
Abstract
Hemoglobin is a key factor in the production of cerebral vasospasm. Metabolism of hemoglobin involves breakdown of heme by heme oxygenase (HO) and sequestration of the released iron in ferritin. We determined whether subarachnoid hemorrhage induces these proteins in cerebral arteries and, if so, in which cells they are produced. Whether the changes correlated with vasospasm also was investigated. Subarachnoid hemorrhage was created in monkeys, and vasospasm was assessed by angiography in cohorts of animals killed 3, 7, or 14 days after the hemorrhage. Ferritin and HO-1 messenger ribonucleic acid (mRNA) and protein were measured by competitive reverse transcription-polymerase chain reaction and Western blotting in hemorrhage-side and control-side cerebral arteries and brain tissue. The location of these proteins was determined by immunohistochemistry. There was significant vasospasm 3 and 7 days but not 14 days after subarachnoid hemorrhage. There were no significant changes in mRNA for HO-1 or ferritin in cerebral arteries or brain tissue at any time. There was a significant increase in HO-1 and ferritin protein in hemorrhage-side compared with control-side cerebral arteries at 3, 7, and 14 days. The increase in HO-1 protein was maximal at 3 days, whereas the increase in ferritin protein was maximal at 7 days. There was no detectable increase in HO-1 or ferritin protein in brain tissue at any time. Immunohistochemistry localized HO-1 protein and ferritin to cells in the adventitia of the arterial wall. We show that subarachnoid hemorrhage is associated with a significant increase in HO-1 and ferritin proteins in cerebral arteries that begins at least as early as 3 days after the hemorrhage and that persists for up to 14 days.
Hemoglobin has been implicated in the pathogenesis of cerebral vasospasm after subarachnoid hemorrhage (SAH)(Macdonald et al., 1991; Mayberg et al., 1990). The mechanisms by which hemoglobin causes vasospasm are uncertain but may involve cytotoxic reactions induced by hemin or iron released from the hemin and acting on arterial smooth muscle or endothelial cells (Everse and Hsia, 1997; Fujii and Fujitsu, 1988). Hemin is normally degraded by enzymes called heme oxygenases (HOs). HO-1, or heat shock protein 32, is an inducible enzyme that metabolizes hemin to biliverdin, carbon monoxide, and iron (Maines, 1997). It is induced in many cell types in response to hemin, heavy metals including iron, ultraviolet light, H2O2, sodium arsenite, heat shock in rats but not in man, and numerous other stimuli. There is a constitutively expressed HO-2 that is found in some cell types including neurons (Maines, 1997). It is induced by glucocorticoids. A third form of HO, HO-3, was isolated from a variety of tissues including brain (McCoubrey et al., 1997). The induction of HO-1 in response to some stimuli, such as exposure of cells to hemin, is followed by an increase in ferritin, an intracellular iron storage protein (Balla et al., 1995). Because hemoglobin is believed to be important in the genesis of vasospasm, these responses might be important in protecting cerebral arteries from hemoglobin-induced reactions such as vasospasm (Kuroki et al., 1998; Macdonald et al., 1997, 1998; Matz et al., 1996a, b ; Suzuki et al., 1999; Turner et al., 1998). Several investigations have documented increases in HO-1 in brain tissue after SAH in rats (Matz et al., 1996a, b ; Suzuki et al., 1999). The rat model of SAH has major limitations to the study of human SAH, however; and changes in cerebral arteries themselves have not been examined in detail (Suzuki et al., 1999). Furthermore, the time course of changes in HO-1 and the cellular localization of HO-1 in cerebral arteries have not been examined. Changes in ferritin, which may be as important as HO-1 in terms of cytoprotection, have not been examined after SAH (Balla et al., 1995). Therefore, the purpose of this investigation was to determine the time course and location of changes in HO-1 and ferritin messenger ribonucleic acid (mRNA) and protein in cerebral arteries and brain tissue adjacent to SAH and to correlate these changes, if any, with angiographic vasospasm.
MATERIALS AND METHODS
Protocol
Fourteen cynomolgus monkeys (Macaca fascicularis) underwent baseline (day 0) angiography and unilateral SAH. The diameter of the intracranial arteries was measured at angiography performed on day 0 (the day of clot placement) and then 3, 7, or 14 days later (n = 4 to 6 per time). After the second angiogram, animals were killed and the brains and middle cerebral arteries were removed and placed in liquid N2. The anterior cerebral arteries and adjacent brain were processed for immunohistochemistry. The mRNA and protein were extracted from right (hemorrhage side) and left (control side) middle cerebral arteries and adjacent brain, and levels of HO-1 and ferritin mRNA were assessed by competitive reverse transcription polymerase chain reaction (RT-PCR) and the respective proteins were assessed by Western blotting. Immunohistochemistry was used to examine the distribution of HO-1 and ferritin in the anterior cerebral arteries and adjacent brain at each time. The animals were operated on in random order and the assessment and analysis of the results was performed in blinded fashion. The Animal Care and Use Committee of the University of Chicago approved procedures on animals.
Angiography and SAH
Angiography, craniotomy, and production of SAH were performed as described previously (Hino et al., 1995; Kim et al., 1996). On day 0, animals were sedated with ketamine, 6 to 10 mg/kg intramuscularly, weighed, intubated, and ventilated (Harvard Apparatus, Millis, MA, U.S.A.) on 1% to 2% isofluorane. Body temperature was monitored and maintained with a water-heated blanket (Gaymar model TP-200; Gaymar, Orchard Park, NY, U.S.A.). Arterial Pa
Repeat angiography and euthanasia
Angiography was repeated through the right axillary artery with the animals under general anesthesia, as described earlier, 3 (n = 6), 7 (n = 4), or 14 (n = 4) days after creation of the hemorrhage. Monkeys that were to be killed on day 14 underwent angiography on day 7. After completion of the final angiogram, animals were exsanguinated while under anesthesia and the brains were removed. The right and left middle cerebral arteries and underlying brain tissue were carefully dissected and frozen in liquid N2 until processing within 24 hours. The anterior cerebral arteries and underlying brain were fixed for 24 hours in 4% paraformaldehyde in 0.1 mol/L phosphate-buffered saline (PBS), placed in 30% sucrose in 0.1 mol/L PBS for 24 hours, and then embedded in tissue-embedding medium (Triangle Biomedical, Durham, NC, U.S.A.) and stored at −20°C until they were sectioned.
Specimen processing, competitive RT-PCR, Western blotting
Total RNA and protein was isolated from middle cerebral artery and brain tissue by using T
For RNA extraction, the aqueous phase was mixed with isopropyl alcohol (0.5 mL/mL T
The mRNA was quantified by competitive RT-PCR (Hino et al., 1996). Specific primers for PCR for each target cDNA were selected by using human cDNA sequences published in the GenBank (Table 1). Competitors were prepared for HO-1 and ferritin heavy chain by constructing eight overlapping 40-mer primers. Each competitor contained two 20-mer primers that are for different target cDNAs. PCR was performed by using a pair of 40-mer primers to add 20 nucleotides to each end of the template. PCR was performed in the presence of 1 μCi [32P]dCTP (3,000 Ci/mmol/L; Amersham, Arlington Heights, IL, U.S.A.). The PCR conditions were 94°C for 1 minute, 60°C for 1 minute, and 72°C for 1 minute for 32 cycles. The PCR products were separated by electrophoresis on a 5% polyacrylamide gel. The intensity of the bands corresponding to each specific PCR product were analyzed by using a computed phosphoimage analyzer (PhosphoImager; Molecular Dynamics, Sunnyvale, CA, U.S.A.).
