsible for induction of vasospasm by autologous blood (Cook and Vollrath, 1995; Macdonald and Weir, 1994). The current results demonstrate that in isolated cerebral arteries OxyHb impairs endothelium-dependent relaxation to bradykinin and reduces basal cGMP production. In contrast, OxyHb does not affect endothelium-independent relaxations to forskolin and DEA-NONOate. These findings strongly suggest that OxyHb selectively impairs endothelial function. The mechanisms underlying this effect of OxyHb may include chemical inactivation of endothelium-derived NO, the siphoning off of endothelial sources of NO, and generation of reactive oxygen species due to autooxidation of OxyHb (Misra and Fridovich, 1972). In addition, a recent study demonstrated that OxyHb induces apoptosis in cultured bovine aortic endothelial cells (Ogihara et al., 1999). Matz et al. (2000) also demonstrated that the presence of blood in subarachnoid space is associated with neuronal apoptotic cell death (Matz et al., 2000). The mechanisms responsible for the adverse effects of OxyHb on endothelial function are beyond the scope of the current study.
Heme oxygenase-1 gene transfer protected the vessels from OxyHb-induced injury. This protection was lost after coincubation with SnPP9 (10−5 mol/L), a selective HO-1 inhibitor, which strongly suggests that protection is caused by enzymatic activity of HO-1. It has been reported that higher concentration of SnPP9 (10−4 mol/L) significantly inhibits eNOS activity (Zakhary et al., 1996). To exclude the possibility of this nonselective effect, the authors used lower concentration of SnPP9 (10−5 mol/L), which had no effect on the relaxation to bradykinin in control of β-Gal–transduced rings. Endothelium-independent relaxation to DEA-NONOate and forskolin also were not affected by SnPP9 (10−5 mol/L) in control and OxyHb-treated vessels. Based on these findings, the authors concluded that SnPP9 exerted its effect through inhibition of recombinant HO-1 expressed in the transduced vessels.
Observations from several laboratories support the hypothesis that HO-1 induction provides cellular protection against both heme-mediated and non–heme-mediated oxidant injuries (Abraham et al., 1995; Choi and Alam, 1996; Nath et al., 1992). However, the exact mechanisms responsible for protection by HO-1 are not understood. Currently, four major mechanisms are considered to be involved in the protection provided by HO-1. First, HO-1 restrains the elevation in intracellular levels of heme, the latter representing a potent oxidizing toxicant. Second, HO-1 promotes iron sequestration by increasing the synthesis of ferritin (Nath et al., 1992; Vile and Tyrrell, 1993). Moreover, HO-1 modulates intracellular iron levels by augmenting iron efflux from cells (Ferris et al., 1999). This minimizes the deleterious effects of reactive oxygen species (such as superoxide and H2 O2) whose toxicity depends on the presence of iron (Camhi et al., 1995). Third, bilirubin, a byproduct of heme degradation, is a powerful chain-breaking antioxidant (Stocker et al., 1987 a). In vitro, bilirubin scavenges peroxyl radicals as efficiently as α-tocopherol, which is regarded to be the most potent antioxidant of lipid peroxidation (Stocker et al., 1987 b). This antioxidant effect of bilirubin also has been demonstrated in vivo (Llesuy et al., 1994). Fourth, carbon monoxide (CO), the other byproduct of heme degradation, can activate soluble guanylate cyclase to produce cGMP and vasodilation (Morita and Kourembanas, 1995). However, unlike NO, CO is not a free radical and does not react with superoxide anion to produce a potent oxidant, peroxynitrite (Stamler et al., 1992).
Evidence presented in this study suggests that CO-cGMP pathway does not play an important role in endothelial protection against OxyHb toxicity, or at least, effects of CO that are mediated through cGMP. This conclusion is based on the fact that the authors did not detect a significant increase of basal cGMP production in HO-1–transduced vessels in the absence or presence of OxyHb. Recently, Petrache et al. (2000) reported that HO-1 inhibited cytokine-mediated apoptosis in cultured fibroblasts, apparently mediated by increased formation of CO (Petrache et al., 2000). It is possible that HO-1 exerts its protective effect against OxyHb-mediated endothelial injury through both antioxidant and antiapoptotic effects. However, further studies are needed to characterize the exact mechanisms of cellular protection by HO-1.
Heme or hemoglobin are well known to induce HO-1 in various types of cells (Applegate et al., 1991; Tenhunen et al., 1970; Vercellotti et al., 1994). Balla and coworkers (1993) demonstrated that endothelial cells in vitro respond to heme/hemoglobin by induction of HO and the production of ferritin (Balla et al., 1993). Recent studies also demonstrated increased expression of endogenous HO-1 and ferritin in cerebral arterial wall after subarachnoid hemorrhage in rats and monkeys (Suzuki et al., 1999; Ono et al., 2000). In contrast, the authors could not detect up-regulation of HO-1 protein expression in isolated cerebral arteries exposed to OxyHb (Western blot analysis, data not shown). This was not because of technical problems or the inability of antibodies to recognize canine HO-1 protein. The authors could detect up-regulation of HO-1 protein expression in basilar arteries obtained from dogs with subarachnoid hemorrhage (unpublished observation). Furthermore, an inhibitor of HO-1, SnPP9, did not affect endothelial function in arteries incubated with OxyHb, suggesting that endogenous HO-1 was not expressed despite prolonged contact of the vascular tissue with OxyHb. The reason for the apparent absence of endogenous HO-1 induction by OxyHb in isolated cerebral arteries is unclear. The authors do not have an explanation for this observation other than the fact that ex vivo experimental conditions may somehow alter the ability of the tissue to increase expression of HO-1. Species differences also may be responsible for the absence of endogenous HO-1 induction.
The authors reported that in isolated cerebral arteries expression of recombinant HO-1 does not affect vasomotor function of cerebral arteries. However, HO-1 provides protection against OxyHb. Although the incubation with OxyHb does not precisely mimic alterations of vascular reactivity observed after SAH, the current results suggest that HO-1 may have a therapeutic application in the prevention and treatment of vasospasm after SAH.
Footnotes
Acknowledgments:
The authors thank Leslie Smith for technical assistance and Janet Beckman for preparing the manuscript.