
Abstract
Clearance of infarct tissue would be an important process for tissue repair after a stroke. Delayed clearance may hamper reconstitution of the blood–brain barrier and glial boundary formation. Recent growing evidence has indicated that apolipoprotein E (APOE), a major apoprotein, plays an important role in lipid transport and homeostasis in the brain. The tissue in the infarction contains abundant lipids must be removed for tissue clearance. In the current study, the authors investigated APOE expression after focal ischemia and the functional role of APOE in tissue clearance using APOE-knockout mice. Expression of APOE was delayed, but marked, in immunohistochemistry and immunoblotting 7 days after permanent focal ischemia. Macrophages were found to express APOE in the infarct center. Infarct size was similar after focal ischemia between wild-type and APOE-knockout mice, although there was no APOE protein expression in knockout mice. However, clearance of infarct tissue 2 weeks after ischemia was significantly delayed in APOE-knockout mice compared with wild-type mice. The current study supports current thinking that APOE is a key molecule for tissue remodeling in the brain. Clearance of damaged tissue may be one of the important functions of APOE in the brain.
Apolipoprotein E (APOE), a 34-kDa protein, is well known to play an important role in plasma cholesterol metabolism (Mahley, 1988). Apolipoprotein E also is a major apolipoprotein in the brain (Elshourbagy et al., 1985), where it is produced predominantly by astrocytes (Boyles et al., 1985). Because APOE is a ligand for several receptors found in the brain, such as the low density lipoprotein receptor (LDLR), very low density lipoprotein receptor (VLDLR), and low density lipoprotein receptor-related protein (LRP), it is presumably involved in the redistribution of lipids among cells and in the regulation of cholesterol homeostasis for plasma membrane biosynthesis (Beffert et al., 1998). The role of APOE in the brain is not known, but its expression appears to be correlated with nerve injury and synaptic remodeling (Ignatius et al., 1986; Snipes et al., 1986). Elevated levels of APOE have been detected in the brain after transient forebrain ischemia (Hall et al., 1995; Kida et al., 1995; Horsburgh and Nicoll, 1996; Ishimaru et al., 1996; Ali et al., 1996), kainic acid administration (Ong et al., 1997; Grootendorst et al., 2000), and subdural hematoma (Horsburgh et al., 1997), although immunohistochemical labeling of APOE in these studies focused only on astrocytes and degenerating neurons. The function of APOE induced after insults is largely unknown, but it has been suggested that APOE expression shows the inherent protective response of cells. Alternatively, APOE is synthesized and released by astrocytes after injury and may be taken up by degenerating neurons. Mice deficient in APOE have worse outcomes with both transient focal and global ischemic insults than wild-type mice (Laskowitz et al., 1997; Sheng et al., 1999; Horsburgh et al., 1999), which supports the notion that APOE in the brain plays a protective role against neuronal insults, possibly through an antioxidant action (Miyata and Smith, 1996), an antiinflammatory action (Laskowitz et al., 1998), or a neurotrophic effect (Nathan et al., 1994). However, the role of APOE in tissue repair after cerebral infarction has not been examined. After completion of infarction, macrophages continue to accumulate in the infarct tissue for about a week (Mabuchi et al., 2000). It is well known that differentiation of monocytes into macrophages is accompanied by APOE secretion (Werb and Chin, 1983). In the atheromatous plaque, APOE is expressed predominantly in macrophages (Rosenfeld et al., 1993), and in the peripheral nervous system, nerve injury induces expression of APOE in invading macrophages (Ignatius et al., 1986; Boyles et al., 1989). Therefore, the authors hypothesized that macrophages in the infarct tissue express APOE and that APOE production is involved in tissue clearance. The authors used a mouse focal ischemia model to investigate APOE expression and APOE-knockout mice to elucidate the role of APOE in tissue clearance after cerebral infarction.
MATERIALS AND METHODS
C57BL/6 strain mice were obtained from Charles River (Yokohama, Kanagawa, Japan). APOE-knockout mice, originally produced by Zhang et al. (1992), were purchased from the Jackson Laboratory (Bar Harbor, Maine, U.S.A.). The authors first backcrossed them with C57BL/6 mice to produce genetic backgrounds of homozygote and wild-type mice. After mating heterozygotes, the authors selected homozygous and wildtype mice by polymerase chain reaction amplification of genomic DNA extracted from the tail. All mice used in the current experiments were mature males aged 12 to 16 weeks. Mice were given free access to food and water before surgery. The experimental procedures involving laboratory animals have been approved by the Institutional Animal Care and Use Committee of the Osaka University Graduate School of Medicine.
Surgery
General anesthesia was induced with 4.0% halothane and maintained with 0.5% halothane with an open face mask. A polyacrylamide column with an inner diameter of 0.8 mm for measurement of cortical microperfusion by laser–Doppler flowmetry (Advance Laser Flowmetry, Model ALF-21; Advance Co., Tokyo, Japan) was attached to the intact skull, 3.5 mm lateral to the bregma with dental cement. Body temperature was monitored with a rectal thermometer and maintained at 36.0° to 37.5°C using a heat lamp. For a comparison of infarct size and tissue clearance between APOE-knockout and wild-type mice, a femoral artery was cannulated with a PE-10 polyethylene tube connected to a pressure transducer attached to a polygraph. Blood pH, Pao2, and Paco2 were measured using the Acid-Base Laboratory system (ABL550; Radiometer, Copenhagen, Denmark). The left middle cerebral artery (MCA) was permanently occluded using electrocoagulation with a slight modification (Welsh et al., 1987; Kitagawa et al., 1998). Mice were placed in the recumbent position, and a vertical skin incision was made in the midpoint between the left orbit and the external auditory canal. The masseter muscles were incised at the inferior edge of the zygoma, and the mandible was pulled downward to expose the skull base. Using a dental drill, a small burr hole was made in the skull over the left MCA. The dura was opened and the MCA was picked up. Using a microbipolar electrocoagulator, the MCA was permanently occluded just proximal to the point where the olfactory branch came off. Cortical microperfusion was monitored under halothane anesthesia for 30 minutes after MCA occlusion. After discontinuation of halothane anesthesia, each mouse was allowed to recover for 2 hours in a chamber where the ambient temperature was maintained at 35°C to prevent hypothermia, and then each mouse was kept at room temperature. At 2 hours, 2, 7, and 14 days after MCA occlusion, mice were evaluated by a blinded observer for their neurologic deficits. The neurologic deficit score assignment of 0 to 4 was based on the methods described previously by Yang et al. (1994): 0, no neurologic deficit; 1, failed to extend right forepaw while held by tail; 2, circled to the right; 3, fell to the right; 4, unable to walk spontaneously.