Sequences of PCR primers
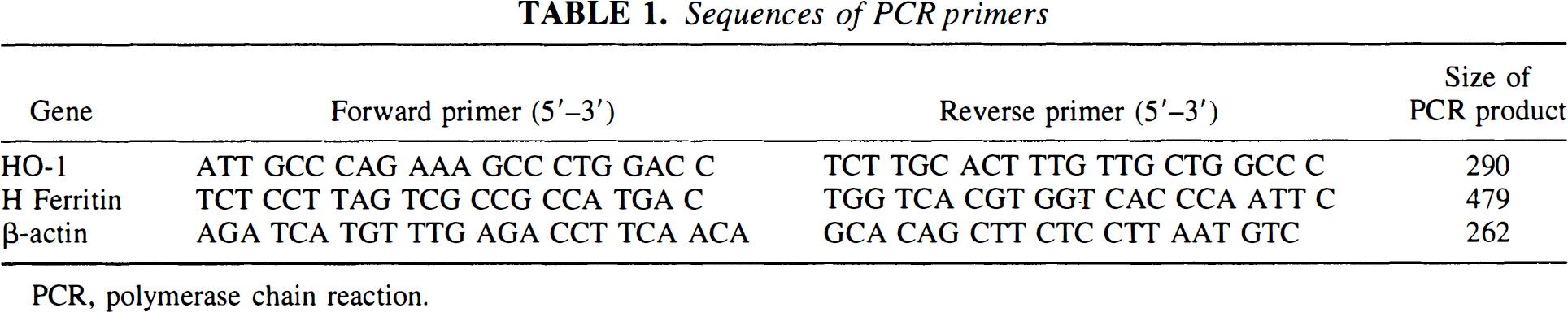
PCR, polymerase chain reaction.
For Western blotting, proteins prepared as described were separated by electrophoresis on 12% sodium dodecyl sulfate—polyacrylamide gels. They were electrotransferred onto nitrocellulose membranes. HO-1 was detected with rabbit polyclonal anti-rat HO-1 antibody (1:500; StressGen Biotechnologies, Victoria, British Columbia, Canada). This antibody detects on Western blotting a single band of heat-shocked rat brain (Ewing and Maines, 1993). Ferritin was detected with rabbit polyclonal anti-horse spleen ferritin antibody (1:2,000 dilution; Sigma, St. Louis, MO, U.S.A.). This antibody detected ferritin heavy and light chains on Western blotting. The membranes were then incubated with peroxidase-conjugated pig anti-rabbit immunoglobulins (1:2,000 dilution; DAKO, Carpinteria, CA, U.S.A.) and reaction products visualized by chemiluminescence (NEN Life Science Products, Boston, MA, U.S.A.). Proteins were quantified by using a computing densitometer and ImageQuant software program (Molecular Dynamics) and standardized to the total amount of protein loaded per lane.
Immunohistochemistry
Five-micrometer-thick sections of anterior cerebral artery and underlying brain were cut on a cryostat (Leica Cryocut 1800; Nussloch, West Germany) and mounted on clean glass slides. Endogenous peroxidase activity was blocked by exposure of sections to H2O2, 0.6%, for 30 minutes followed by washing three times in 0.1 mol/L PBS, pH 7.4. They were permeabilized with 0.2% Triton X-100 in PBS containing 1% bovine serum albumin for 30 minutes and then incubated in 0.5% bovine serum albumin and 0.5% goat serum in PBS. They were incubated for 24 hours at 4°C with the primary rabbit polyclonal antibodies to rat HO-1 (1:500; Stressgen) and horse spleen ferritin (1:500; Sigma) that were used for Western blotting. These times and concentrations produced minimal background staining. After washing three times in PBS, slides were exposed to biotinylated goat, anti-rabbit secondary antibody (1:200; Vector Laboratories, Burlingame, CA) for 1 hour. They were washed three times in PBS, incubated for 1 hour at room temperature with ABC reagent (Vector), washed three times in PBS, and then incubated in a solution of 2 μmol/L diaminobenzidine and 0.05% H2O2 for 10 to 20 minutes. Sections were rinsed in distilled water, counterstained with hematoxylin, dehydrated in a graded series of alcohol solutions, washed in xylene, coverslipped, and examined by light microscopy. Negative controls were processed by using bovine serum albumin instead of the primary antibody.
Data analysis
To calculate changes in mRNA levels, the ratio of intensity of bands of products of cDNA and the corresponding internal standard DNA (cDNA:standard DNA) was determined. Relative mRNA changes in the right (hemorrhage side) arteries and brain were calculated as the ratio compared with the level of mRNA in the corresponding left (control side) tissues from the same animal after normalization for the amount of β-actin in the sample. For Western blotting, changes in levels of proteins were expressed as a ratio of the amount of protein, as measured by band intensity on the hemorrhage side, compared with the control side in the same animal. The amount of protein loaded was the same for each pair of samples, and β-actin was used as a control to confirm that equal amounts of protein were loaded. All assays were performed in triplicate. Immunohistochemical staining for each protein was examined by two experienced investigators (R.L.M., B.Y.) who did not know the time after SAH when the samples were obtained. The types of cells that contained staining was recorded and an impression of the intensity of staining was given.
Data are expressed as mean ± SD values. Statistical analysis between groups was by analysis of variance for multiple comparisons, followed by pairwise comparisons by using Fisher's test if significant variance was found. Paired or unpaired t-tests were used for comparisons between two measurements. Linear regression was used to study the relation between angiographic vasospasm and protein levels of HO-1 and ferritin. Significance was taken at P < 0.05.
RESULTS
Physiologic variables and angiography
The only significant difference in weight, blood pressure, body temperature, heart rate, and arterial Pa
Angiography showed that there was a 45 ± 13% (P < 0.0005, paired t-test) reduction between day 0 and day 3 and a 41 ± 23% (P < 0.05, paired t-test) reduction between day 0 and day 7 in right middle cerebral artery diameter (Fig. 1). On day 7, there was a 35 ± 10% (P < 0.01, paired t-test) reduction in right middle cerebral artery diameter in animals that were killed on day 14. By day 14, there was a statistically insignificant 6 ± 14% reduction in middle cerebral artery diameter. Angiography of the right intradural internal carotid and anterior cerebral arteries showed similar changes (Table 2). There was no significant reduction in arterial diameter in the left-side arteries that were not exposed to blood clot (Fig. 1; Table 2).
