
Abstract
Hyperlipidemia is a highly prevalent risk factor for coronary and cervical atherosclerosis and stroke. However, even in the absence of overt atherosclerosis, hyperlipidemia disrupts endothelial and smooth muscle function. We investigated the impact of hyperlipidemia on resting-brain perfusion, fundamental cerebrovascular reflexes, and dynamic perfusion defect during acute focal ischemia in hyperlipidemic apolipoprotein E knockout mice before the development of flow-limiting atherosclerotic stenoses. Despite elevated blood pressures, absolute resting cerebral blood flow was reduced by 20% in apolipoprotein E knockout compared with wild type when measured by [14C]-iodoamphetamine technique. Noninvasive, high spatiotemporal resolution laser speckle flow imaging revealed that the lower autoregulatory limit was elevated in apolipoprotein E knockout mice (60 vs. 40 mm Hg), and cortical hyperemic responses to hypercapnia and functional activation were attenuated by 30% and 64%, respectively. Distal middle cerebral artery occlusion caused significantly larger perfusion defects and infarct volumes in apolipoprotein E knockout compared with wild type. Cerebrovascular dysfunction showed a direct relationship to the duration of high-fat diet. These data suggest that hyperlipidemia disrupts cerebral blood flow regulation and diminishes collateral perfusion in acute stroke in the absence of hemodynamically significant atherosclerosis.
Keywords
INTRODUCTION
Hyperlipidemia promotes extracranial atherosclerosis and the risk for atherothrombotic and cardioembolic stroke.1,2 However, its impact on cerebrovascular regulation in normal or ischemic brain is less well understood. Independent of atherosclerotic stenoses, hyperlipidemia impairs endothelial and smooth muscle function in both systemic and cerebral arteries. For example, hyperlipidemia disrupts endothelium-dependent relaxations in carotid and basilar arteries in vitro, and in pial arterioles in situ.3–6 Endothelial function is restored by free radical scavengers, genetic deletion of NADPH oxidase subunit gp91(phox), and
Cerebral blood flow (CBF) is tightly regulated during changes in perfusion pressure and arterial pCO2, and to match the increased demand during functional metabolic activation. Moreover, upon focal arterial occlusion, collaterals play a critical role in sustaining tissue perfusion and survival in ischemic penumbra. The impact of hyperlipidemia on these complex cerebrovascular responses that maintain tissue homeostasis is not known. Using non-invasive and high-resolution laser speckle imaging of CBF through intact skull, we investigated cerebrovascular dysfunction in hyperlipidemic ApoE knockout (ApoE−/−) mice, a frequently used model of hyperlipidemia and atherosclerosis. 10 Mutants develop subendothelial foam cells at as early as 8 weeks of age, progressing to form fatty streaks and frank atherosclerotic plaques over several months, accelerated by high-fat diet (HFD).11,12 Here, we show reduced resting CBF, impaired physiologic cerebral vasodilator reflexes, and progressively worse CBF deficits after focal cerebral arterial occlusion in hyperlipidemic mice at an age before the development of flow-limiting atherosclerotic stenoses in cervical or intracranial arteries.
MATERIALS AND METHODS
Experimental Animals and Diet
All experimental procedures were carried out in accordance with the Guide for Care and Use of Laboratory Animals (NIH Publication No. 85-23, 1996), and were approved by the institutional review board (MGH Subcommittee on Research Animal Care, SRAC). A total of 144 mice were used. Male wild-type mice (C57BL/6J) were compared with ApoE−/− mice (Jackson Laboratories, Bar Harbor, ME, USA) backcrossed on a C57BL/6J background for more than 10 generations. All mice were fed regular chow or ’western’-type HFD that contained 42% of total calories from fat and 0.15% cholesterol (Harlan-Teklad, Indianapolis, IN, USA) for up to 12 weeks starting at 4 weeks of age.
