
Abstract
Heme oxygenase (HO) is the rate-limiting enzyme in the degradation of heme to produce bile pigments and carbon monoxide. The HO-1 isozyme is induced by a variety of agents such as heat, heme, and hydrogen peroxide. Evidence suggests that the bile pigments serve as antioxidants in cells with compromised defense mechanisms. Because hypoxia-ischemia (HI) increases the level of oxygen free radicals, the induction of HO-1 expression in the brain during ischemia could modulate the response to oxidative stress. To study the possible involvement of HO-1 in neonatal hypoxia-induced ischemic tolerance, we examined the brains of newborn rat pups exposed to 8% O2 (for 2.5 to 3 hours), and the brain of chronically hypoxic rat pups with congenital cardiac defects (Wistar Kyoto; WKY/ NCr). Heme oxygenase-1 immunostaining did not change after either acute or chronic hypoxia, suggesting that HO-1 is not a good candidate for explaining hypoxia preconditioning in newborn rat brain. To study the role of HO-1 in neonatal HI, 1-week-old rats were subjected to right carotid coagulation and exposure to 8% O2/92% N2 for 2.5 hours. Whereas HO enzymatic activity was unchanged in ipsilateral cortex and subcortical regions compared with the contralateral hemisphere or control brains, immunocytochemistry and Western blot analysis showed increased HO-1 staining in ipsilateral cortex, hippocampus, and striatum at 12 to 24 hours up to 7 days after HI. Double fluorescence immunostaining showed that HO-1 was expressed mostly in ED-1 positive macrophages. Because activated brain macrophages have been associated with the release of several cytotoxic molecules, the presence of HO-1 positive brain macrophages may determine the tissue vulnerability after HI injury.
Hypoxia induces the expression of a set of stress proteins called oxygen-regulated proteins, which have been implicated in the development of drug and radiation resistance in tumor cells (Heacock and Sutherland, 1990). Among these proteins, heme oxygenase-1 (HO-1; also called HSP32) has received increasing attention (Maines, 1992). Heme oxygenase catalyzes the degradation of heme molecules derived from hemoproteins such as cytochrome P-450, nitric oxide synthase, tryptophan pyrrolase, and several peroxidases and catalases, to biliverdin, ferrous iron, and carbon monoxide (CO). Biliverdin is then converted to bilirubin by biliverdin reductase (Maines, 1992). Two different gene products that both contribute to HO enzymatic activity (microsomal isozymes HO-1 and HO-2) have been characterized (Maines et al., 1986; Shibahara et al., 1993). Whereas HO-2 activity is refractory to most types of stress or injury, HO-1 is induced by various stimuli. In healthy unstressed adult rat brain, most of HO activity has been attributed to the HO-2 isozyme whereas the HO-1 isozyme seems to be present only at very low levels (Sun et al., 1990; Ewing and Maines, 1991, 1992; Maines, 1992).
The 5′-flanking promoter region of HO-1 gene contains several important regulatory elements including at least one copy of the following: a heat shock element (Shibahara et al., 1987; Okinaga and Shibahara, 1993), a site for nuclear factor-kappa B (NFkB) (Lavrosky et al., 1994), an AP-l-like binding site (Müller et al., 1987; Alam and Zhining, 1992), and a metal regulatory element (Müller et al., 1987) that may modulate the responses to heat shock or denatured proteins, hypoxia or oxidative stress, Fos/Jun immediate early genes, and heme, respectively. Indeed, after global and focal ischemia (Paschen et al., 1994; Takeda et al., 1994; Nimura et al., 1996), hyperthermia (Ewing and Maines, 1991) and subarachnoid administration of lysed blood or hemoglobin (Matz et al., 1996; Turner et al., 1997) in adult rats, as well as heat or hydrogen peroxide exposure in primary cultures of neurons and astrocytes (Dwyer et al., 1995), the induction of HO-1 messenger RNA and protein is observed mainly in nonneuronal cells. Increased HO-1 messenger RNA and protein have also been reported in rat brain after glutathione depletion (Ewing and Maines, 1993). There is evidence to suggest that the bilirubin produced by HO activity may serve as an anti-oxidant in cells with compromised defense mechanisms (Stocker et al., 1987). This may be important for neurons which have low levels of the common antioxidants glutathione and ascorbate (Raps et al., 1989). Because hypoxia and ischemia have been associated with increased free oxygen species (Siesjö et al., 1989), the production of antioxidants by HO-1 could help protect the brain from oxidative injury.
Hypoxia pretreatment has been shown to confer neuroprotection against hypoxic-ischemic (HI) injury in newborn rats (Gidday et al., 1994). Though the molecular mechanism underlying this hypoxic preconditioning is still poorly understood, HO-1 could play a role because it is an oxygen-regulated protein. Indeed, protection against subsequent lethal insults has been shown after prior induction of HO-1 either by UVA exposure in skin fibroblasts (Vile et al., 1994), hemoglobin pretreatment in rat (Otterbein et al., 1995), or overexpression in rabbit coronary microvessel endothelial cells (Abraham et al., 1995). To elucidate the role of HO-1 in neonatal hypoxia and ischemia, the present study describes the effect of acute and chronic hypoxia and the effect of ischemia on HO-1 expression in newborn rat brain. In view of the suggested protective effects of hypoxia preconditioning against ischemic brain damage in the neonatal rat (Gidday et al., 1994), we also investigated the role of oxygen-regulated HO-1 protein as a potential candidate responsible for neonatal hypoxia-induced ischemic tolerance.
MATERIALS AND METHODS
Animal preparation
All procedures were performed in accordance with the National Institutes of Health Guide for Care and Use of Laboratory Animals, and all protocols were approved by the University of California at San Francisco Committee on Animal Research. Male and female Sprague-Dawley rats (Bantam Kingman, Fremont, CA, U.S.A.) were used. Seven litters each containing 10 Sprague-Dawley pups at postnatal day 7 (P7), 4 litters each containing 7 to 10 normotensive chronically hypoxic Wistar Kyoto pups at P7 (WKY/NCr) bearing various combinations of cardiac anomalies including hypertrophic cardiomyopathy, defects of the aortic arch system, and Tetralogy of Fallot (Kuribayashi et al., 1990) and, 3 litters of 10 P7 Wistar pups as the control strain were used as previously described (Rice et al., 1981; Ferriero et al., 1990). Rat pups at P7 were used because their brain maturity is grossly comparable to that of a late-term gestation human fetus or newborn infant (Dobbing and Sands, 1979). After anesthesia with a gas mixture containing 1% halothane in 70% N2O and 30% O2, rat pups underwent the right common carotid artery coagulation through a ventral midline neck incision. The wound was sutured and pups returned to their dam for 2 hours. Pups were then placed in an 8% O2/92% N2 humidified atmosphere in a chamber partially submerged in a water bath maintained at a constant temperature of 37°C (HI group; coagulation, hypoxia). Sham-operated animals underwent the same operative procedure except that the carotid artery was not ligated (hypoxia-treated group; no coagulation, hypoxia). Control untreated group (no coagulation, no hypoxia) animals and a group of coagulation-only animals (with no hypoxia; n = 5) were also studied. Because the latter two groups showed no difference in cell integrity and HO-1 expression, the control animals shown in the present study refer to the untreated control group (no coagulation, no hypoxia). In general, animals from each litter were divided into control (1 to 2 per litter), hypoxia-treated (2 to 3 per litter), and HI groups (6 to 7 per litter). The systemic oxygen saturation in live P7 animals (untreated WKY/NCr and normal Wistar) was determined with an oxygen transducer probe (Oxisensor II N-25, Nellcor Inc., Hayward, CA, U.S.A.) wrapped around the pups abdomen and connected to a Nellcor Pulse Oximeter.