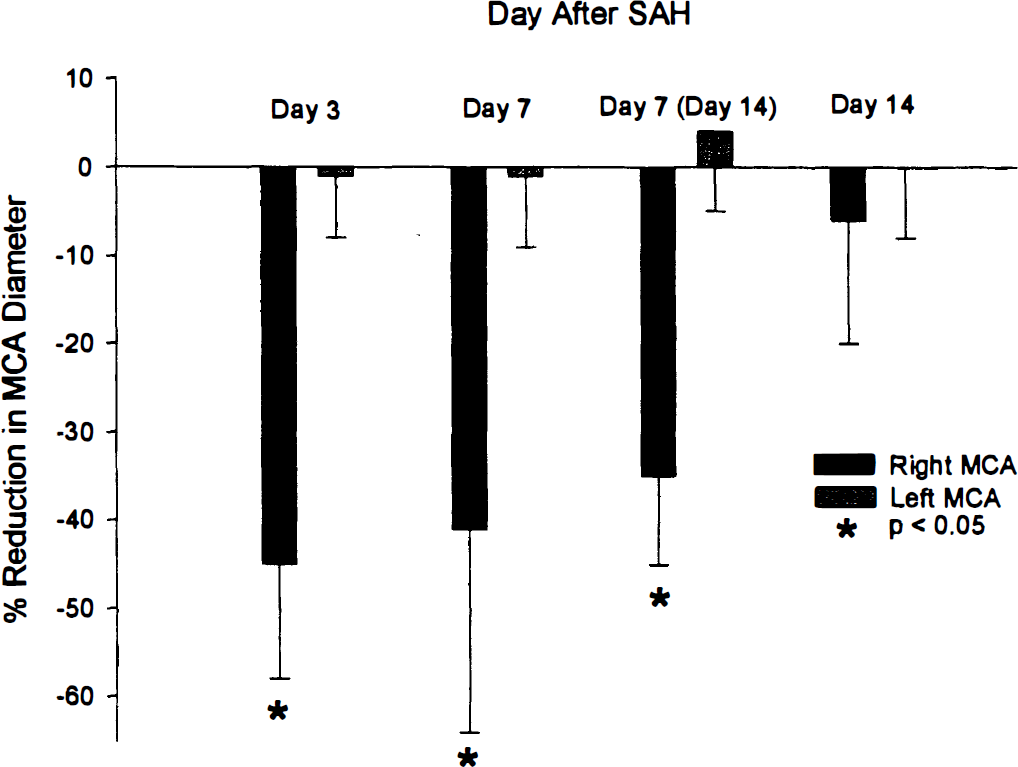
Bar graph showing percent reduction compared to before subarachnoid hemorrhage (SAH) in right (hemorrhage-side) and left (control-side) middle cerebral artery (MCA) diameters 3, 7 or 14 days after creation of right SAH. There was significant reduction in hemorrhage-side MCA diameter 3 and 7 days after SAH (P < 0.05, paired t-tests) with no difference at day 7 between animals killed on day 7 and those killed on day 14 (values are means ± SD, n = 4 [day 7 and 14 groups] or 6 [day 3 group]).
Percent reduction in angiographic arterial diameter for anterior cerebral arteries

Values are means ± SD.
P < 0.005.
Changes in cerebral arteries
Western blotting showed that there was a statistically significant 5.5 ± 2.7-fold increase in HO-1 on day 3 in the vasospastic middle cerebral arteries compared with control-side arteries (Figs. 2 and 3). By day 7, there was a significant 2.9 ± 1.0-fold (P < 0.05, paired t-test) increase in HO-1 protein, and this persisted at a lower level until 14 days after SAH, when there was a 1.4 ± 0.6-fold increase in HO-1 (P < 0.05, paired t-test). Ferritin protein also was significantly elevated 3 (2.0 ± 0.7-fold; P < 0.005), 7 (3.7 ± 1.1-fold; P < 0.05), and 14 (3.6 ± 1.2-fold; P < 0.05) days after SAH (Fig. 2). Ferritin H chain was the major ferritin chain detected in cerebral arteries.
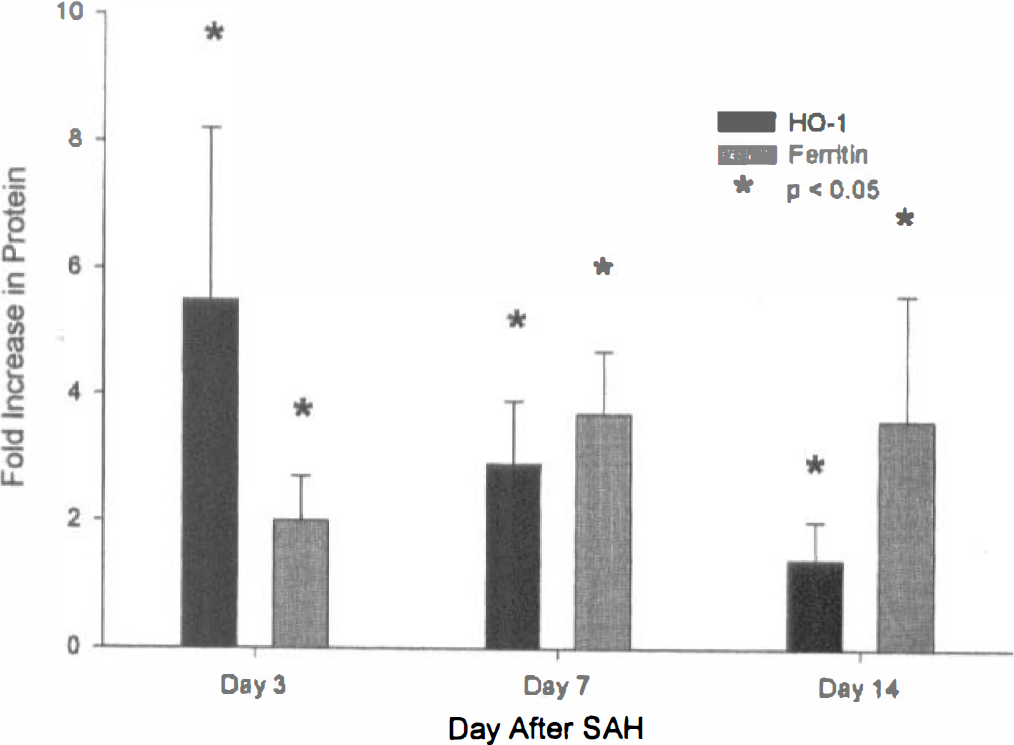
Bar graph of ratio of HO-1 and ferritin protein, measured by Western blotting, in hemorrhage- compared to control-side middle cerebral arteries. There is a statistically significant increase in HO-1 protein 3 days after subarachnoid hemorrhage (SAH) that continues through day 7 and day 14 (P < 0.05, analysis of variance, values are means ± SD, n = 3 per group). There is a significant increase in ferritin protein at all times after SAH, peaking 7 and 14 days after SAH.