Immunohistochemistry
For histologic analyses, 4 nonoperated C57BL/6 mice, 8 ischemic mice 2 (n = 4) and 7 (n = 4) days after MCA occlusion, and 4 sham-operated mice (2 mice per ischemic period) were studied. Two nonoperated, wild-type and 2 APOE-knockout mice, and 4 of each type 7 days after MCA occlusion also were studied. The mice were killed under deep pentobarbital anesthesia through transcardiac perfusion with cold saline followed by a 4% paraformaldehyde solution dissolved in 0.1 mol/L phosphate buffer (pH 7.2). Brains were removed, postfixed in the same fixative at 4°C overnight, and then placed in 30% sucrose in phosphate-buffered saline for 2 days. The brain was frozen in dry ice and 10-μm cryostat sections were cut. For immunohistochemistry for APOE, glial fibrillary acidic protein (GFAP), mouse MAC-2, and mouse immunoglobulin G sections were treated with 0.3% H2 O2 in methanol to eliminate endogenous peroxide. Sections were washed in 0.8% NaCl and 50 mmol/L Tris-HCl, pH 7.4, and nonspecific sites were blocked with 10% normal rabbit or goat serum, or horse serum in the buffer, and then incubated overnight with the primary antibody (APOE, 1:3000, Chemicon, Temecyla, CA, U.S.A.; GFAP, 1:400, Sigma-Aldrich, Tokyo, Japan; MAC-2; 1:100, Cedarlane, Hornby, Ontario, Canada) or a biotinylated anti-mouse IgG antibody (1:1000). After washing, sections for APOE and GFAP were incubated with biotinylated secondary antibodies and all sections were processed with a Vectastain ABC elite kit. An anti–MAC-2 monoclonal antibody recognizes a 32,000 dalton surface antigen found on a subpopulation of mouse macrophages.
Double-immunostaining was performed using immunofluorescence and confocal microscopy (Carl Zeiss, LSM410, Oberkochen, Germany). The sections were incubated with an anti-APOE antibody and an anti-MAC-2 antibody at 4°C overnight. After washing, sections were incubated with an aminomethylcoumarin-labeled donkey anti-goat IgG antibody (Chemicon, 1:200) and rhodamine-labeled donkey anti-rat IgG antibody (Chemicon, 1:200) for 2 hours at 25°C. After washing, the sections were examined using confocal microscopy.
Immunoblotting
For immunoblotting, 4 nonoperated C57BL/6 mice, 4 sham-operated mice, 8 ischemic mice 2 (n = 4) and 7 (n = 4) days after MCA occlusion, and 4 APOE-knockout mice 7 days after MCA occlusion were studied. After decapitation under deep pentobarbital anesthesia, each brain was removed and sectioned coronally into 2-mm slices using a mouse brain matrix (Muromachi Kikai, Tokyo, Japan). The infarcted cortex and caudoputamen were dissected out from the left hemispheres and stored at −70°C. Each sample was homogenized in 10 vol of a buffer containing 4% sodium dodecyl sulfate in 50 mmol/L Tris-Cl, pH 6.8, and boiled for 5 minutes. After chromosomal DNA was sheared by sonication, the lysate was centrifuged at 15,000 g at 4°C for 30 minutes. The protein concentration of the resulting supernatant was determined with bovine serum albumin as the control (Bio-Rad protein assay kit; Bio-Rad Laboratories, Hercules, CA, U.S.A.). Each sample was diluted to 1 mg/mL with a Laemmli buffer containing 62.5 mmol/L Tris-HCl, pH 6.8, 2% sodium dodecyl sulfate, 10% glycerol, and 5% 2-mercaptoethanol. Each sample (3 μg) was electrophoresed in a 12.5% sodium dodecyl sulfate-polyacrylamide gel, transferred to a polyvinylidene difluoride sheet (Immobilon P; Millipore, Bedford, MA, U.S.A.), immunoblotted with a goat polyclonal antibody against APOE (1:3000) or with a rat monoclonal antibody against mouse MAC-2 (1:400), and developed using the enhanced chemiluminescence technique (Amersham International, Buckinghamshire, U.K.).
Evaluation of cerebral infarction and tissue clearance
For evaluation of infarct size, six animals each of wild-type and APOE-knockout mice were used and processed as described previously (Kitagawa et al., 1998). Seven days after permanent MCA occlusion, mice were killed with an overdose of pentobarbital. The whole brains were removed, fixed by immersion into an alcohol-5% acetic acid solution for 5 hours at 4°C, dehydrated, and embedded in paraffin. Tissue sections (5 μm) were obtained every 1 mm, beginning at the frontal pole, and were examined after conventional staining with hematoxylin and eosin. The volume of ischemic infarction was measured using an MCID image analysis system (Imaging Research, St. Catharine's, Ontario, Canada). The infarction areas were calculated by tracing the areas on a computer screen. The volume (mm3) was determined by integrating the appropriate area and the section thickness.
For evaluation of tissue clearance after infarction, nine animals each of the wild-type and APOE-knockout mice were used. Fourteen days after permanent MCA occlusion, mice were killed with an overdose of pentobarbital. The whole brains were removed and immediately frozen in powdered dry ice. Ten-micrometer frozen sections 3 mm caudal to the frontal pole were stained with hematoxylin and eosin. To evaluate the degree of tissue clearance, the percent residual infarct volume was obtained by dividing the area of infarct debris with the expected infarct area, which was calculated as the area of the contralateral hemisphere minus the intact area of the ipsilateral hemisphere.
Statistical analysis
All values reported here are expressed as mean ± SD. Statistical significance of differences among groups was tested by unpaired t-test. P < 0.05 was considered significant.
RESULTS
Apolipoprotein E expression in cerebral infarction
Apolipoprotein E expression in the whole brain after MCA occlusion is presented in Figs. 1A to 1C and 2A to 2C. Immunoreactivity was faint in the control brain. Two days after MCA occlusion, when development of infarction was almost complete and massive extravasation of IgG was observed in the affected hemisphere (Fig. 1D), only weak immunoreactivity for APOE was observed within the infarction (Figs. 1B and 2B). However, seven days after MCA occlusion, marked expression of APOE was observed in the infarction (Figs. 1C and 2C). Within the infarction, large round cells showed a strong immunoreaction. Reactive astrocytes were observed in the infarct rim (Fig. 2D), but disappeared in the infarct core (Fig. 2E). Alternatively, immunohistochemistry with an anti–MAC-2 antibody showed macrophage accumulation in the infarction seven days after MCA occlusion (Fig. 1E). Macrophages with large nuclei were abundant in the infarct core (Fig. 2F). Double-immunostaining for APOE and MAC-2 demonstrated that macrophages expressed APOE in the infarct core (Fig. 3). Immunoblotting also showed elevated level of APOE in the ischemic hemisphere 2 days after MCA occlusion and marked induction of APOE 7 days later (Fig. 4A). Immunoblotting with an anti–MAC-2 antibody showed marked accumulation of macrophages in the ischemic hemisphere 7 days after MCA occlusion (Fig. 4B).