Absolute Resting CBF
We measured absolute resting CBF in the cortex, striatum, and cerebellum using the [14C]-iodoamphetamine technique (n = 4 each, wild type and ApoE−/−). Mice were initially anesthetized with isoflurane (2% induction, 1% maintenance), intubated via tracheostomy, and mechanically ventilated (70% N2O, 30% O2; SAR 830/P, CWE, Ardmore, PA, USA). Ventilation parameters and the gas mixture were adjusted to maintain normal arterial pCO2 and pO2. Femoral artery and external jugular vein were catheterized. Anesthesia was then switched to α-chloralose (50 mg/kg, intraperitoneal). Body temperature was kept at 37 °C using a thermostatic heating pad (FHC, Bowdoinham, ME, USA). After confirming normal blood pressure and blood gases, arterial blood was withdrawn continuously from the femoral artery at a rate of 0.3 ml/min. One μCi of
Laser Speckle Flowmetry (LSF)
We measured CBF changes non-invasively through intact skull using LSF, as described in detail elsewhere. 14 Briefly, a charge-coupled device camera (Cohu, San Diego, CA, USA) was positioned above the head (imaging field 5.24 × 7 mm2), and a laser diode (780 nm) was used to illuminate the intact skull surface in a diffuse manner. The penetration depth of the laser is approximately 500 μm. Raw speckle images were used to compute speckle contrast, which is a measure of speckle visibility related to the velocity of the scattering particles, and therefore CBF. The speckle contrast is defined as the ratio of the standard deviation of pixel intensities to the mean pixel intensity in a small region of the image. Ten consecutive raw speckle images were acquired at 15 Hz (an image set), processed by computing the speckle contrast using a sliding grid of 7 × 7 pixels, and averaged to improve the signal-to-noise ratio. Speckle contrast images were converted to images of correlation time values, which represent the decay time of the light intensity autocorrelation function. The correlation time is inversely and linearly proportional to the mean blood velocity. Relative CBF images (percentage of baseline) were calculated by computing the ratio of a baseline image of correlation time values to subsequent images. In addition, to compare resting CBF among groups and confirm the findings obtained by [14C]-iodoamphetamine technique, we determined the laser speckle contrast inverse correlation time (1/τc) values in dorsal cortex at baseline resting state, and normalized this to the resting CBF measured in the same way in a separate group of mice without HFD, as previously described. 15
Cerebrovascular Physiology
We assessed functional neurovascular coupling, hypercapnic hyperemia, and autoregulation in the same mouse in this order (n = 11 and 7, wild type and ApoE−/−, respectively). Mice were anesthetized with isoflurane (2% induction, 1% maintenance), intubated via a tracheostomy, and mechanically ventilated (70% N2O, 30% O2; SAR 830/P, CWE). Femoral artery was catheterized for continuous blood pressure recording (ETH-400 transducer amplifier, ADInstruments, Medford, MA, USA), and blood gas and pH measurements at least every 30 minutes (30 μl samples, Blood Gas Analyzer 248, CIBA/Corning, New York, NY, USA). Ventilation parameters and gas mixture were adjusted to maintain normal arterial pCO2 and pO2. Body temperature was kept at 37 °C using a thermostatic heating pad (FHC). Mice were paralyzed (pancuronium, 0.4 mg/kg/h, intraperitoneal) and placed in a stereotaxic frame (David Kopf, Tujunga, CA, USA), and the scalp and periosteum were pulled aside. Anesthesia was then switched to α-chloralose (50 mg/kg, intraperitoneal, repeated every 45–60 minutes), which is known to better preserve cerebrovascular reflexes. 14 The adequacy of anesthesia was regularly checked by the absence of blood pressure response to tail pinch. Data were continuously recorded and stored using a data acquisition and analysis system (PowerLab, AD Instruments, Medford, MA, USA).
Functional coupling was tested using whisker stimulation. Whiskers were trimmed (5 mm) and manually stimulated using a cotton tip applicator (5–6 Hz vertically for 30 seconds). Laser speckle flowmetry images were obtained every 7.5 seconds. A region of interest (500 × 500 μm2) was placed within the whisker barrel field (1.3 mm posterior, 3.7 mm lateral from Bregma), using speckle contrast images to avoid large pial vessels. The peak CBF increase was taken as the response amplitude in each animal. Whisker stimulation paradigm was repeated 2–3 times in each experiment to determine the maximum response. After obtaining an arterial blood gas sample and baseline CBF images, hypercapnia was induced for 5 minutes (5% CO2 in 30% O2, 65% N2). Arterial blood gas measurement was repeated before the termination of hypercapnia. A total of 20 images were obtained every 30 seconds, and CBF changes were quantified using a polygonal region of interest that spanned the entire right hemisphere. Data were expressed as percent of baseline. To normalize the hyperemic responses to the magnitude of pCO2 increase in each mouse, hypercapnia index was also calculated (ΔCBF/ΔpCO2 in %/mm Hg). Autoregulation was tested at least 30 minutes after the completion of hypercapnia to allow normalization of arterial blood gases and pH. Mean arterial blood pressure was then lowered in 10 mm Hg steps by controlled exsanguination. One to two CBF images were obtained at least 4 minutes after each stable blood pressure level was attained. Baseline CBF at a blood pressure of 80 mm Hg was taken as 100%, and subsequent changes were expressed relative to this value. The lower limit of autoregulation was defined as the blood pressure below which CBF was statistically significantly lower compared with baseline CBF at a blood pressure of 80 mm Hg.