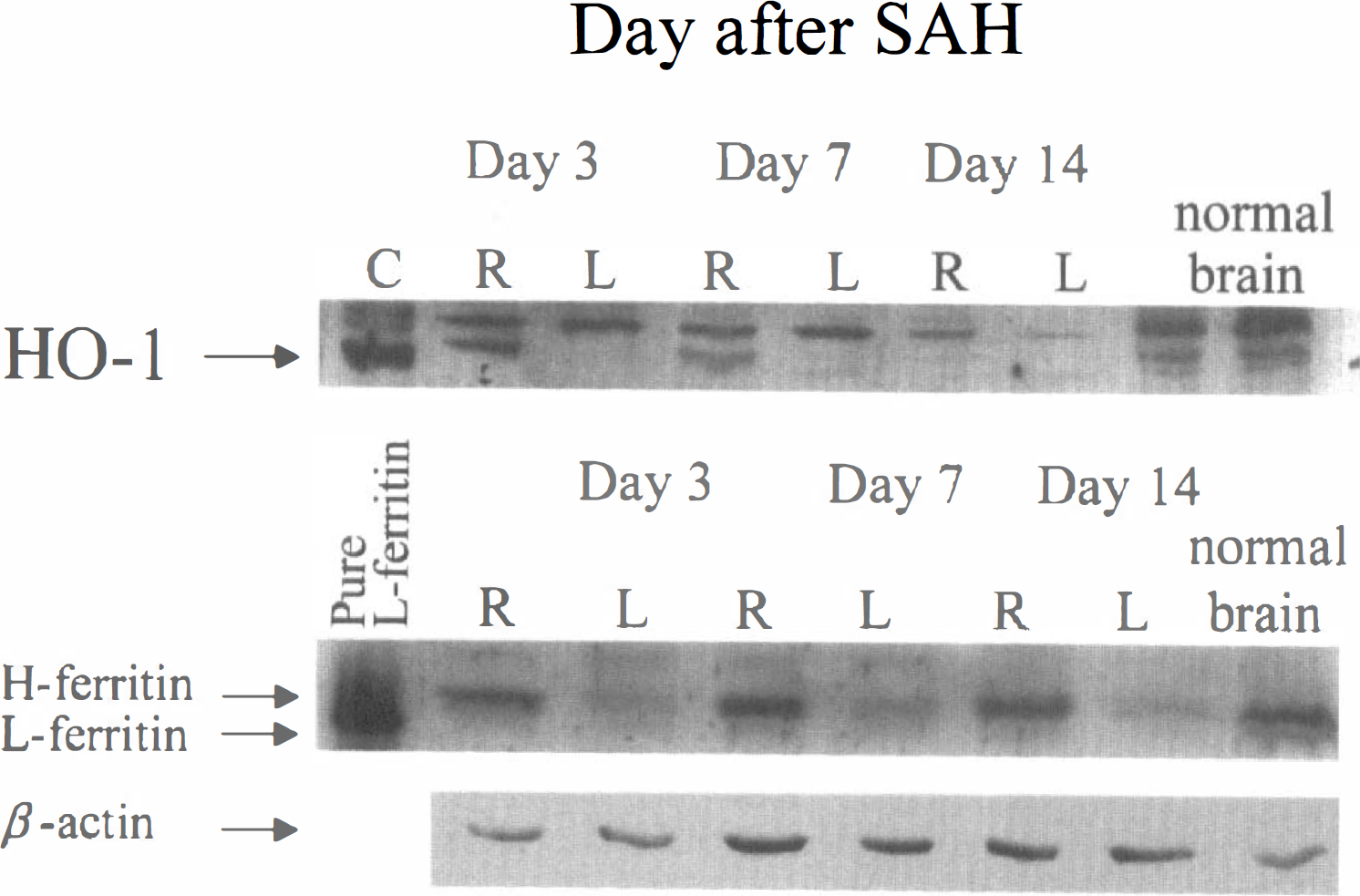
Western blotting of right-(hemorrhage-) and left- (control-) side middle cerebral arteries 3, 7, or 14 days after subarachnoid hemorrhage (SAH) showing that there is an increase in HO-1 protein (top) that is most marked 3 days after SAH and that persists for 7 days. Control is cultured smooth muscle cells exposed to hemoglobin for 48 hours. There is an increase in ferritin protein (middle) at all times after SAH with the maximal increase at 7 and 14 days after SAH. The amount of β-actin protein is relatively constant (lower).
Competitive RT-PCR showed that there was a transient but statistically insignificant increase in ferritin H chain mRNA 7 days after SAH (Fig. 4). The mRNA for HO-1 could not be detected in cerebral arteries at any time after SAH. The concentration of mRNA, therefore, was less than approximately 10−12 mol/L. The PCR product that was amplified by the ferritin H chain primers was sequenced and was consistent with ferritin H chain. In a similar manner, the PCR product that was obtained by using the HO-1 primers on mRNA isolated from cultured monkey cerebrovascular smooth muscle cells exposed to hemoglobin was sequenced and was consistent with HO-1 (unpublished observations).
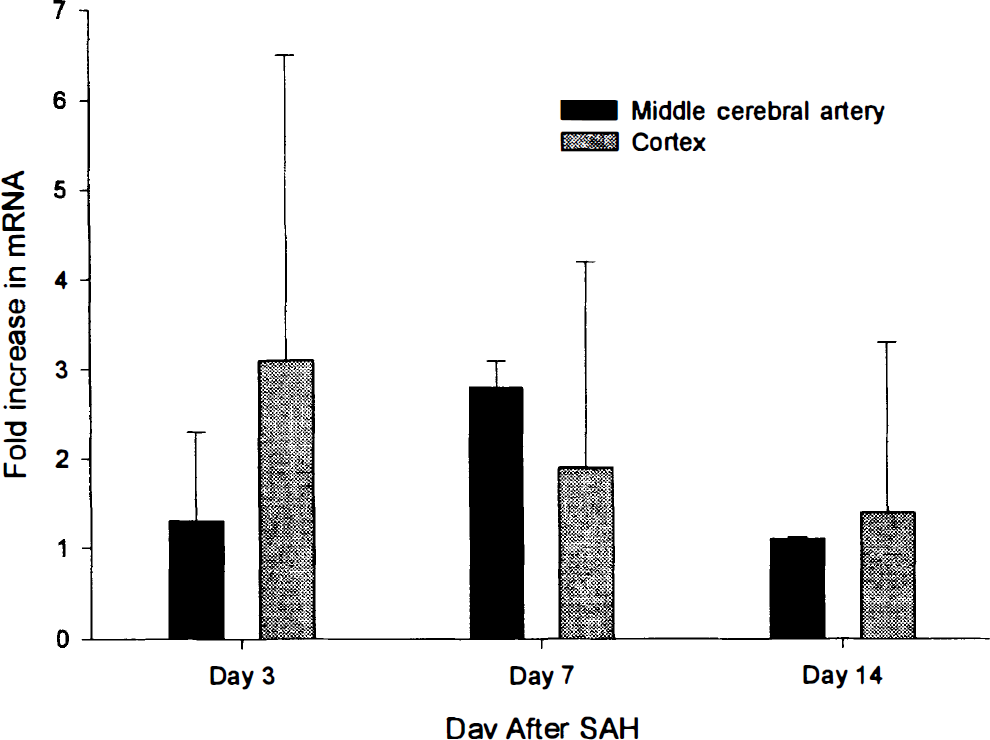
Bar graph of ratio of ferritin mRNA, measured by reverse transcription polymerase chain reaction, in hemorrhage- compared to control-side middle cerebral arteries and cortex. There are no statistically significant changes in ferritin mRNA in either tissue at any time after subarachnoid hemorrhage (SAH). Values are means ± SD; n = 3 per group.