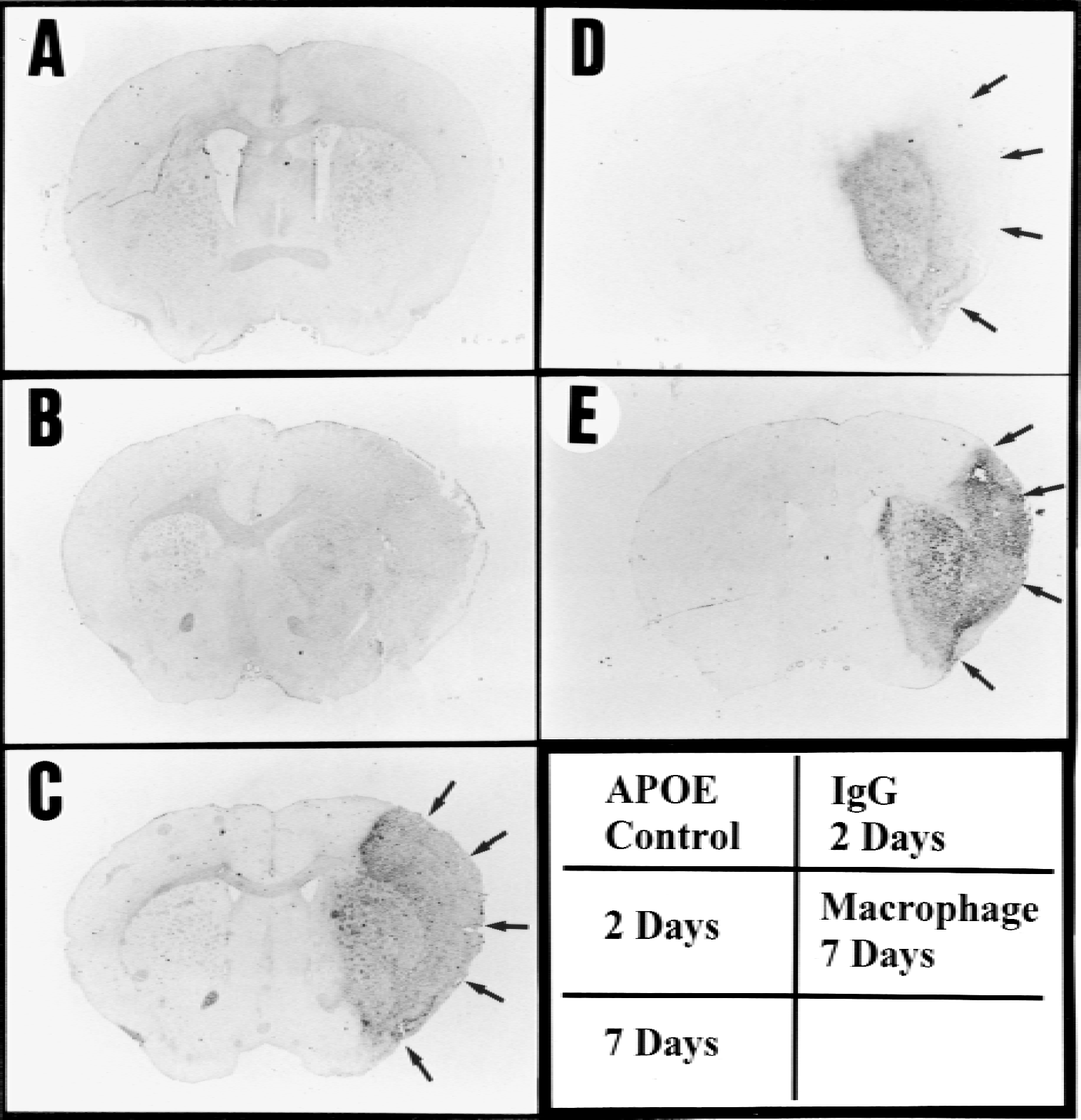
Apolipoprotein E (APOE) expression in the whole brain after permanent middle cerebral artery (MCA) occlusion. Immunoreactivity for APOE was faint in the control
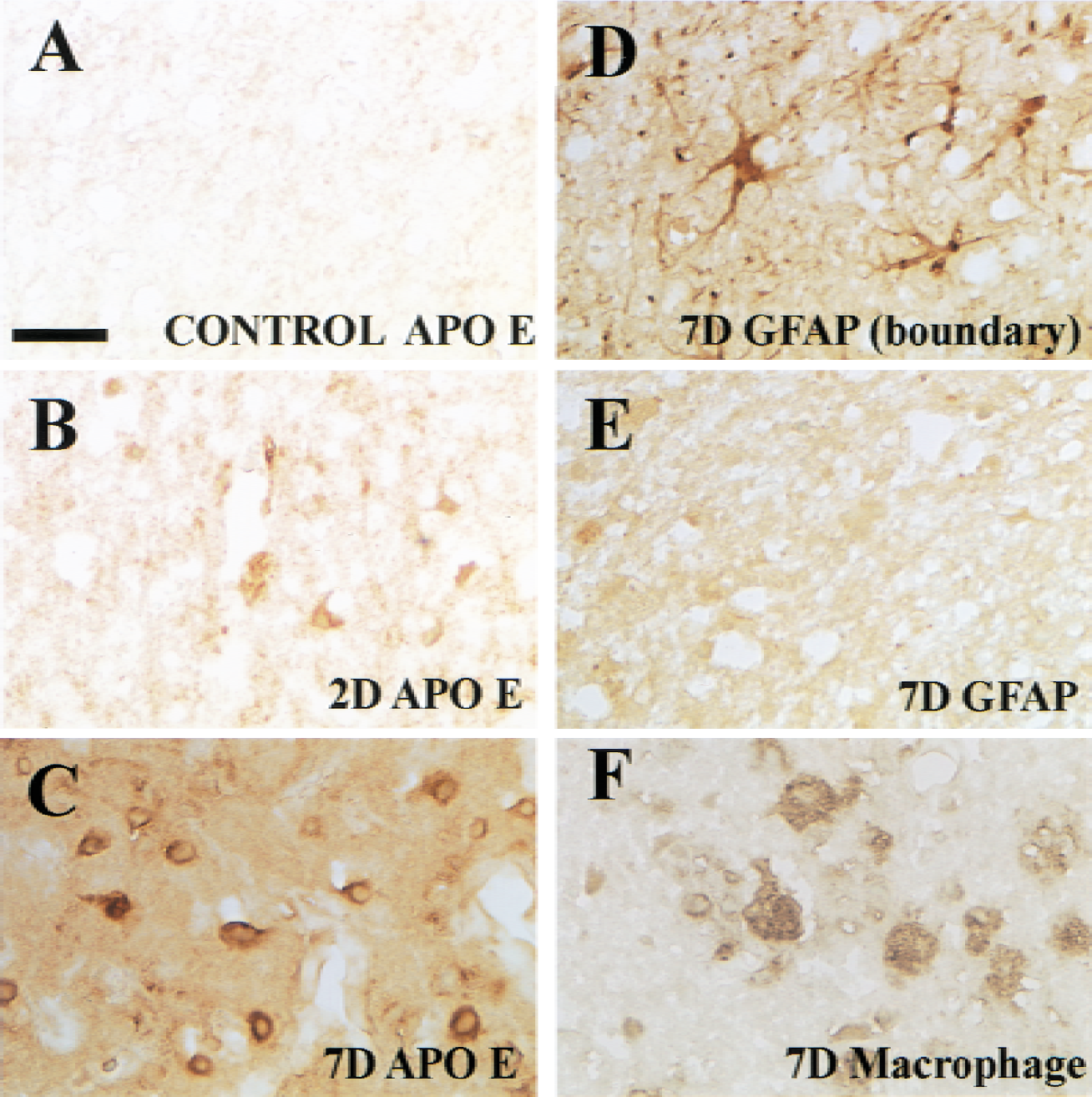
Apolipoprotein E (APOE) expression in the infarct core after permanent middle cerebral artery (MCA) occlusion. Immunoreactivity for APOE in the center of the MCA territory in the control
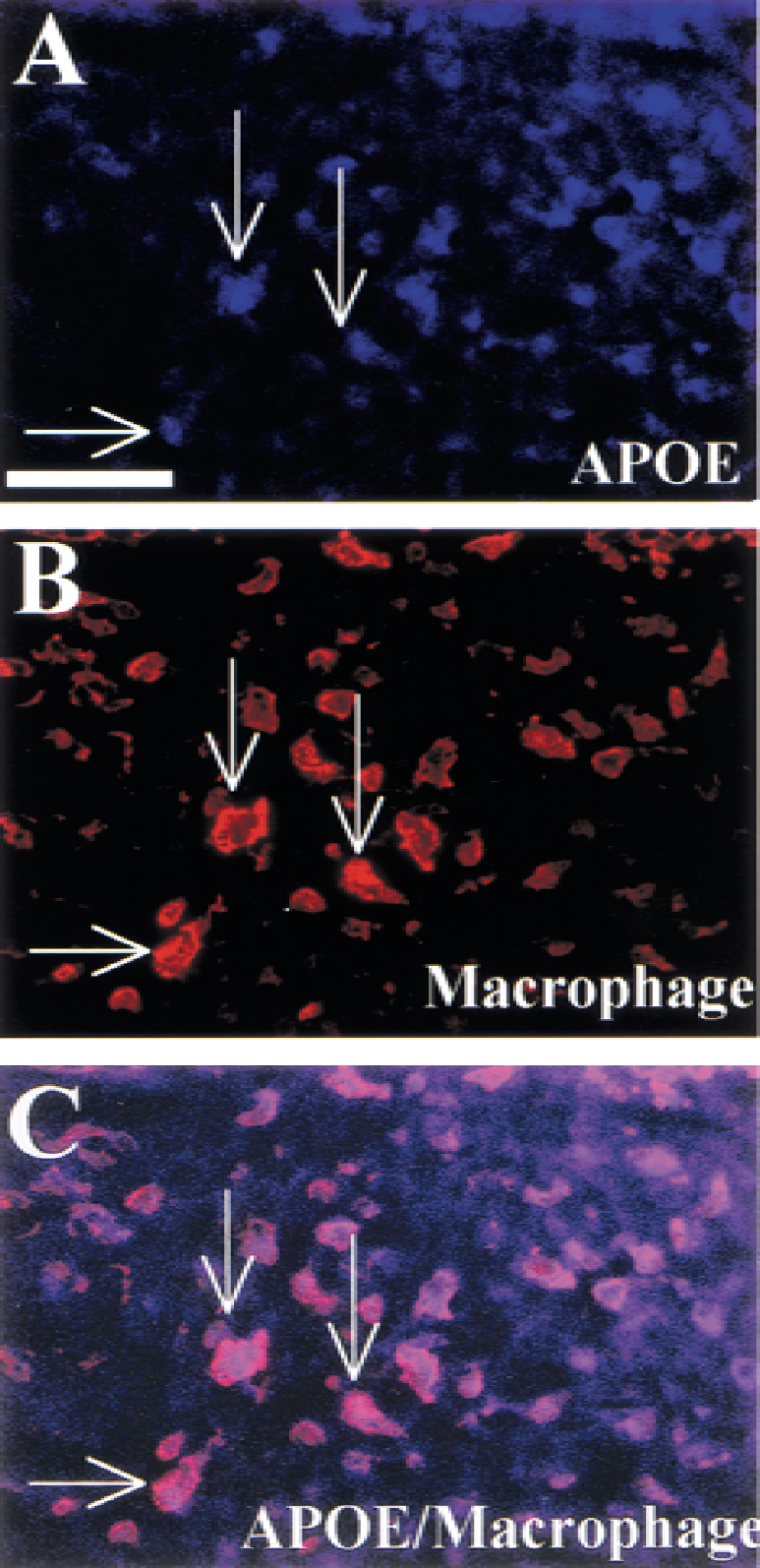
Apolipoprotein E (APOE) expression in macrophages in the infarct core. Double immunostaining
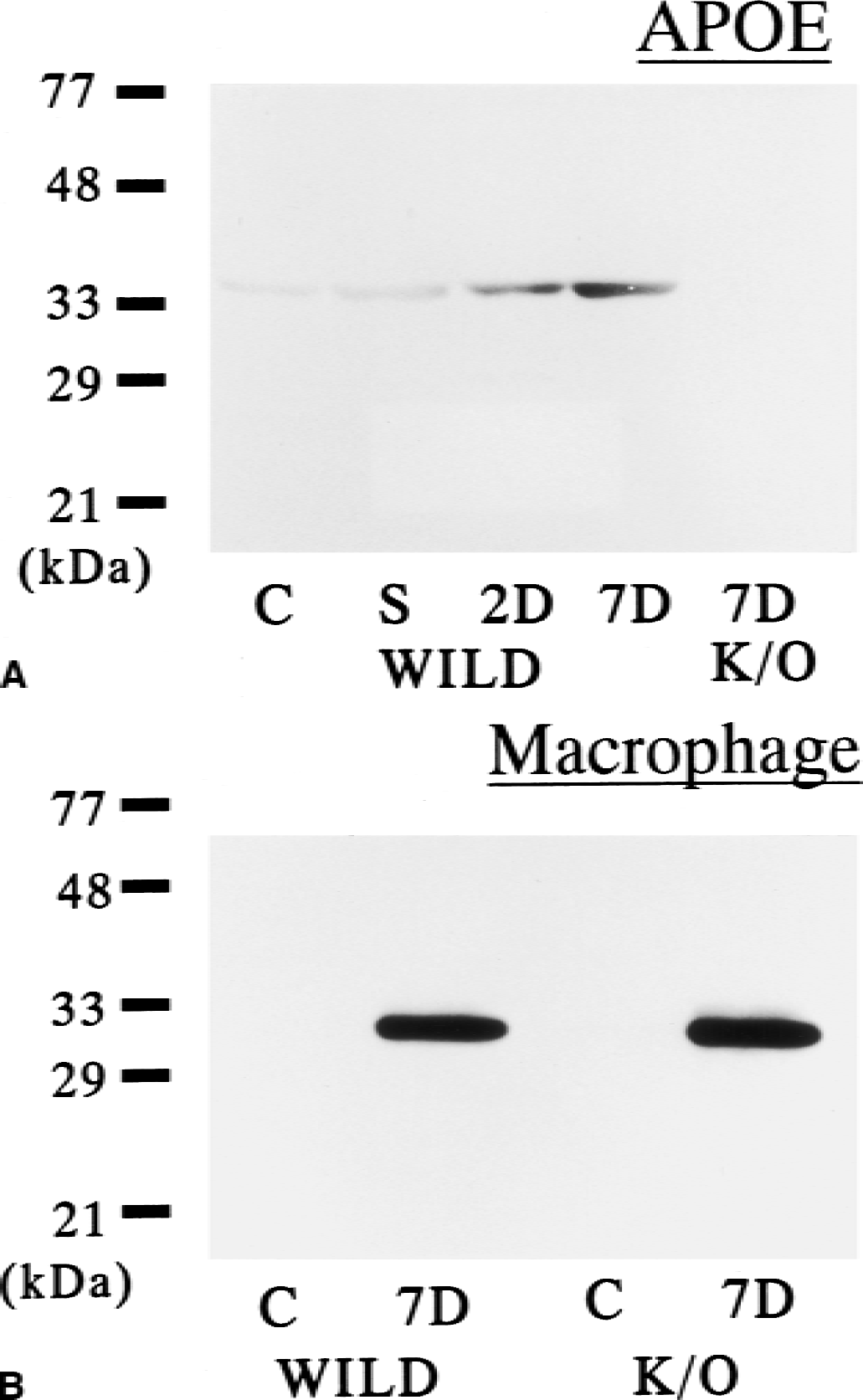
Immunoblotting for apolipoprotein E (APOE)
Infarction size and tissue clearance in apolipoprotein E–knockout mice
The physiologic variables, while allowing spontaneous breathing under halothane anesthesia, were determined for Paco2 (wild type, 37.2 ± 5.3 mm Hg; knockout, 40.0 ± 5.4 mm Hg), Pao2 (wild type, 111.1 ± 9.3 mm Hg; knockout, 118.7 ± 21.2 mm Hg), and pH (wild type, 7.26 ± 0.02; knockout, 7.22 ± 0.04) for wild-type (n = 6) and APOE-knockout (n = 6) mice. There were no significant differences in any of the variables between the wild-type and APOE-knockout mice. Cortical microperfusion in the ischemic center after MCA occlusion was 20% to 30% of the baseline value, and the values were similar between knockout and wild-type mice (Table 1). Other physiologic variables, including blood pressure and body temperature, were maintained at similar levels in both types of mice.