In a separate group of mice (n = 9 and 6, wild type and ApoE−/−, respectively), a craniotomy was carefully drilled under saline cooling to keep the dura intact. Two subcutaneous steel needle electrodes placed 5 mm apart in the contralateral whisker pad delivered single square wave current pulses every 10 seconds (0.2 ms, 700 μA; S48, Grass Instruments, West Warwick, RI, USA; A395 Linear Stimulus Isolator, WPI, Sarasota, FL, USA). Cortical evoked field potentials were recorded using a glass micropipette (200 mM NaCl, 40 μm tip) inserted into layer IV of the whisker barrel cortex (1.3 mm posterior, 3.7 mm lateral, 0.5 mm deep from Bregma), a subcutaneous Ag/AgCl reference electrode in the neck, and a differential amplifier (EX-1, Dagan Corporation, Minneapolis, MN, USA). Approximately 30–40 evoked field potentials were averaged to improve the signal-to-noise ratio, and the peak latency, amplitude, area-under-curve, and onset slope were measured.
Perfusion Deficit in Ischemic Stroke
We examined the ischemic perfusion defect using LSF during distal middle cerebral artery occlusion (dMCAO; n = 27 and 25, wild type and ApoE−/−, respectively, on various durations of HFD). Mice were anesthetized with isoflurane (2% for induction, 1% for maintenance, in 70% N2O and 30% O2) and intubated via a tracheostomy. The depth of anesthesia was adjusted to suppress cardiovascular responses to the tail pinch. Femoral artery was catheterized for the measurement of blood pressure. Rectal temperature was kept at 37 °C using a thermostatically controlled heating mat. Mice were paralyzed (pancuronium bromide, 0.4 mg/kg/h, intraperitoneal), mechanically ventilated, and placed in a stereotaxic frame. Arterial blood gases and pH were measured every 30 minutes and ventilation adjusted to maintain them within physiologic range. Data were continuously recorded and stored using a data acquisition and analysis system. After general surgery, mice were placed in a stereotaxic frame, and the skull surface was prepared for imaging as described above. Temporalis muscle was then separated from the temporal bone and removed, and a burr hole (2 mm diameter) was drilled under saline cooling in the temporal bone overlying the middle cerebral artery (MCA) just above the zygomatic arch. Dura was kept intact. Mice were allowed to stabilize for 30 minutes after surgical preparation, and then MCA was occluded using a microvascular clip confirmed in realtime using LSF. Imaging was started 1 minute before dMCAO and continued for 60 minutes (one image every 7.5 seconds).
We analyzed the spatiotemporal dynamics of the CBF deficit using three different approaches. First, based on the initial perfusion defect immediately after dMCAO, we placed an ROI (250 × 250 μm2) within the hemodynamic penumbra (steep portion of CBF gradient between the core and non-ischemic cortex). CBF changes within the ROI were then followed over time and expressed as percent of pre-ischemic baseline. This approach provided excellent temporal resolution but limited spatial information. Therefore, we next plotted the CBF gradient along a profile between lambda and the occluded MCA branch, both of which served as fixed and objective anatomic landmarks in each animal. When placed in this manner, the profile started from the non-ischemic parietal and occipital territories medially and passed through the penumbra into the deeply ischemic core in proximity to the occluded MCA branch. This approach accurately assessed the magnitude and the spatial extent of CBF reduction, but represented only a segment of the entire perfusion defect. We further enhanced the spatial analysis by using a thresholding paradigm to identify pixels with less than a certain amount of residual CBF (e.g. ≤20% or ≤30% of pre-ischemic baseline), and calculated their total area in mm2 based on the known scale factor. This parameter served as a spatially integrated measure of the entire perfusion defect.
Infarct Volume Determination
We determined the infarct volume after 4 or 8 weeks of HFD in a separate group of mice (n = 12 each, wild type and ApoE−/−) without intubation or mechanical ventilation, since these interventions significantly increase morbidity and mortality during the survival period. After 60 minutes of dMCAO as described above, the microvascular clip was carefully removed and reperfusion confirmed using LSF. Mice were excluded if reperfusion was not established, or if hemorrhage occurred at the clip site. Mice were killed 48 hours after ischemia and whole brains were incubated in 2,3,5-triphenyl-tetrazolium chloride (TTC) for 60 minutes. After obtaining images of the dorsal and lateral hemispheric surfaces separately, brains were cut into 1-mm-thick coronal sections, and infarct area at each section was measured and integrated. Because of the relatively small infarct volumes after dMCAO, indirect infarct volumes for edema correction were not reliable and therefore not calculated.