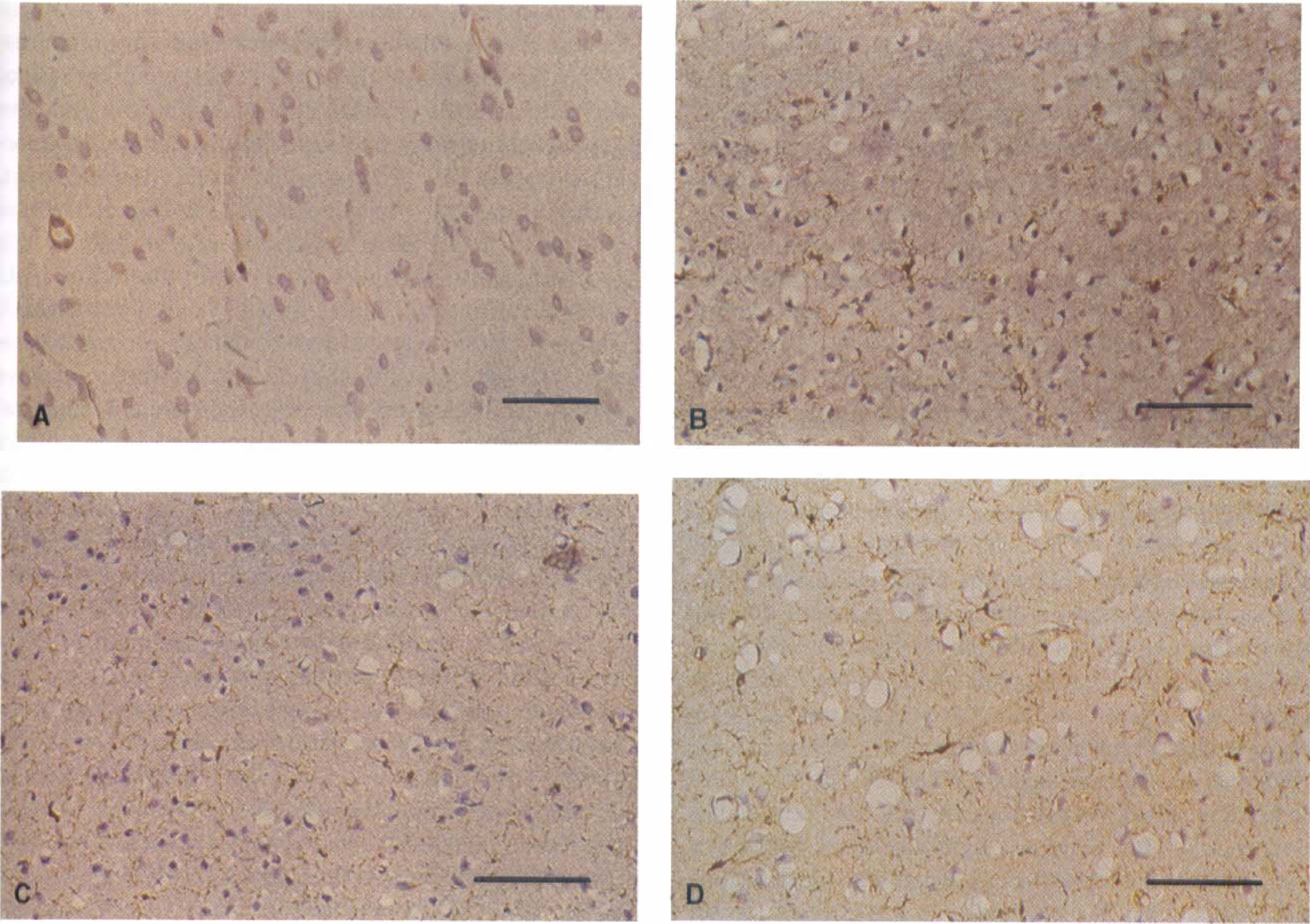
Immunohistochemistry for HO-1 showed HO-1 protein in cells in the fibroblasts in the adventitia of the wall of the right, hemorrhage-side anterior cerebral artery (Fig. 5). There was, qualitatively, more staining in these cells and a greater number of cells stained 3 days after SAH compared with 7 and 14 days. Only very occasional positively stained cells were observed 3 and 7 days after SAH in the left, control-side anterior cerebral arteries, but the cells were always less abundant than on the right, hemorrhage side. HO-1 immunoreactivity was not observed within the smooth muscle cells of the arterial wall at any time. By 14 days after SAH, there were abundant macrophages and cells derived from the arachnoid in the subarachnoid space around the right-sided cerebral arteries, and some of these cells contained HO-1 immunoreactivity. This immunoreactivity was qualitatively less intense than that observed 3 days after SAH. The time course of the changes was consistent with the results of Western blotting.
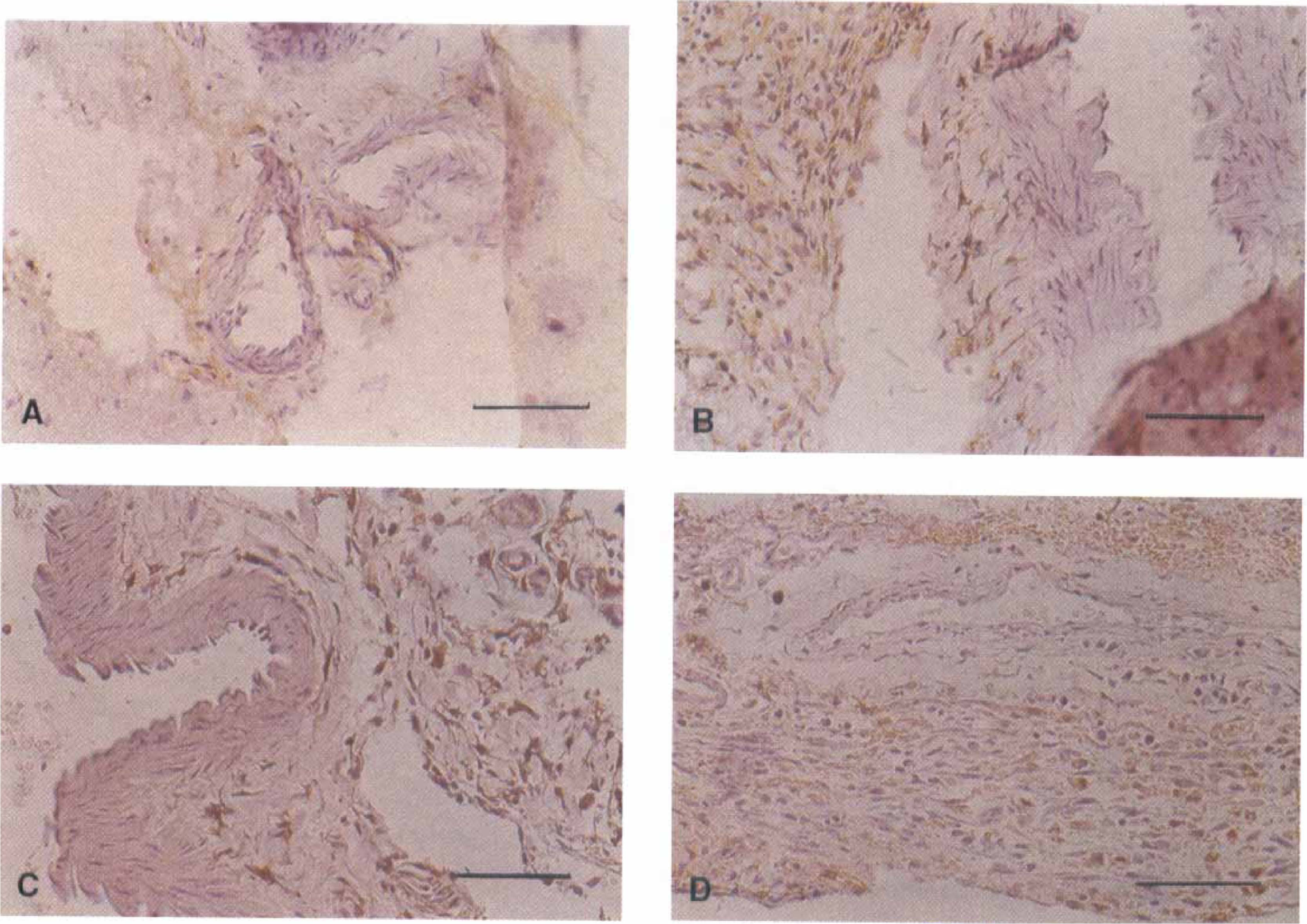
Photomicrographs of immunohistochemical staining for HO-1 of hemorrhage- and control-side anterior cerebral arteries 3, 7, and 14 days after subarachnoid hemorrhage (SAH). A control-side artery 3 days after SAH
Immunohistochemistry for ferritin was consistent with the results of Western blotting in that there was more abundant ferritin immunoreactivity in the hemorrhage-side, compared with the control-side, anterior cerebral arteries at all times after SAH (Fig. 6). The immunoreactivity was confined to fibroblasts in the arterial adventitia and to cells in the subarachnoid space around the anterior cerebral artery. Three days after SAH, there was less immunoreactivity than at later times, and that which was present was within fibroblasts in the adventitia of the arterial wall. At 7 and 14 days after SAH, there was an increase in intensity of staining within the fibroblasts in the arterial wall adventitia, and there also was development of immunoreactivity within the cells that appeared in the subarachnoid space. These cells consisted of macrophages and proliferating cells derived from the arachnoid. No immunoreactivity was observed in the smooth muscle or endothelial cells in the tunica media and intima of the arteries.