Western blot analysis
At 24 hours after hypoxia or HI, rats were deeply anesthetized with an intraperitoneal injection (0.3 g/kg) of Nembutal (pentobarbital sodium; Abbott Lab, North Chicago, IL, U.S.A.) and killed by decapitation. Brains were quickly removed and dissected on ice. The cerebral cortex, hippocampus, and striatum from each hemisphere were placed in Laemmli solubilizing buffer (2.5% sodium dodecyl sulfate, 10% glycerol, 62.5 mmol/L Tris-HCl, pH 6.8, 5% 2-mercaptoethanol) and boiled for 10 minutes. The whole tissue extract was then frozen at −70°C. Western immunoblot analysis was performed as described previously (Bergeron et al., 1996) with modifications. Protein concentration was determined using the bicinchoninic acid method (Pierce, Rockford, II, U.S.A.). Equal amounts (55 µg) of protein per sample were separated on 12% sodium dodecyl sulfate polyacrylamide gels with 4.5% stacking gel. After electrotransfer onto a nitrocellulose membrane (0.2 µm; Schleicher and Schuell, Keene, NH, U.S.A.), immobilized proteins were stained with Ponceau solution to verify equal protein loading. After a brief rinse in deionized water, the membranes were incubated overnight at 4°C in 0.1 mol/L sodium phosphate buffer (PB) pH 7.4, containing 5% nonfat dry milk, 1% bovine serum albumin (BSA) and 0.1% Tween-20, rinsed briefly in 0.1 mol/L PB containing 1% BSA and 0.1% Tween-20, then incubated for 2 hours with a 1:3500 dilution of rabbit polyclonal anti-rat HO-1 antibody (StressGen, Victoria, BC, Canada). This polyclonal antibody, raised against rat liver purified HO-1 protein, was originally described by Maines et al., (1986). After three washes, membranes were incubated with a 1:2500 dilution of anti-rabbit Ig-horseradish peroxidase antibody (Amersham, Arlington Heights, IL, U.S.A.) for 1.5 to 2 hours. Finally, the membranes were washed three times and the bound antibody was visualized with the ECL chemiluminescence system according to the manufacturer's protocol (Amersham). A computer-based imaging system (MCID, Imaging Research, St-Catherines, Ontario, Canada) was used to measure the areas of HO-1 protein on Western immunoblot autoradiograms. The relative density of HO-1 protein (32 kDa) bands was analyzed after subtraction of the film background.
Immunocytochemistry
At the appropriate times after each treatment, rats were anesthetized with Nembutal and perfused through the left ventricle with cold 4% paraformaldehyde made up in 0.1 mol/L PB, pH 7.4. Brains were removed from the skulls, postfixed in 4% paraformaldehyde for 1 to 4 hours and stored in a 30% sucrose overnight at 4°C. Fifty micrometer-thick coronal sections were cut on a vibratome and washed twice with 0.05 mol/L PB. After 1 hour incubation in a peroxidase-inhibiting solution (0.65% sodium azide and 0.2% hydrogen peroxide in 0.05 mol/L PB, pH 7.4), sections were incubated for 2 hours in a blocking solution (5% nonfat dry milk, 2% goat serum, 1% BSA, 0.1% Triton X-100, and 0.1% rat serum, made up in 0.1 mol/L PB, pH 7.4). The sections were then incubated for 12 to 48 hours at 4°C with the same anti-rat HO-1 antibody used for Western blotting (StressGen, Victoria, BC, Canada) diluted 1:4000 in 2% goat serum, 1% BSA, 0.1% Triton X-100, made up in 0.1 mol/L PB, pH 7.4. Alternate sections from each brain were incubated without primary antibody (as negative controls). After three 10 minute-washes in 0.05 mol/L PB, sections were incubated at room temperature for 2 hours with a 1:200 dilution of biotinylated goat anti-rabbit IgG antibody (Vector Laboratories, Burlingame, CA, U.S.A.). Sections were then incubated in an avidin-horseradish peroxidase solution (Elite Vectastain, Vector Laboratories) for 2 hours, followed by three washes with PB. Staining was visualized with 0.015% diaminobenzidine (Sigma, St-Louis, MO, U.S.A.) and 0.001% hydrogen peroxide. Sections were then washed, mounted on gelatin-coated slides and coverslipped.
Double immunofluorescence labeling
To identify which cell type stained for HO-1, some sections were coincubated with rabbit polyclonal anti-rat HO-1 antibody (1:4000) and either the mouse monoclonal anti-rat antibody ED-1 (uncharacterized cytoplasmic antigen expressed by all cells of the rat monocyte/macrophage lineage; Serotec, Oxford, U.K.; 1:2000), OX-42 (complement type 3 receptor found on both resting and activated monocytes, macrophages, neutrophils, and microglia; Serotec, Oxford, U.K.; 1:4000), or glial fibrillary acidic protein (GFAP) present in astrocytes (ICN, Costa Mesa, CA, U.S.A; 1:4000). All dilutions were performed in 2% goat serum, 1% BSA, 0.1% Triton X-100, made up in 0.1 mol/L PB, pH 7.4. After 24 to 48 hours incubation at 4°C, sections were washed three times for 10 minutes in PB and incubated for 2 hours in the dark with a Texas-Red-conjugated, goat anti-rabbit IgG antibody (1:150; Vector Laboratories) together with a biotinylated goat anti-mouse IgG antibody (1:150; Vector Laboratories). After three 10 minute washes, labeled sections were incubated for 2 hours with avidin-conjugated fluorescein isothiocynate (FITC; Vector Laboratories; 1:150 made up in a solution containing 0.1 mol/L sodium bicarbonate and 0.15 mol/L sodium chloride, pH 8.2). Sections were mounted onto slides and immediately coverslipped with Fluoromount-G (Southern Biotech. Assoc. Inc., Birmingham, AL, U.S.A.). Sections were photographed on a Leitz varioorthomat microscope using a Ploemopak 2.1 fluorescence illuminator. The same area of representative sections was photographed with interchangeable filters for Texas-Red and FITC fluorescence.
Histopathological evaluation
Histopathological scoring of each newborn rat brain was performed blindly on alternate coronal sections stained with cresyl violet (Nissl). Because increased OX-42 and GFAP staining occurs in areas of neuronal loss and brain damage after HI (Sheldon et al., 1996), alternate sections stained with OX-42 and GFAP antibodies were also examined. The scoring scale was as follows: 0, no injury or no detectable neuronal loss; 1, minimal neuronal loss with occasional gliosis; 2, columnar damage in cortex involving predominantly layers II through IV, moderate cell loss with areas of infarction and concomitant gliosis; and 3, severe cell loss and gliosis associated with extensive tissue infarction.