Physiologic variables after permanent MCA occlusion
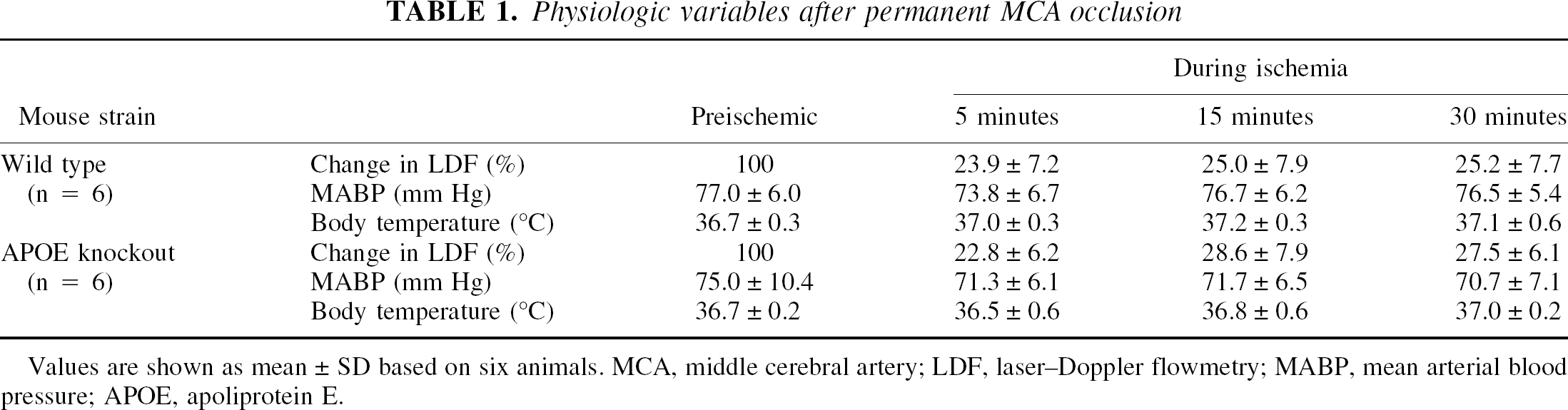
Values are shown as mean ± SD based on six animals. MCA, middle cerebral artery; LDF, laser–Doppler flowmetry; MABP, mean arterial blood pressure; APOE, apoliprotein E.
Infarct areas 7 days after MCA occlusion were almost the same for both types of mice on the coronal slices 3 mm from the frontal pole (Fig. 5A and 5B). The total infarct volume also was similar between both types of mice (wild type, 30.6 ± 5.2 mm3, n = 6; K/O, 34.9 ± 6.5 mm3, n = 6;P = 0.23) (Fig. 6). Although marked induction of APOE was observed within the infarction 7 days after MCA occlusion in wild-type mice (Figs. 5C and 7), there was no APOE induction in the infarct tissue in the knockout mice with immunohistochemistry (Figs. 5D and 7) or immunoblotting (Fig. 4A). However, macrophage accumulation within the infarction was similar in both types of mice with immunohistochemistry (Fig. 7) and immunoblotting (Fig. 4B).
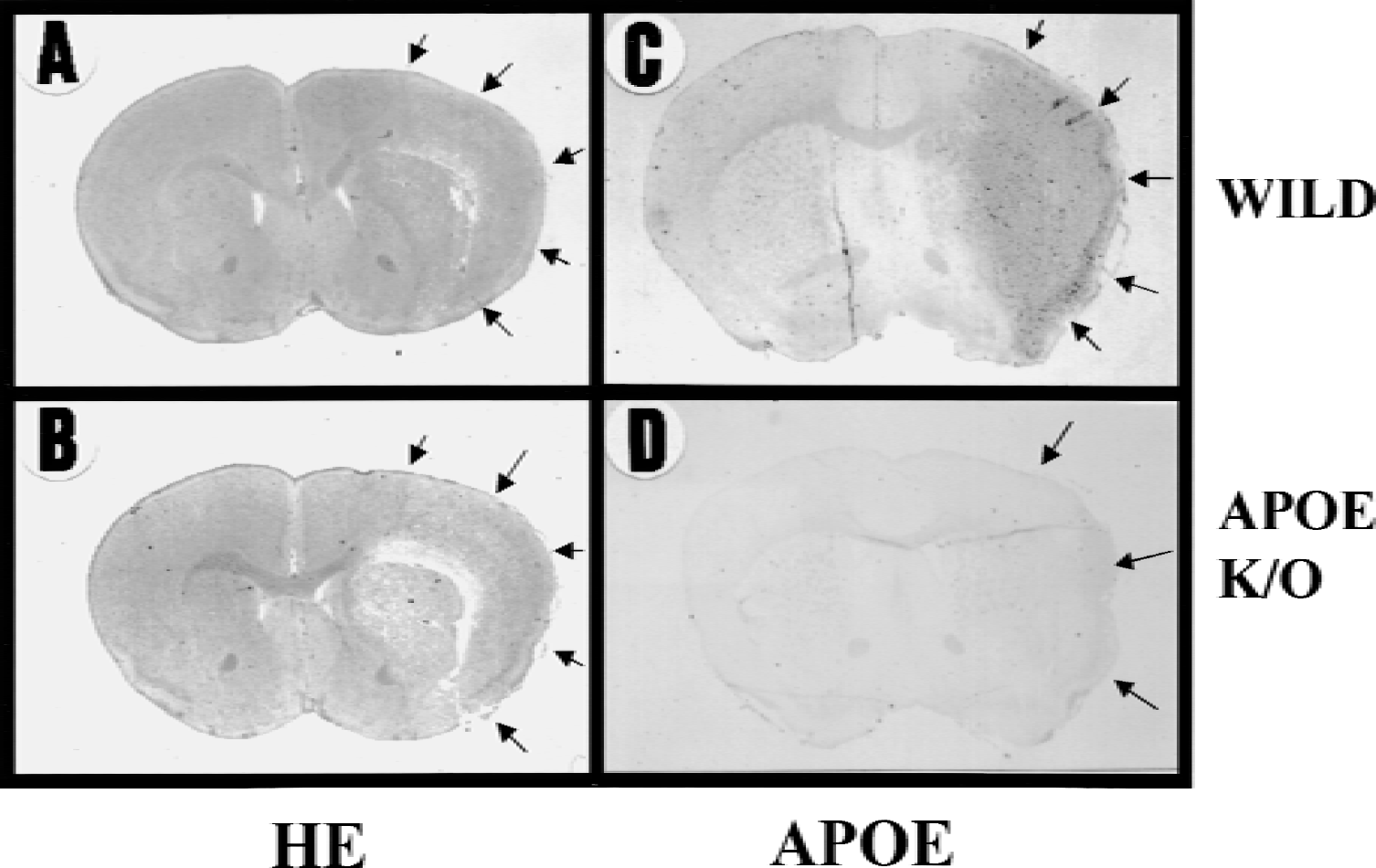
Hematoxylin and eosin (HE) staining and immunoreaction for apolipoprotein E (APOE) in the whole brain of wild-type and APOE-knockout (K/O) mice 7 days after middle cerebral artery (MCA) occlusion.