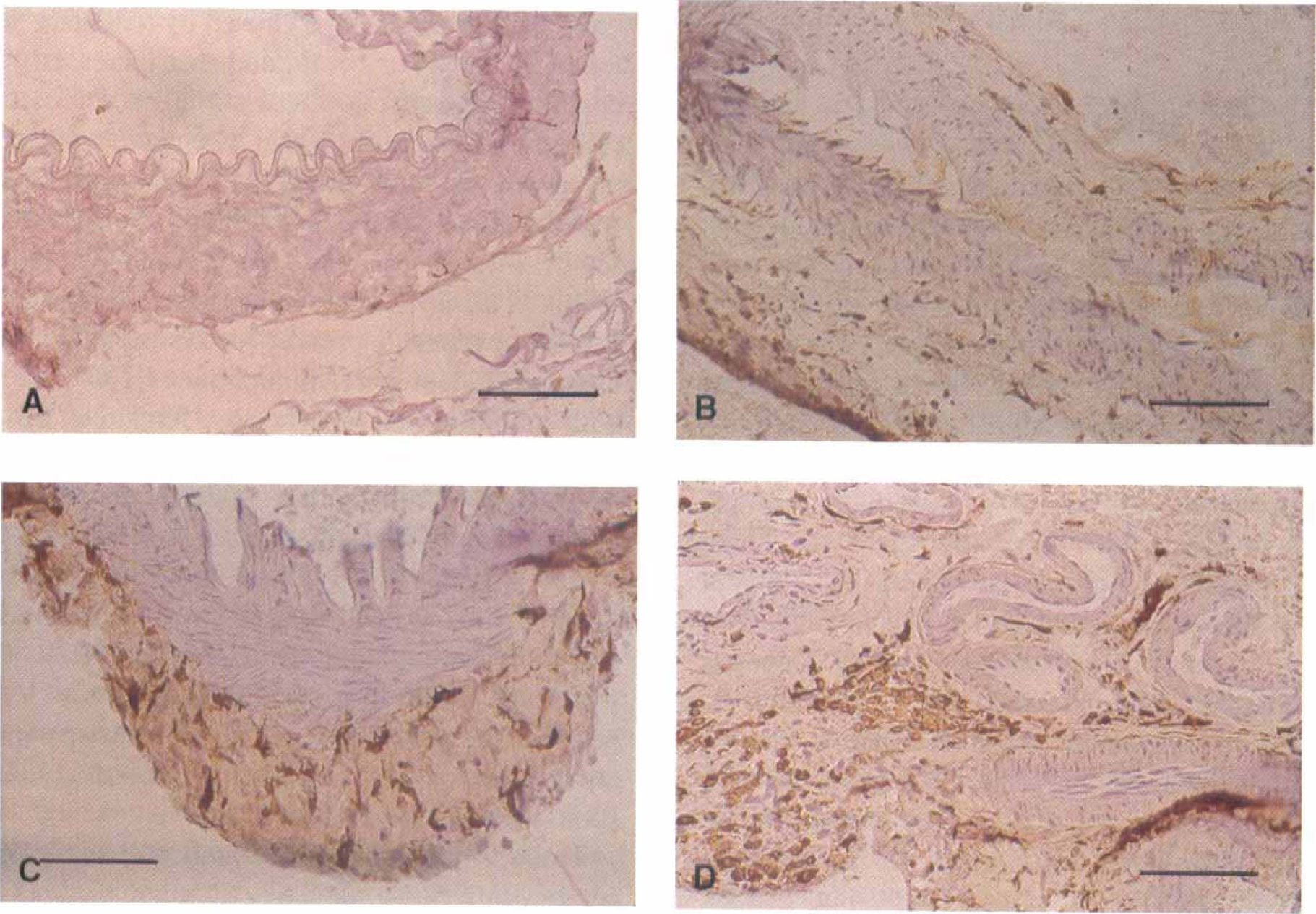
Photomicrographs of immunohistochemical staining for ferritin of hemorrhage- and control-side anterior cerebral arteries 3, 7 and 14 days after subarachnoid hemorrhage (SAH). There is minimal ferritin immunoreactivity in the control-side artery 3 days after SAH
Changes in brain tissue
Western blotting of the cerebral cortex and white matter adjacent to the middle cerebral arteries failed to detect HO-1 protein. In a similar manner, the mRNA for HO-1 could not be detected at any time after SAH, which suggested that the concentration of mRNA was therefore less than approximately 10−12 mol/L.
Western blotting of brain tissue for ferritin showed higher levels of ferritin than in arterial tissue but no significant changes between the right and left sides at any time after SAH (Fig. 7). In a similar manner, competitive RT-PCR showed that there were no significant changes in ferritin mRNA at any time after SAH (Fig. 4).
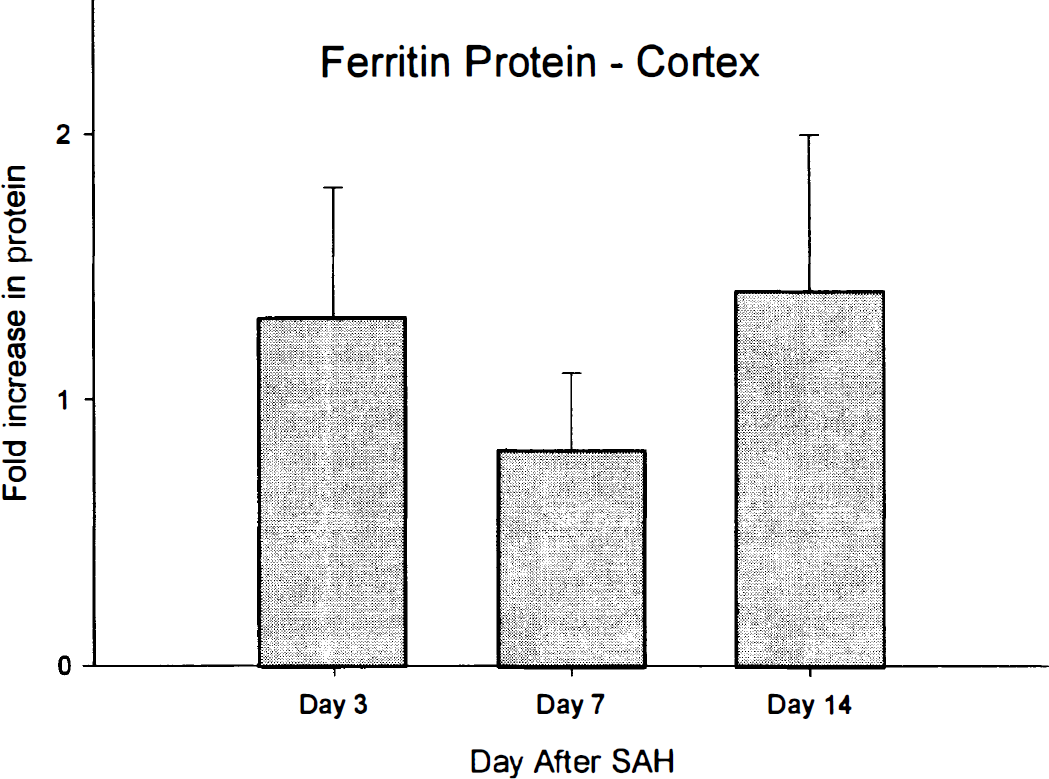
Bar graph of ratio of ferritin protein, measured by Western blotting, in hemorrhage- compared with control-side brain tissue. There were no significant changes in ferritin protein at any time after subarachnoid hemorrhage (SAH).
Immunohistochemistry for ferritin showed abundant ferritin immunoreactivity in cells throughout the brain on both hemorrhage sides and control sides at all times after SAH (Fig. 8). Most cells that contain ferritin immunoreactivity had morphology that was consistent with microglia. They had small, round or rod-shaped cell bodies with round nuclei and two to four short cell processes that branched only a few times. Immunoreactivity to HO-1 was not observed in the brain (Fig. 8).