Determination of heme oxygenase enzymatic activity
Twenty-four hours after HI, rat pups were anesthetized with Nembutal and decapitated. Brains were removed and dissected on ice. The cerebral cortex and a subcortical region comprised of hippocampal, striatal, thalamic, and hypothalamic tissue were isolated from each hemisphere. To account for the CO that may be bound to erythrocytes in the cerebral blood vessels, we perfused some animals with cold saline (0.9% sodium chloride) before processing the tissue and found no difference in HO activity compared with nonperfused animals. The tissue was weighted and homogenized by sonication (4 pulses of 1 second; Branson Sonifier Cell Disrupter 185 with microtip) in 4 volumes of ice-cold 0.1 mol/L potassium phosphate buffer, pH 7.4. The protein concentration was determined using the bicinchoninic acid method (Pierce, Rockford, IL, U.S.A.). Heme oxygenase activity was determined using a gas chromatographic method as described previously (Vreman and Stevenson, 1988) with some modifications. Briefly, 20 µL of brain homogenate was reacted with 20 µL of 4.5 mmol/L NADPH (reduced form of nicotinamide adenine dinucleotide phosphate) and 20 µL of 150 µmol/L heme in a septum-sealed, amber-colored vial at 37°C in the dark. For blank values, the NADPH solution was replaced by an equal volume of potassium phosphate buffer. For negative controls, 1 µL of 600 µmol/L chromium mesoporphyrin (potent inhibitor of HO activity) was added to the complete reaction mixture (Vreman et al., 1993). This procedure inhibited the HO-induced CO production down to the blank value, suggesting that the entire CO production in the reaction vials was derived from HO activity. As a positive control, rat pup brain HO activity was induced by a subarachnoid (intracisternal) injection of purified hemoglobin, which has been shown to markedly increase HO-1 expression in adult rat brain (Turner et al., 1997). Twenty-four hours after injection, HO activity values in these brain homogenates ranged from 1.02 to 1.33 nmol CO produced/h/mg protein which is about two- to threefold higher than the activity measured in normal newborn brain (Table 1). After 5 minutes of preincubation at 37°C in the dark, vials were purged with CO-free air and allowed to incubate for an additional 15 minutes. The reaction was stopped by addition of 2 µL of sulfosalicylic acid solution (60% weight-to-volume ratio) and by cooling in wet ice. The amount of CO generated by the enzyme in the vial headspace was analyzed by gas chromatography (Vreman and Stevenson, 1988). Carbon monoxide concentration in tissue extracts was calculated from the peak area of the sample compared with that of CO external standards. The chromatographic assay was linear in the range of CO values obtained with brain tissue homogenates.
HO activity in homogenates of contralateral and ipsilateral cerebral cortex and subcortical regions of P7 newborn rats 24 hours after hypoxia-ischemia

Values represent the mean ± SD of triplicate determinations from “n” animals in each group and are expressed in nanomole of carbon monoxide produced/h/mg protein. Intergroup comparisons (between controls and the contralateral and ipsilateral hemispheres) determined by analysis of variance showed no significant difference.
Statistical analysis
Histopathological scores are reported as median score values. Other data represent the mean ± SD expressed as percent of normal (oxygen saturation data) and nanomole of CO produced/h/mg protein (HO activity data). Intergroup comparisons for HO activity data were performed by one-way analysis of variance. All other data were analyzed by unpaired two-tailed Mann-Whitney nonparametric test.
RESULTS
Heme oxygenase-1 expression in untreated controls
Because there was no difference in the intensity and the profile of HO-1 expression between the brain of untreated controls and the cerebral hemisphere contralateral to the carotid occlusion (Fig. 1, Lanes 1–3 and 4–6, respectively), only the results obtained from the contralateral hemisphere were included in Fig. 2 to Fig. 4. In untreated P7 rats, constitutive HO-1 immunoreactivity was detectable throughout the brain (Figs. 2C,E 3A,B, and 4A) with more intense staining in areas of myelinogenesis of the white matter such as the cingulum, corpus callosum, internal and external capsule, and around the periventricular ependyma (not shown). Double fluorescence staining of normal P7 rat white matter showed that almost all HO-1 positive cells were ED-1 positive macrophages (Fig. 5E,F) and occasionally OX-42 positive microglia (not shown). In addition to the white matter staining, constitutive HO-1 expression was also found in the endothelium throughout the brain, in glia-like cells and some neurons in the cortex (Fig. 4A) and in portions of the hippocampus such as the hilus of the dentate gyrus, the stratum oriens, the pyramidal cell layer and the stratum lucidum of the CA1 to CA3 subfields of Ammons horn (Figs. 2E and 3B).
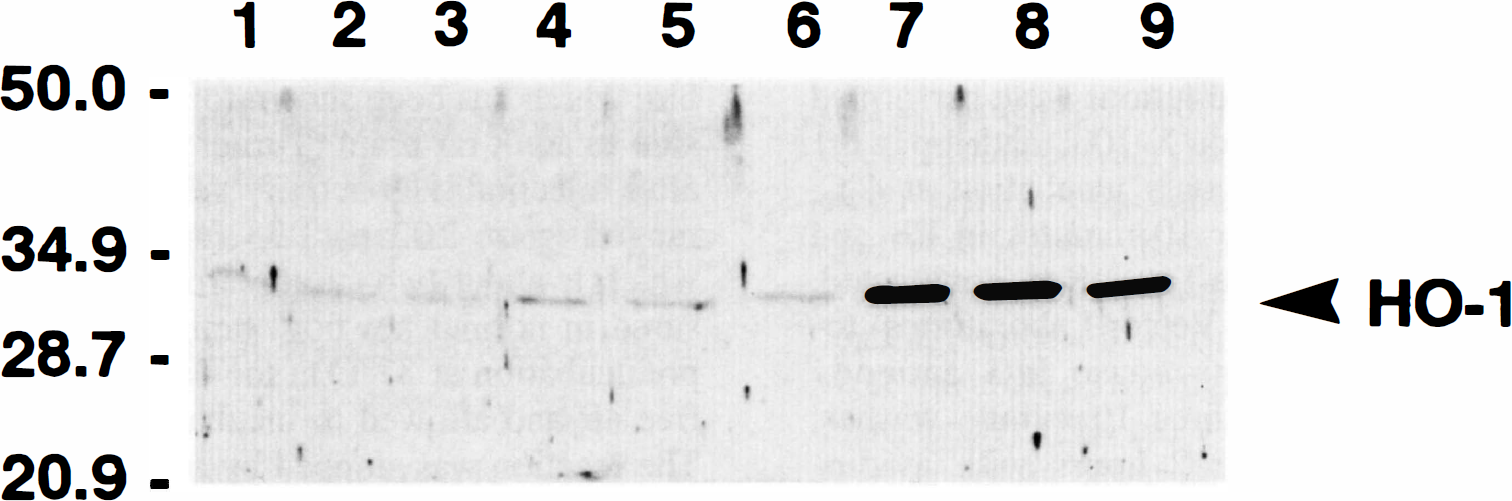
Western immunoblot analysis of rat brain heme oxygenase-1 (HO-1) protein levels, 24 hours after a hypoxic-ischemic insult induced at postnatal day 7 (P7). Whole tissue extracts of striatum (Lanes 1, 4, and 7), hippocampus (Lanes 2, 5, and 8), and cerebral cortex (Lanes 3, 6, and 9) were prepared as described in Materials and Methods and equal protein samples (55 µg) were analyzed by sodium dodecyl sulfate polyacrylamide gel electrophoresis and immunoblotting using rabbit anti-rat HO-1 antibody. Lanes 1 to 3: untreated control rat. Lanes 4 to 6: contralateral hemisphere exposed to hypoxia (8%O2 for 2.5 hours) without the common carotid artery coagulation. Lanes 7 to 9: ipsilateral hemisphere with both the common carotid artery coagulation and exposure to hypoxia (8%O2 for 2.5 hours). Molecular weight markers in kilodalton are indicated on the left. This experiment was repeated four times with similar results. HO-1, heme oxygenase-1.