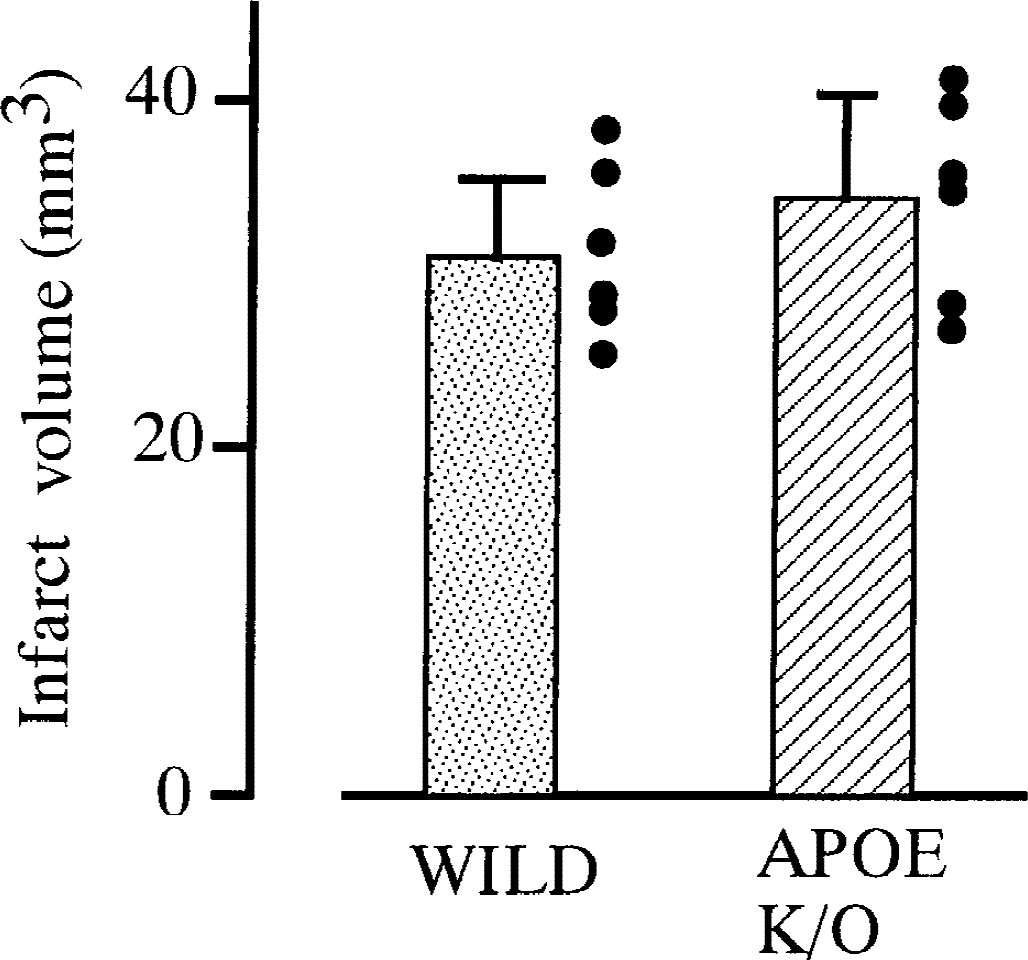
The total volume of infarct lesions 7 days after permanent middle cerebral artery occlusion. The lesion volume for wild-type (stippled column) and APOE-knockout (K/O) mice (hatched column). No differences (P = 0.23) were found between the two types of mice.
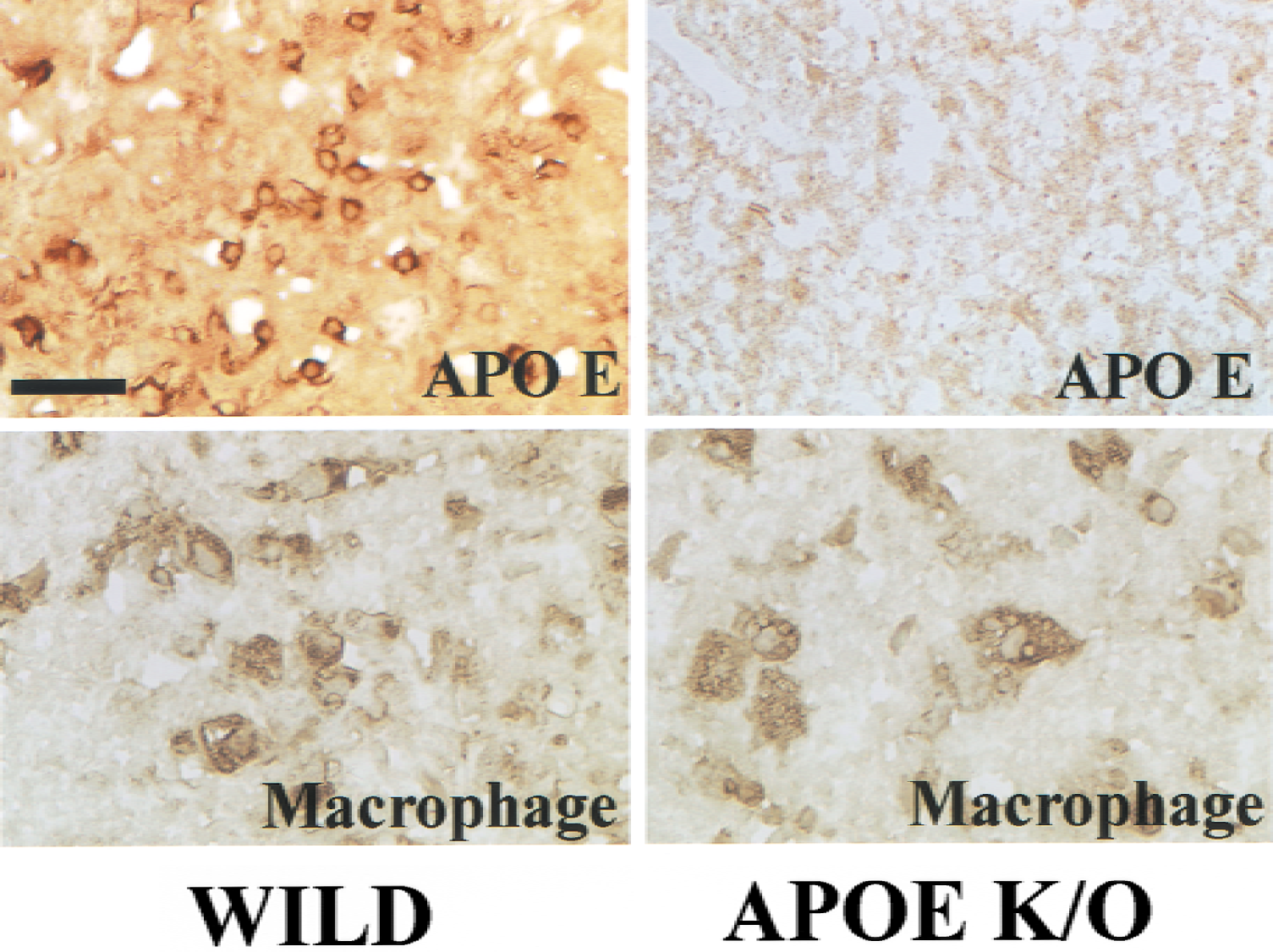
Immunoreaction for apolipoprotein E (APOE) in the infarct core 7 days after middle cerebral artery occlusion of wild-type and APOE-knockout (K/O) mice. (left column) Wild-type. (right column) APOE K/O mice. (top row) Immunoreaction for APOE. (bottom row) Immunoreaction for macrophage surface antigen, MAC-2. A strong immunoreaction for APOE was observed in the wild-type mice, but there was no immunoreaction in the APOE K/O mice. Macrophages with large nuclei were abundant in both types of mice. Scale bar = 25 μm.