Photomicrographs of immunohistochemical staining for HO-1
Correlation with vasospasm
Comparisons over time showed that there was a significant decrease in HO-1 protein as time after SAH increased (P < 0.05, analysis of variance), with there being significantly less of an increase in HO-1 protein 14 days after SAH compared with 3 days after SAH. Regression of the reduction in hemorrhage-side compared with control-side middle cerebral artery diameter at the time of death, against the fold difference in HO-1 protein, showed that the more severe the arterial narrowing, the greater the increase in HO-1 protein (r2 = 0.34, P = 0.10). There was no relation between time after SAH or middle cerebral diameter reduction and ferritin protein levels.
DISCUSSION
The novel findings of this study are as follows: (1) Placement of a subarachnoid blood clot in monkeys caused significant vasospasm 3 and 7, but not 14, days later; (2) this is the first study of HO-1 mRNA and protein, and ferritin mRNA and protein, in a large-animal model of vasospasm and subarachnoid hemorrhage; (3) vasospasm was associated with a pronounced increase in HO-1 protein by 3 days after SAH, which decreased but remained significantly greater than in control arteries 7 and 14 days after SAH; (4) there was some correlation between the change in HO-1 protein and vasospasm, with both being maximal 3 days after SAH and then decreasing with increasing time; (5) changes in ferritin have not been described after SAH, and we report that there was an increase in ferritin protein that was slightly delayed compared with HO-1 protein, which became significant by day 7 and persisted for at least 14 days; (6) the increase in ferritin did not correlate directly with the time course of vasospasm but, in fact, remained elevated after the vasospasm resolved; and (7) the increases in HO-1 and ferritin proteins were confined to cells in the adventitia of the cerebral arteries.
Suzuki et al. (1999) reported that HO-1 mRNA was increased in the basilar artery of rats after SAH and that the increase correlated with the time course of vasospasm. HO-2 mRNA was not increased. Our measurements of concentrations of HO-1 protein are similar to and extend the findings of Suzuki et al. (1999), by showing that the increase in HO-1 mRNA is translated into HO-1 protein and that the time course is similar to that of angiographic vasospasm in a model that replicates the disease more closely than the rat model. Suzuki et al. (1999) administered antisense HO-1 oligonucleotide to rats with SAH. This prevented the SAH-induced increase in HO-1 mRNA and aggravated angiographic vasospasm, which suggests that induction of HO-1 acts to decrease vasospasm. This is consistent with experiments that show that increases in HO-1 protect several different types of cultured cells from hemoglobin (Abraham et al., 1995), hemin (Balla et al., 1993), and oxidative stress (Lee et al., 1996; Motterlini et al., 1996; Suttner et al., 1999; Yachie et al., 1999). Mice that lack HO-2 exhibit enhanced hyperoxia-induced lung damage (Dennery et al., 1998). An increase in HO-1 protects the kidney from rhabdomyolysis in vivo (Nath et al., 1992), the lung from hyperoxia (Otterbein et al., 1999), and the pleural cavity from inflammation (Willis et al., 1996). Overexpression of HO-1 protects against permanent focal cerebral ischemia in mice (Panahian et al., 1999). In contrast, several studies showed that HO-1 induction does not protect cultured cells from damage caused by oxidative stress (Nutter et al., 1994) or hemin and that increased ferritin that occurs in response to these stimuli is the cytoprotective response (Balla et al., 1992). Changes in ferritin were not measured in some cases where cytoprotective effects were attributed to HO-1 (Abraham et al., 1995; Lee et al., 1996; Otterbein et al., 1995; Takizawa et al., 1998; Willis et al., 1996), which suggests that the relative contributions of each protein to the protective effects are undecided. However, induction of HO-1 is associated with oxidative stress; whereas, ferritin is not altered in hamster fibroblasts (Dennery et al., 1996), and adenoviral-mediated transfection of lung tissue in vivo with HO-1 protects from hyperoxia without altering ferritin (Otterbein et al., 1999). Therefore, both HO-1 and ferritin may mediate cell resistance to oxidative stress, hemin, and hemoglobin (Nath et al., 1992). The results presented herein in a primate model show a correlation between the induction of HO-1 and vasospasm but do not allow a conclusion to be drawn about whether this response is detrimental or protective.
This study is the first to examine changes in ferritin in cerebral arteries after SAH. One previous investigation showed that patients with “cerebral bleeding” had increased cerebrospinal fluid ferritin concentrations that peaked 4 to 6 days after the ictus (Hällgren et al., 1980). We have shown that hemoglobin increases ferritin protein in cultured rat cerebrovascular smooth muscle cells (Macdonald et al., 1997). Ferritin is an iron-binding protein whose expression may increase in a delayed fashion by stimuli that first induce HO-1 (Balla et al., 1992; Vile and Tyrrell, 1993; Vile et al., 1994). Not all stimuli that increase HO-1 expression are associated with increased ferritin expression, however; and ferritin may increase independent of HO-1 (Carraway et al., 1998; Dennery et al., 1996). After SAH, however, we report that ferritin protein increased progressively for up to 14 days after SAH and that the increase follows the increase in HO-1. SAH was associated with ferritin H chain expression in cerebral artery adventitial fibroblasts and in periadventitial cells. The proportion of ferritin H and L chains varies in different cells and tissues (Harrison and Arosio, 1996). Ferritin L chain is expressed predominately in spleen and liver. The L chain is involved primarily in iron storage. Ferritin H chain is important in intracellular iron transport and possesses ferroxidase activity that converts ferrous to ferric iron, a critical step in the sequestration of iron in ferritin. Ferritins rich in H chains predominate in tissues such as heart and pancreas. The selective increase in ferritin H chain after SAH may be particularly important, because the H chain was reported to be specifically involved in the antioxidant activity of ferritin (Balla et al., 1992).
We report that the increase in HO-1 protein is most pronounced earlier after SAH and then declines, whereas ferritin protein tends to increase with time. This is consistent with patterns seen in other studies and with the concept that HO-1 metabolizes heme and releases iron that then induces ferritin protein expression (Carraway et al., 1998; Eisenstein et al., 1991). The times examined in this study were selected based on the time course of vasospasm in humans. Previous studies that used this model measured angiographic vasospasm at 7 and 14 days after SAH and found similar vasospasm to that in humans (Espinosa et al., 1984; Macdonald et al., 1992). We assumed that because vasospasm starts 3 days after SAH in humana, a similar time course would occur in monkeys. This study shows, however, that vasospasm is already maximal or near maximal 3 days after SAH in monkeys. Studies in humans suggest that vasospasm may have an earlier onset after severe SAH (Disney et al., 1988). We speculate that the large volume of SAH produced in the monkey model causes vasospasm to develop fairly rapidly. We report that HO-1 protein already is significantly elevated 3 days after SAH. The time course of vasospasm and of elevations in HO-1 mRNA and protein at earlier times in the onset of vasospasm may need to be defined in additional studies. Another consideration is that we compared changes on the side of surgery plus subarachnoid hemorrhage to a normal, nonmanipulated artery. Therefore, we cannot exclude the possibility that some of the changes were the result of surgery themselves. This seems unlikely because other stressful manipulations, such as saline injection into the cisterna magna, did not increase HO-1 in rats (Matz et al, 1996a). HO-1 may be induced by stress, although this has been shown to be a transient response that resolves within 48 hours (Motterlini et al., 1998).
What is the significance of alterations in HO-1 and ferritin after SAH? In previous studies, we found that 5 or more days after SAH in a monkey model, blood clot is still present but vasospasm is resolving (M. Stoodley, unpublished data). We postulate that induction of ferritin may be important in the resolution of vasospasm because it persists during the resolution of vasospasm, in contrast to induction of HO-1, which is diminishing during this time. Further studies are needed to determine if purposely increasing HO-1, ferritin, or both after SAH will affect vasospasm.