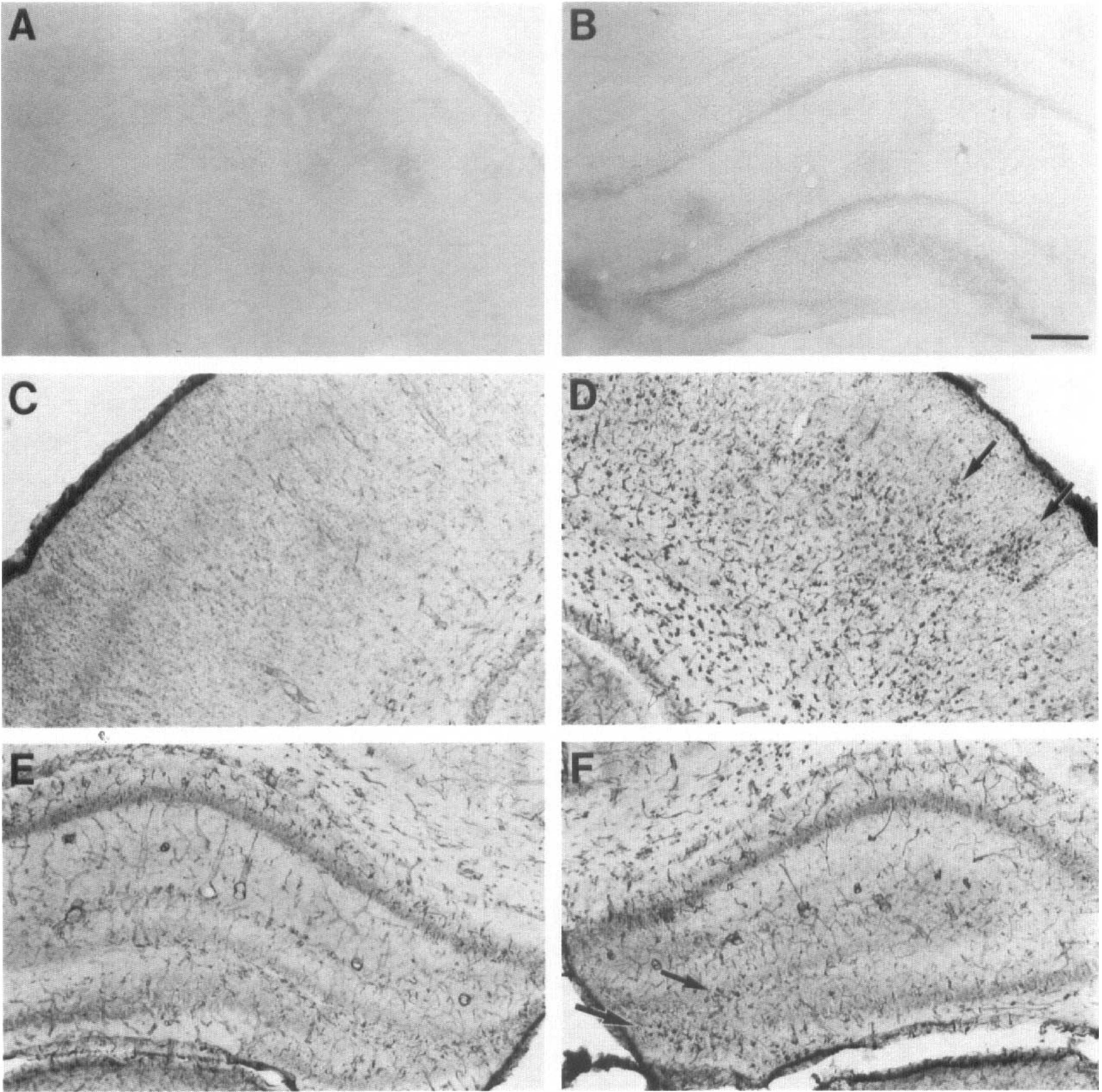
Effect of hypoxia and hypoxia-ischemia on brain HO-1 expression in newborn Sprague Dawley rat. Twenty-four hours after the hypoxic-ischemic insult induced at P7, increased HO-1 immunostaining was observed throughout the cortex (D) and in the hilus of the dentate gyrus of the hippocampus (arrows in
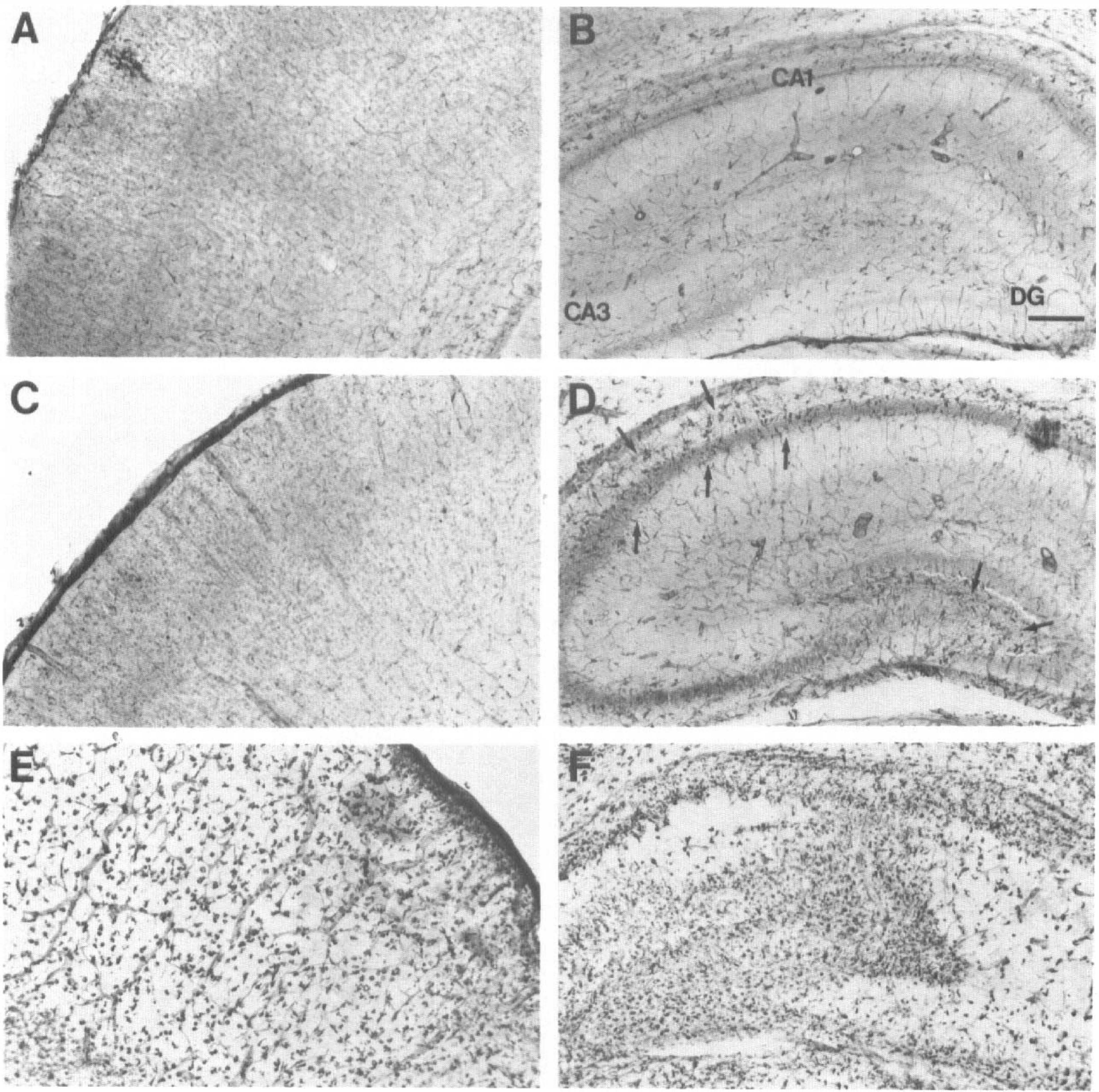
Effect of hypoxia and hypoxia-ischemia on brain HO-1 expression in chronically hypoxic rat pups bearing congenital cardiac defects (Wistar Kyoto, normotensive; WKY/NCr). Normoxic Wistar rats at P7 were used as untreated controls. The pattern of constitutive HO-1 expression found in Wistar rat cortex (
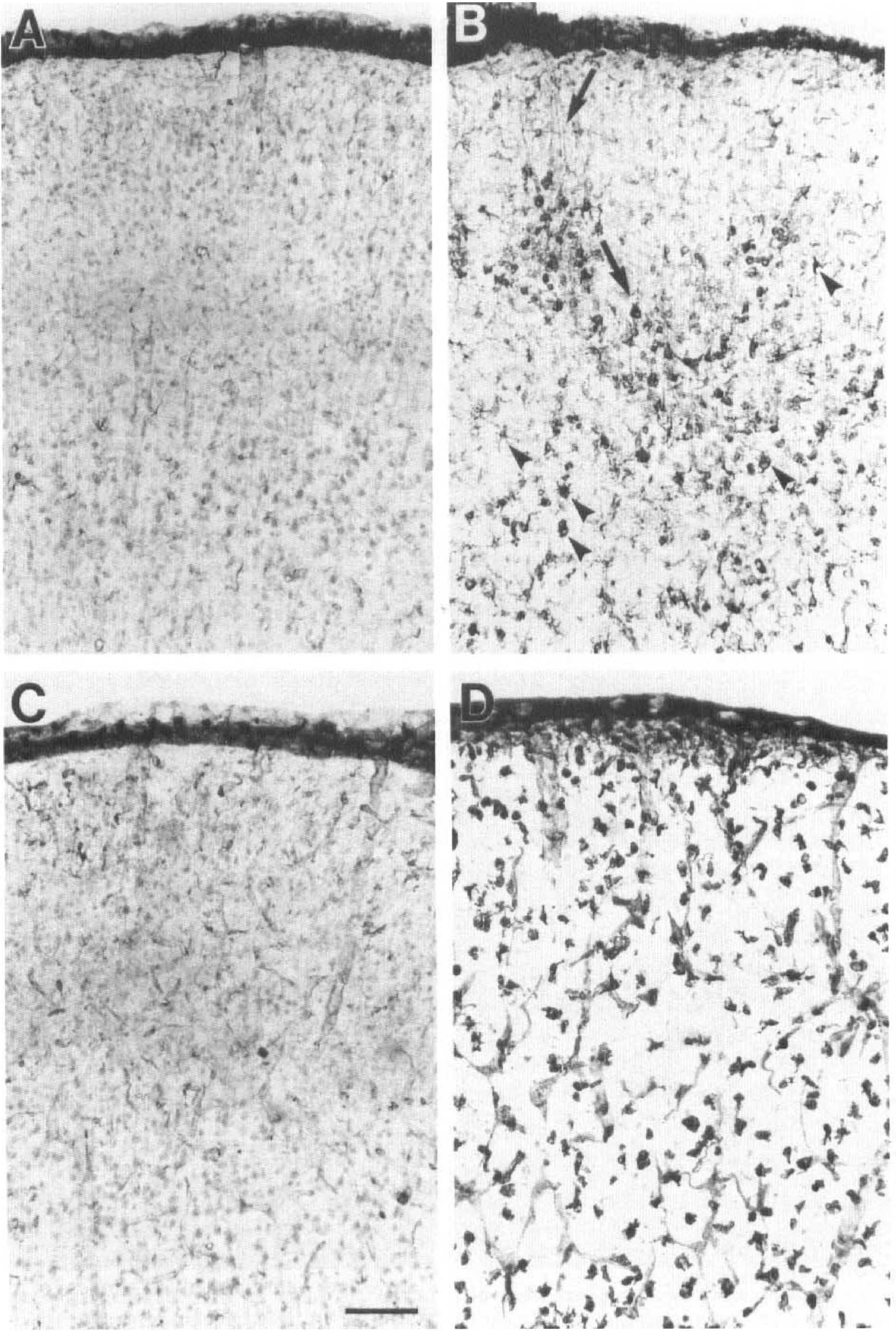
High-power photomicrographs of contralateral (
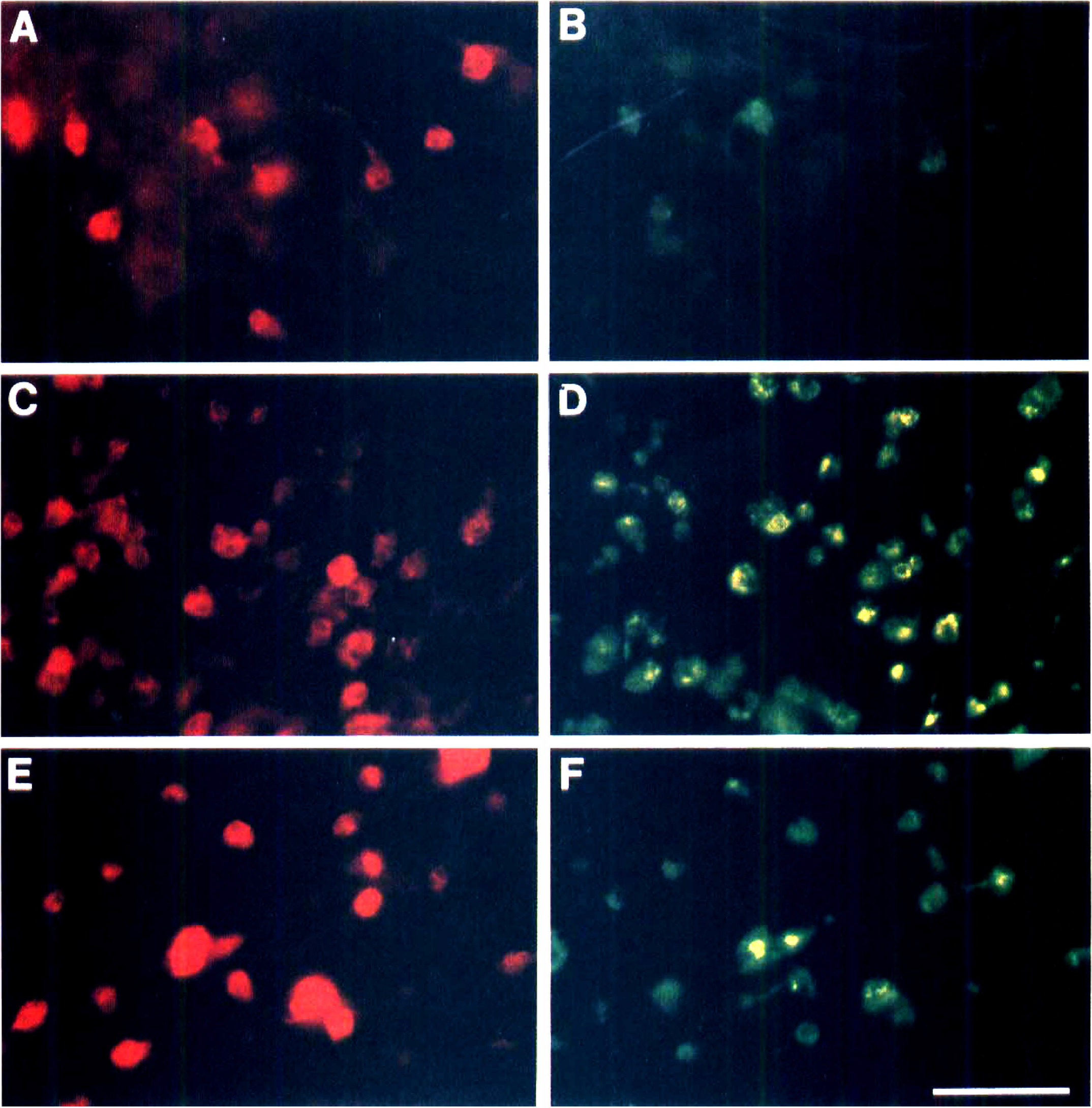
Double fluorescence labeling of cerebral cortex sections from newborn Sprague-Dawley rats 24 hours and 4 days after hypoxicischemic injury. Brain coronal sections (50 µm) were stained with HO-1 antibody (Texas Red, red) and ED-1 antibody (FITC, green/yellow) as described in Materials and Methods. Panels
Heme oxygenase-1 expression after acute and chronic hypoxia
Acute exposure to 8% O2 (for 3 hours) 1 day before ischemia has been shown to confer tolerance to the newborn Sprague Dawley rat pup brain against ischemic injury (Gidday et al., 1994). Using the same experimental protocol, we studied the possible role of HO-1 in the phenomenon of hypoxia-induced ischemic tolerance by examining brains from rat pups 24 hours after exposure to 8% O2 for 3 hours. Western immunoblot analysis of crude brain extracts (Fig. 1) showed that the rabbit anti-rat HO-1 antibody used in the present study recognized a characteristic ∼32 kDa band that is consistent with the rat HO-1 protein (HSP32) described previously (Maines et al., 1986; Ewing and Maines, 1991). In all regions investigated (striatum, hippocampus, and cortex), there was no significant difference