The infarct area, especially the infarct cortex, began to diminish 14 days after MCA occlusion in wild-type mice (Fig. 8A). However, in the APOE-knockout mice, clearance of infarct tissue was delayed and the infarct cortex with debris was still present in the ischemic hemisphere (Fig. 8B). The percentage of residual infarct areas was significantly greater in the APOE-knockout mice (n = 9, 90.4% ± 24.2%) than in the wild-type mice (n = 9, 54.7% ± 18.5%) (Fig. 9).
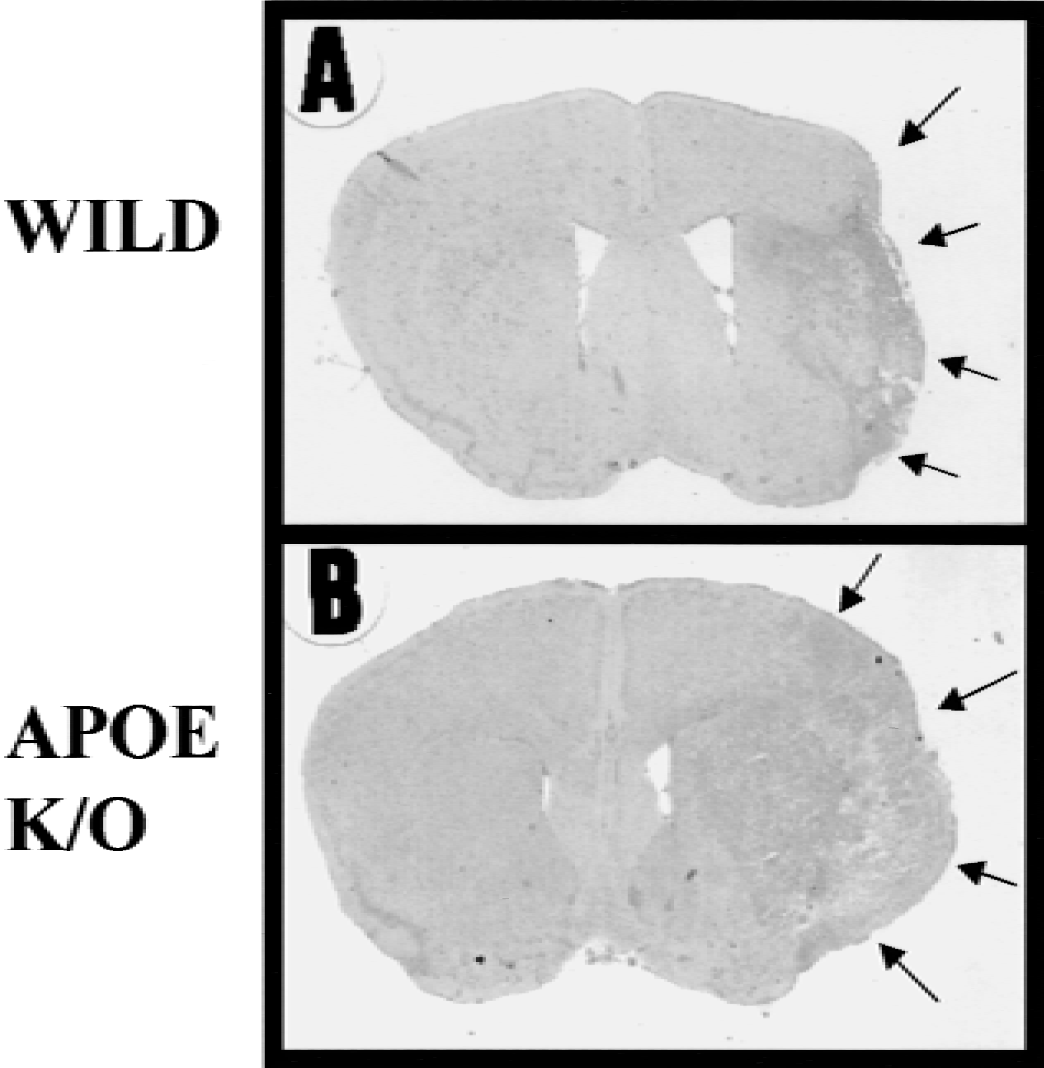
Hematoxylin and eosin (HE) staining in the whole brain of wild-type
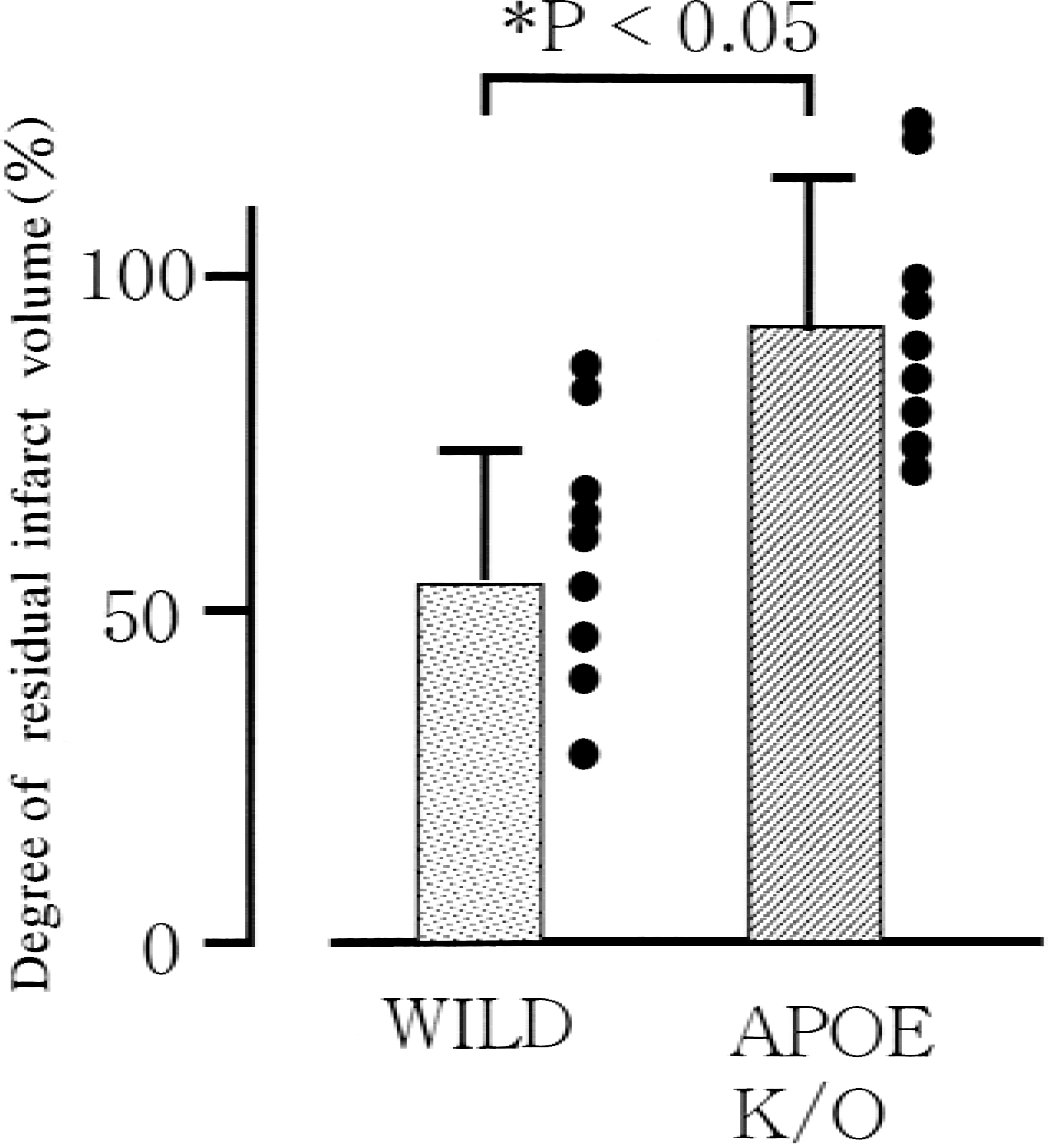
Degree of residual infarct volume 14 days after middle cerebral artery occlusion. The percentage of residual infarct volume for wild-type (stippled column) and APOE-knockout (K/O) mice (hatched column) was obtained by dividing the area of infarct debris with the expected infarct area, as described in the text. Significant differences were found between the two types of mice.
DISCUSSION
Previous studies demonstrated that APOE-deficient mice showed more severe brain damage in a transient ischemia model (Laskowitz et al., 1997); however, the current study showed almost the same infarct volume after permanent MCA occlusion between wild-type and APOE-knockout mice. Because the neuroprotective effect of APOE after metabolic stress has been discussed as an antioxidant (Miyata and Smith, 1996; Laskowitz et al., 1997), whether ischemia is followed by reperfusion that produces a burst of free radical formation (Peters et al., 1998) may explain the difference in the effects of APOE deficiency in permanent and transient focal ischemia.
The first novel finding in the current study was the delayed, but marked, induction of APOE within the infarct core. Previous studies concerning APOE expression after ischemia were limited to findings after transient and brief global ischemia (Hall et al., 1995; Kida et al., 1995; Horsburgh and Nicoll, 1996; Ishimaru et al., 1996; Ali et al., 1996), when APOE expression was localized in reactive astrocytes and degenerating neurons. However, in these models, macrophage accumulation was much less than in the permanent MCA occlusion model (Morioka et al., 1991; Gregersen et al., 2000). There has been no study clearly demonstrating APOE expression in macrophages after brain insult. Macrophages begin to accumulate in the infarction one day after MCA occlusion (Gregersen et al., 2000; Mabuchi et al., 2000). However, APOE expression was faint until 2 days after MCA occlusion in this study (Figs. 1 and 2). Although relatively little is known about the regulation of APOE expression in macrophages, several inflammatory cytokines, such as tumor necrosis factor α and interleukin 1β, have been known to be produced mainly in the microglia/macrophages after MCA occlusion (Wang et al., 1994; Davies et al., 1999; Mabuchi et al., 2000; Gregersen et al., 2000), and most of these could inhibit APOE expression in macrophages (Larkin et al., 2000). Thereafter, the level of proinflammatory cytokines subsided within 1 week after MCA occlusion. Delayed expression