The importance of the increase in HO-1 and ferritin protein is predicated on the theory that hemoglobin and/or its breakdown product(s) cause vasospasm and that a direct effect of these substances on cells is important in the pathogenesis of vasospasm. This theory is not proven and there are other possibilities (Guan et al., 1998; Wang et al., 1999; Zhang et al., 1995). Pure hemoglobin does not increase intracellular calcium in isolated cerebral smooth muscle cells, yet it contracts cerebral arteries in vitro (Zhang et al., 1995). The mechanism of contraction may be largely indirect and mediated, at least in part, by abrogation of the effect of vasodilating NO. HO-1 is an intracellular protein that is not known to be secreted and would not protect against indirect effects of hemoglobin such as binding of NO. This study is the first to localize histologically HO-1 and ferritin proteins in cerebral arteries after SAH. We found that HO-1 and ferritin proteins are increased in cells in the adventitia of cerebral arteries and in the subarachnoid space around the vessels. Normal cerebral blood vessels have not been studied in detail, but systemic arteries have HO-2 in their smooth muscle and endothelial cells and have none or low concentrations of HO-1 in the unstressed state (Maines, 1997). No protein was detected in smooth muscle cells of the arterial walls. Ferritin is secreted and might have extracellular effects, although HO-1 is not known to occur extracellularly (Harrison and Arosio, 1996; Maines, 1997). In contrast, vasospasm did develop despite an increase in HO-1 protein that was of a magnitude that protects cells in vivo (Lee et al., 1996; Motterlini et al., 1996; Suttner et al., 1999; Yachie et al., 1999), which suggests that increases in HO-1 are not sufficient to prevent vasospasm in primates. It is noteworthy that we used the anterior cerebral arteries for immunohistochemistry. Although they develop vasospasm in this model (Table 2), it is not as severe as the spasm of the middle cerebral artery, and it remains possible that examination of more severely affected arteries would show induction of HO-1 and/or ferritin in other cells in the arterial wall. Our preliminary results with the use of middle cerebral arteries, however, have shown similar locations of HO-1 and ferritin, 7 and 14 days after SAH.
Induction of HO-1 may protect against vasospasm by metabolizing hemin released from hemoglobin to biliverdin, carbon monoxide, and iron. Biliverdin may be metabolized to bilirubin, which may have spasmogenic (Duff et al., 1988; Macdonald et al., 1991) and antispasmogenic effects inasmuch as it is an antioxidant (Stocker et al., 1987). Carbon monoxide can activate guanylate cyclase and thereby induce vascular relaxation, although whether this occurs in cerebral arteries is controversial (Brian et al., 1994). Iron may be toxic, but the attendant increase in ferritin may sequester it and, thus, prevent its ability to catalyze injurious oxidant reactions (Balla et al., 1992; Nutter et al., 1994). That HO-1 is an intracellular protein that breaks down hemin has important implications for the role of increased HO-1 in the prevention of vasospasm. Hemoglobin breakdown products such as hemin that act intracellularly theoretically could be involved in vasospasm (Letarte et al., 1993). If this were true, then induction of HO-1 would have to occur specifically in these cells to decrease vasospasm. Our results indicate that HO-1 and ferritin were increased in adventitial fibroblasts but not in smooth muscle cells of vasospastic arteries. This implies that hemoglobin and hemin do not reach the smooth muscle cells directly after SAH and is consistent with immunohistochemical studies that show that hemoglobin penetrates into the arterial wall after SAH but is most abundant in the adventitia not the media (Foley et al., 1993). Turner et al. (1998) suggested that heme was released from hemoglobin and transported into microglia. In view of the possible lack of exposure of arterial smooth muscle to hemoglobin, an indirect effect of HO-1 metabolites such as CO and bilirubin, which may be released to act to relax smooth muscle cells or prevent oxidant effects on them, may be important. These possibilities must be addressed when considering the use of HO-1 protein for the prevention of vasospasm.
The lack of significanct change in ferritin mRNA is consistent with known mechanisms of regulation of ferritin (Eisenstein et al., 1991; Hentze and Kühn, 1996). Ferritin expression is regulated mainly posttranscriptionally. We did not detect HO-1 mRNA at any time after SAH. There was already a pronounced increase in HO-1 protein 3 days after SAH. Because HO-1 is believed to be regulated mainly at the transcriptional level and this seems to be the case in cerebral smooth muscle cells (Macdonald et al., 1997), it may be that HO-1 mRNA was increased earlier than 3 days after SAH and had already returned to low concentrations by this time. Technical problems may also explain our inability to detect HO-1 mRNA in these samples.
We did not find an increase in HO-1 protein in brain tissue adjacent to the SAH. Normally, there is minimal or no HO-1 immunoreactivity in human and rat brain neurons and astrocytes and some reactivity in ependymal cells (Ewing et al., 1992; Maines, 1997; Schipper et al., 1995). HO-2 is abundant in many normal neurons (Maines, 1997). Other studies, however, have documented an increase in HO-1 immunoreactivity predominately in microglia but also in occasional astrocytes for up to 2 days after SAH in rats (Matz et al., 1996a, b ). Injection of lysed blood or oxyhemoglobin into the cisterna magna produced similar results, and Western blotting confirmed the presence of HO-1 protein after oxyhemoglobin injection, which suggests that the increase in HO-1 is caused by oxyhemoglobin (Kuroki et al., 1998; Matz et al., 1996a, b ; Turner et al., 1998). Explanations for why we did not detect an increase in HO-1 include a lack of sensitivity of Western blotting to induction of HO-1 focally in microglia. This is unlikely because we did not observe microglial activation in the brain adjacent to the SAH. Technical problems with the antibody also are possible. Another possibility is that there are differences between changes induced by clotted blood placement in the subarachnoid space and injection of lysed blood products into the cisterna magna. The intracellular contents of fresh erythrocytes are different, and the effects of application of these hemolysate solutions are different from those of erythrocytes that have been incubated in vivo or in vivo for days (Guan et al., 1998; Zhang et al., 1995). The concentration of adenosine triphosphate declines rapidly, hemoglobin oxidizes, and there are probably many other changes, some of which might influence responses of the brain tissue to the erythrocyte contents. Roost et al. (1972) noted that bilirubin was formed in cerebrospinal fluid within hours of injection of methemalbumin, hemoglobin, or whole blood into the cisterna magna of rats. HO activity was found to be induced in the arachnoid and choroid plexus but not in the brain. This result is consistent with our observations based on Western blotting and immunohistochemistry. The most pronounced increase in HO-1 protein was not in the underlying brain but in cerebral arteries that may be removed with the arachnoid during such dissections in rats.
We did not find significant effects of SAH on ferritin in the cerebral cortex adjacent to the SAH. Intraventricular hemorrhage in neonates was reported to be associated with SAH over the cerebellum (Ozawa et al., 1994). In neonates dying with such SAH, 5 to 30 days after birth, ferritin-positive microglia were noted in the molecular layer of the cerebellum. We did find ferritin immunoreactivity in microglia, which is consistent with the known location of this protein, although no induction of microglia and of ferritin was noted in the brain after SAH (Grundke-Iqbal et al., 1990).
In conclusion, HO-1 and ferritin are increased in cerebral arteries after SAH. The increase in HO-1 is transient, whereas the increase in ferritin in arterial adventitial fibroblasts and cells in the subarachnoid space persists during the resolution of vasospasm. Further experiments are necessary to determine whether modulation of these responses would affect the severity and time course of the vasospasm that occurs after SAH.