between untreated controls (Fig. 1, lanes 1 to 3), the contralateral side that received the hypoxia without the coagulation (in animals subjected to HI) (Fig. 1, lanes 4 to 6), or sham-operated animals with hypoxia only (not shown). In agreement with these observations, immunocytochemistry experiments showed no detectable change in HO-1 expression in newborn brain 24 hours after hypoxia (Figs. 2,E and 4A), compared with untreated control animals (not shown). Heme oxygenase-1 expression in pup brain was similar to controls whether animals had been exposed to 8% O2 for 2.5 hours up to 3.5 hours or whether brain tissue was analyzed 0, 1, 5, 24, 48, or 72 hours or 4 days after hypoxia (not shown).
Induction of HO-1 has been reported after chronic in vitro hypoxia in tumor cells (Heacock and Sutherland, 1990). Rat pups (P7) generally survive a maximum of 3.5 to 4 hours in 8% O2 before dying from cardiac arrest related to severe hypotension and hypoglycemia (Rice et al., 1981; Vannucci and Yager, 1992). In the present study, because brain HO-1 expression was not induced by 3 hours of in vivo hypoxia at 8% O2, we examined the effect of chronic hypoxia on brain HO-1 expression by studying rats with congenital cardiac defects (WKY/ NCr; normotensive, chronically hypoxic Wistar rats). The rats of this inbred strain spontaneously develop various heart anomalies including Tetralogy of Fallot, ventricular septal defect, pulmonary valve stenosis and/or hypertrophic cardiomyopathy in association with hypoplasia of the ductus arteriosus, and occasional anomalies in the aortic arch systems (Kuribayashi et al., 1990). Because the WKY/NCr rats are from a Wistar rat lineage, we used normal Wistar rats as untreated controls. The percentage of oxygen saturation in live untreated WKY/NCr and normal Wistar P7 rat pups was 82.1 ± 7.5% and 95.0 ± 3.0%, respectively (values represent the mean ± SD of n = 10 animals; P = 0.0015: significantly different by unpaired two-tailed Mann-Whitney nonparametric test).