of APOE in the macrophages in the infarction may be explained by the expression profile of inflammatory cytokines after MCA occlusion. Lipid and cholesterol released from necrotic tissue could also enhance APOE expression in the macrophages (Rouis et al., 1990). It is highly unlikely that plasma APOE entered the infarction and was taken by the macrophages, because extravasation of immunoglobulin G was marked 2 days after MCA occlusion, when the APOE immunoreaction was still very faint (Fig. 1).
The second novel finding was the delayed clearance of infarct tissue after MCA occlusion in APOE-knockout mice. Fagan et al. (1998) demonstrated the persistence of axon degeneration products in the deafferented hippocampus in APOE-knockout mice after entorhinal cortex lesions, and suggested the importance of APOE in reactive astrocytes for clearance of neurodegeneration products after brain injury. The current study suggests that APOE production in macrophages contributed to clearance of infarct debris that could be monitored in stroke patients with computed tomography or magnetic resonance imaging. Persistence of infarct debris may disturb the recovery of the blood–brain barrier and the formation of glial boundary, and can be the focus of bacterial infection. Therefore, marked induction of APOE in the infarct core may be a crucial factor for tissue repair after cerebral infarction. The role of APOE in tissue clearance is largely speculative. At first, secreted APOE from macrophages can bind to the lipids released from necrotic tissue and the complex is taken up through several receptors for redistribution or storage by the neurons, astrocytes, and macrophages (Boyles et al., 1985), or it enters the blood circulation for reverse transport of excess cholesterol from the brain to the liver (Pitas et al., 1987). Apolipoprotein E also regulates lipid and cholesterol efflux from macrophages (Basu et al., 1983; Hara and Yokoyama, 1992; Lin et al., 1999) and APOE dysfunction may lead to intracellular accumulation of cholesterol (Cullen et al., 1998), subsequently resulting in delayed clearance.
Recently, there has been growing evidence for dysfunction in the APOE ε4 isoform, as compared with the ε2 and ε3 isoforms, in terms of neurite extension (Holtzman et al., 1995; Arendt et al., 1997), lipid efflux from astrocytes and neurons (Michikawa et al., 2000), and antioxidant activity (Miyata and Smith, 1996). The presence of the APOE ε4 allele is well known as a predisposing factor for Alzheimer disease (Poirier et al., 1993; Corder et al., 1993; van Dujin et al., 1994). Although it is unknown whether the gene of the APOE ε4 isoform results in delayed clearance of damaged brain tissue, as found in the APOE-knockout condition, the current results may lead to another approach to investigate the involvement of APOE dysfunction in the repair process after brain insult in neurologic diseases.
Footnotes
Acknowledgments:
The authors thank Mr. Nobuo Katsube of Ono Pharmaceutical Company for careful management of animals, and Miss Y. Iameda and Miss R. Morimoto for their secretarial assistance.