The distribution and intensity of HO-1 expression in untreated control P7 Wistar pups was similar to that described above for the normal P7 Sprague Dawley rats (compare Figs. 2C with 3A and, 2E with3B). The pattern of HO-1 expression in the brain of chronically hypoxic P7 WKY/NCr rats was similar to that of untreated P7 Wistar controls. In general, chronic hypoxia seemed to have no effect on constitutive HO-1 expression throughout the brain of P7 WKY/NCr pups compared with normal Wistar pups. The only difference in WKY/NCr animals compared with normal Wistar rats (Fig. 3B,D) was a higher density of HO-1 neuronal staining in the stratum oriens and the pyramidal cell layer of CA1 to CA3 subfields of the hippocampus and in the polymorphic layer of the dentate gyrus. Exposure of WKY/NCr rats to 8% O2 for 2.5 to 3.0 hours had no detectable effect on brain HO-1 expression compared with untreated WKY/NCr animals or the contralateral side (nonligated side with hypoxia only) of WKY/NCr pups subjected to HI procedures (Fig. 3C,D).
Heme oxygenase-1 expression after hypoxic-ischemic injury
The perinatal HI model produced in P7 rats is characterized by injury in the hemisphere ipsilateral to the carotid occlusion (Rice et al., 1981). Damage ranges from the loss of a few isolated cells up to gross infarction in cortex, hippocampus, striatum, thalamus, and white matter ipsilateral to the ligation. Moderate (damage score = 2) to severe injury (damage score = 3) is more common than little (damage score = 1) or no damage (damage score = 0) (Table 2). Exposure to hypoxia alone (contralateral side) does not produce any evidence of cellular damage.
Distribution of histopathologic damage scores in Sprague Dawley and WKY/NCr newborn rats 24 hours after hypoxia-ischemia
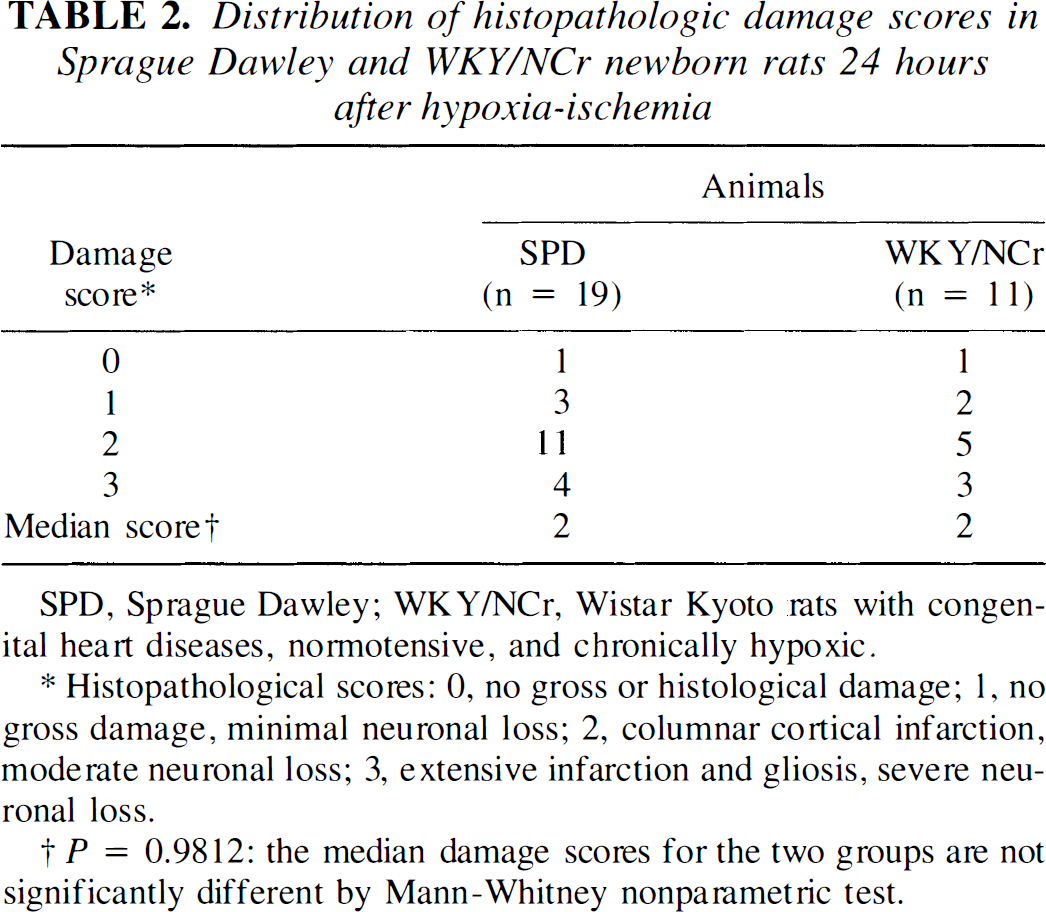
SPD, Sprague Dawley; WKY/NCr, Wistar Kyoto rats with congenital heart diseases, normotensive, and chronically hypoxic.
Histopathological scores: 0, no gross or histological damage; 1, no gross damage, minimal neuronal loss; 2, columnar cortical infarction, moderate neuronal loss; 3, extensive infarction and gliosis, severe neuronal loss.
P = 0.9812: the median damage scores for the two groups are not significantly different by Mann-Whitney nonparametric test.
Western immunoblot analysis of crude brain extracts obtained from Sprague-Dawley rats pups 24 hours after HI showed that HO-1 protein expression was increased 5 to 10-fold in ipsilateral striatum, hippocampus, and cortex (Fig. 1, lanes 7 to 9), compared with the contralateral side (Fig. 1, lanes 4 to 6) or untreated controls (Fig. 1, lanes 1 to 3). In agreement with these observations, increased HO-1 immunoreactivity was detected as early as 6 to 12 hours after HI in the ipsilateral cortex near the corpus callosum (not shown). Markedly increased HO-1 immunostaining was observed in cells from cortex, hippocampus, striatum, and thalamus ipsilateral to the carotid occlusion by 24 hours after HI (Fig. 2D,F). Heme oxygenase-1 immunostaining was still quite prominent at 4 days after HI (Fig. 5C,D), and still detectable in the remaining tissue at 7 days (not shown). Interestingly, the increased HO-1 expression found in ipsilateral cortex often displayed a columnar pattern (Fig. 2D, arrows). Sections incubated without HO-1 antibody showed no endogenous staining (Fig. 2A,B). WKY/NCr pups subjected to HI showed a pattern of brain injury and HO-1 expression very similar to that described above for the Sprague Dawley pups. As shown in Table 2, there was no significant difference in the median histological damage score between the two groups (P = 0.9812, Mann-Whitney nonparametric test). Increased HO-1 expression was observed throughout ipsilateral ischemic hemisphere (Fig. 3E,F) compared with the contralateral hypoxic side (Fig. 3C,D). For all animals investigated in this study, the intensity of HO-1 staining was generally proportional to the degree of brain injury. Whereas moderate injury (Fig. 2D,F) resulted in increased HO-1 expression in some neurons and glia-like cells (Fig. 4B), severe injury (Fig. 3E,F) resulting in large areas of brain tissue infarction revealed an increase in HO-1-positive macrophage-like cells and sustained HO-1 staining in the remaining endothelium (Fig. 4D). Induction of HO-1 protein expression did not increase total HO enzymatic activity in ipsilateral cortex and subcortical regions 24 hours after HI (Table 1).
Double immunofluorescence staining after hypoxia-ischemia
To define the type of cells expressing HO-1 protein, double fluorescence labeling using antibodies against HO-1 and ED-1, OX-42 or GFAP was performed 24 hours and 4 days after HI in newborn Sprague-Dawley rats. Heme oxygenase-1 expression in ipsilateral cortex (Fig. 5A to D) was found mainly in brain macrophages expressing the ED-1 antigen. Double immunolabeling with HO-1 and OX-42 or GFAP antibodies showed occasional costaining of HO-1/OX-42 or HO-1/GFAP (not shown). Twenty-four hours after moderate HI injury, most but not all of the HO-1-positive cells were found to be ED-1 positive (Fig. 5A,B). Almost all HO-1 positive cells were found to be colabeled with the ED-1 antigen 4 days after HI (Fig. 5C,D).
DISCUSSION
Hypoxia preconditioning and heme oxygenase-1 expression
Hypoxia pretreatment (8% O2/3 hours) confers neuroprotection in newborn rats against ischemic injury 24 hours after the initial preconditioning (Gidday et al., 1994). Such hypoxia treatment has relatively no effect on neuronal integrity (Rice et al., 1981) and on several physiological parameters such as regional cerebral blood flow and water content (Vannucci et al., 1988; Mujsce et al., 1990), NADH fluorescence (Welsh et al., 1982), brain protein synthesis (Dwyer et al., 1987), and cerebral calcium uptake (Stein and Vannucci, 1988). In addition, the expression of several genes including the immediate early genes fos and jun (Munell et al., 1994), HSP72 (Ferriero et al., 1990; Munell et al., 1994) and GFAP (Burtrum and Silverstein, 1994) are unaffected by such in vivo hypoxia pretreatment in newborn rat brain.
Recent studies have suggested that the induction of oxygen-regulated proteins could be involved in the mechanism of hypoxia-induced ischemic tolerance. Increased expression of certain proteins including HO-1 has been reported in tumor cell lines (Heacock and Sutherland, 1990), endothelial cells (Zimmerman et al., 1991), and astrocyte cultures (Kuwabara et al., 1996) exposed to hypoxia followed by reoxygenation. A role for HO-1 as a candidate protein responsible for hypoxia preconditioning was suggested because prior induction of HO-1 protein protects against subsequent lethal insults in several biological systems (Vile et al., 1994; Abraham et al., 1995; Otterbein et al., 1995; Vogt et al., 1995). Previous in vitro studies have shown that hypoxia stimulated the DNA binding activity of the transcription factors NFkB (Koong et al., 1994) and heat shock factor (HSF; Benjamin et al., 1990). Although the promoter region of HO-1 gene contains a NFkB binding site (Lavrosky et al., 1994) and a heat shock element (Shibahara et al., 1987; Okinaga and Shibahara, 1993), the present study failed to show an induction of HO-1 protein expression after the 2.5 to 3.5 hour-hypoxia treatment necessary for preconditioning and neuroprotection. The HO-1 induction observed in cultured cells may be caused by low oxygen tensions for long periods of time that cannot be achieved in vivo. For this reason, we investigated the effect of chronic in vivo hypoxia on brain HO-1 expression in rat pups with congenital heart defects (WKY/NCr). These animals also failed to show increased HO-1 protein expression. In contrast to the study of Gidday et al., (1994) which used acute hypoxia (8% O2 for 3 hours) to induce ischemic tolerance, the present study showed that chronic neonatal hypoxia associated with congenital heart defects (WKY/NCr rats) did not protect the brain against subsequent HI injury (Table 2). Taken together, these observations suggest that the induction of HO-1 expression is not the mechanism responsible for hypoxia preconditioning in newborn rat brain.
Effect of hypoxia-ischemia on heme oxygenase-1 expression
Perinatal HI brain damage is associated with increased HO-1 protein expression. The degree of HO-1 expression in the hemisphere ipsilateral to the common carotid artery occlusion was dependent on the severity of the insult. After moderate injury, HO-1 expression was increased mostly in brain macrophages found throughout the focal areas of tissue damage and in scattered neurons, some astrocytes, and endothelial cells ipsilateral to the carotid occlusion. Severe insults resulting in extensive tissue infarction were accompanied by HO-1 staining in ED-1-positive macrophages and the surviving endothelium almost exclusively. Heme oxygenase-1-positive astrocytes were infrequent despite massive and progressive reactive astrogliosis in areas of HI brain damage in the newborn (Burtrum and Silverstein, 1994; Sheldon et al., 1996). In contrast, HO-1/ED-1 positive macrophages increased in number and became the major cellular component expressing HO-1 after HI. The distribution of these HO-1-positive cells was found to be similar to that recently reported for ED-1-positive cells in the same animal model of neonatal HI (Ivacko et al., 1996). The origin of these HO-1-positive macrophages is not known. Some of the HO-1/ED-1 macrophages observed in the present study may have entered the brain from blood vessels possibly in response to the breakdown of blood-brain barrier (Vannucci et al., 1993) and signals from damaged neurons and glia. Alternatively, some of the HO-1/ED-1 macrophages might have been derived from brain-resident macrophages and possibly microglia (Thomas, 1992).
Glutathione depletion (Ewing and Maines, 1993) as well as hyperthermia (Ewing and Maines, 1991) in rats significantly induce HO-1 messenger RNA and protein without concomitant increased brain HO activity. Similarly, the present study showed that despite a 5- to 10-fold increase in HO-1 expression after HI, HO activity remained unchanged compared with controls. Heme oxygenase-2 protein, which is found mainly in neurons (Ewing and Maines, 1992; Maines, 1992), is the most abundant HO isozyme in the brain and is responsible for the bulk of brain HO activity (Sun et al., 1990; Ewing and Maines, 1991). With the ongoing neuronal loss associated with HI injury, it is possible that the increased HO-1 expression observed mainly in proliferating macrophages may compensate for the loss of neuronal HO-2 protein and thus, maintain the overall HO activity at a normal level. Although it is also possible that HO-1 protein becomes inactive after HI, the lack of detectable changes in brain HO activity after neonatal HI does not rule out the possibility for a local effect of HO-1 activity resulting in the release of free iron, bile pigments, and CO.
Carbon monoxide (like nitric oxide) has been proposed as a putative neurotransmitter acting as a physiologic regulator of guanylyl cyclase and cGMP-dependent protein kinase (Maines, 1993; Verma et al., 1993). Recent studies have shown that the activation of metabotropic receptors could modulate neuronal HO activity and in turn, CO could be involved in the signal transduction pathway coupling these receptors to the activity of the NaK-ATPase pump (Glaum and Miller, 1993; Nathanson et al., 1995). In view of this suggested relationship between glutamate receptor activation and increased HO activity in the brain, it is possible that the increased HO-1 expression observed in neurons after moderate HI injury may be caused by, at least in part, the overactivation of glutamate receptors (Rothman and Olney, 1986). Ischemia-induced oxidative stress and cellular protein denaturation may also induce neuronal HO-1 through activation of specific transcription factors such as fos and jun, NFkB, and HSF. Indeed, a region-selective induction of the immediate early genes fos and jun has been reported ipsilateral to the HI injury in newborn rat brain (Munell et al., 1994). Moreover, increased of DNA-binding activity at the AP-1 binding site and the heat shock element has been reported in cerebral cortex after transient focal ischemia in adult rats (Salminen et al., 1995) and in gerbil hippocampus after global ischemia (Nowak and Abe, 1994). However, because neurons only express HSF-2, which is less able than HSF-1 to direct a strong heat shock response (Marcuccilli et al., 1996), a major increase of HO-1 expression in neurons through HSF activation seems less likely. Interestingly, increased NFkB binding activity was noted in ischemic cortex only 5 days after focal ischemia (Salminen et al., 1995), suggesting that this late NFkB surge may be related to postischemic infiltration of inflammatory macrophages.
Increased production of the antioxidant bilirubin (Stocker et al., 1987) after induction of HO-1 expression could protect neurons from subsequent delayed injury. However, this study showed that although there was a clear attempt by neurons to synthesize more HO-1 protein after moderate injury, there was also a clear failure for them to survive more severe HI insults. These observations are consistent with previous reports suggesting that when ischemia is sufficiently severe to produce infarction, transcription and/or translation of the heat shock genes is blocked in neurons and glia destined to die, whereas the surviving endothelial cells, which have been shown to induce both HSP72 and HO-1 after focal ischemia in adult rat (Nimura et al., 1996) or neonatal HI (Ferriero et al., 1990; present study), continue to synthesize stress proteins (Gonzalez et al., 1989; Kinouchi et al., 1993). In addition, studies have shown that neurons in culture have a very limited capacity to induce HO-1 expression even after hydrogen peroxide exposure. In fact, this may contribute to their selective vulnerability to oxidative stress (Dwyer et al., 1995). The protective mechanism suggested for HO-1 activity may also involve the potential lethal effects of ferrous iron, which if unsequestered, may exacerbate HI brain injury by increasing the formation of hydroxyl radicals through the Fenton reaction (Braughler et al., 1986). Almost two thirds of the iron in the brain is stored as ferritin (H and L isoforms). H-ferritin (heavy-chain) is found mainly in neurons and has a low storage capacity consistent with the high iron utilization. L-ferritin (light-chain) which is localized mainly in brain macrophages is involved in long-term iron storage, consistent with the major role of this cell type as a scavenger (Connor et al., 1994). As a result of HI, the increased pro-oxidant levels produced by HO-1 activity combined with the low iron-storage capacity of neurons (H-ferritin) could contribute to HI neuronal injury. In contrast, because of the greater iron-sequestering capacity of L-ferritin, brain macrophages may be able to survive greater increases in iron release and thus, may better benefit from the antioxidant activity of HO-1 induced expression. Because inhibition of phagocytic and secretory functions in mononuclear phagocytes after ischemia has been shown to reduce ischemic injury in the spinal cord (Giulian and Robertson, 1990), it is suggested that HO-l-protected newborn macrophages, besides scavenging iron and removing cellular debris from developing and/or injured brain tissue, may also contribute to neuronal damage after HI and reoxygenation by releasing neurotoxic molecules (Giulian et al., 1993).
Though the bilirubin produced by HO-1 may act as an antioxidant in many tissues, it may also be toxic to the brain. Hyperbilirubinemia is commonly observed during the first week of life in humans and rats (Maines, 1992). Whereas physiological hyperbilirubinemia alone does not cause bilirubin encephalophathy (kernicterus), certain conditions such as asphyxia and acidosis can predispose the brain to bilirubin toxicity by decreasing bilirubin binding to serum albumin and increasing tissue binding of bilirubin (Maines, 1992). Besides its suggested role as an antioxidant at moderate concentrations (Stocker et al., 1987), bilirubin has also been shown to be toxic to cultured astrocytes, neurons, and neural cell lines (Amit and Brenner, 1993). Thus, the elevated levels of bilirubin produced by local increases in HO-1 expression after HI injury could contribute to neuronal damage after ischemia and to the neuronal injury in kernicterus after perinatal asphyxia.
Footnotes
Abbreviations used
Acknowledgements
The authors thank Ronald J. Wang for expert technical assistance with the heme oxygenase activity assay.