CBF Threshold for Tissue Viability
We determined the minimum critical level of residual CBF required to maintain tissue viability by combining the tissue outcome using TTC staining and perfusion deficit using LSF (n = 17 and 12, wild type and ApoE−/−, respectively, on various durations of HFD). Using surface landmarks, the dorsal views of topical TTC-stained brains at 48 hours were co-registered with the LSF images obtained 60 minutes after dMCAO before reperfusion. Minor spatial mismatch was corrected by image warping when necessary. The residual CBF profile between lambda and the occluded MCA branch was then plotted as described above using LSF maps and superimposed on the TTC-stained dorsal brain image showing the infarct. Residual CBF corresponding to the infarct edge was determined as the threshold below which infarction ensued, as reported previously. 16
Measurement of Serum Cholesterol
Blood was collected from the femoral artery at the end of experiments, put on ice for 30 minutes, centrifuged (14,000 r.p.m., 4 °C for 10 minutes), and serum stored at –80 °C until lipoprotein cholesterol distribution was measured by high-pressure liquid chromatography (Liposearch, Skylight Biotech, Akita, Japan). Plasma lipid measurements showed approximately 4–6-fold increase in total cholesterol and low density lipoprotein (LDL) fraction, and 60% to 70% decrease in high density lipoprotein fraction (HDL), in ApoE−/− compared with wild-type mice; triglyceride levels were unchanged (Supplementary Table 1).
Western Blotting
For Western blot analysis, whole brains from wild type (WT) on 8 weeks HFD, ApoE−/− mice on regular diet, and ApoE−/− mice on 8 weeks HFD (n = 3 each) were homogenized by a tissue grinder with lysis buffer containing 10 mM Tris-HCl, pH 6.8, 1 mM EDTA, and 10% SDS with one complete mini protease-inhibitor-cocktail tablet (Roche Diagnostics, Indianapolis, IN, USA) per 50 mL buffer. Fifteen μl of appropriately diluted protein solution and 10 μl of 2 × sample buffer (125 mM Tris-HCl, pH 6.8, 4% SDS, 20% glycerol, 0.05% bromphenol blue and 10% β-2-mercaptoethanol) were boiled for 10 minutes at 95 °C. Equal amounts of 60 μg of protein per sample were separated on 7.5% SDS-polyacrylamide gels and subsequently transferred to polyvinylidene fluoride membrane (Millipore, Billerica, MA, USA). After subsequent washing and blocking with 5% nonfat dry milk, the polyvinylidene fluoride membranes were incubated overnight at 4 °C with antibodies specific for eNOS (1:1,000, BD Bioscience), phospho-eNOS (1:1,000, p-Ser1177 and p-Thr495 human sequence numbering, BD Bioscience) and β-actin (1:2,000, Santa Cruz Biotechnology, Dallas, TX, USA), followed by secondary antibodies, and detected using enhanced chemiluminescence and exposed on autoradiography films.
Histopathology
We examined the degree of atherosclerosis in cerebral and extracerebral arteries using routine hematoxylin/eosin staining of intracardiac perfusion-fixed tissues (4% paraformaldehyde) harvested at the end of LSF experiments in three wild type and five ApoE−/− mice. In addition, three ApoE−/− mice were examined after more than 6 months of HFD starting at 12 months of age. Brains were also assessed for neuropathological changes. The skull base and cervical and thoracic spine were dissected en bloc, with the heart, aortic arch and its major cervical branches in situ, including the petrous portion of internal carotid arteries and cervical vertebral arteries. Tissues were then decalcified, paraffin-embedded, and sectioned (12 μm) en bloc to preserve anatomic relationships. Extracranial and intracranial arteries were examined for the presence of overt (e.g., cholesterol clefts) or occult (e.g., subendothelial foam cells) atherosclerosis.
Data Analysis and Statistics
Experiments were carried out masked to the genotype; however, discernible changes in fur quality over time precluded effective blinding to HFD duration. Absence of a therapeutic intervention obviated the need for randomization. Based on predetermined criteria, animals were excluded if baseline arterial blood pressure was less than 80 mm Hg, pH less than 7.25, pCO2 more than 50 mm Hg, and pO2 less than 100 mm Hg. Data were analyzed in a masked fashion, expressed as mean ± standard error, and statistically tested using t-test or one- or two-way analysis of variance for independent or repeated measures followed by Tukey's, Sidak's, or Dunnet's multiple comparisons tests where appropriate (SPSS v11.0, IBM, Armonk, NY, USA; GraphPad Prism v6, La Jolla, CA, USA). P < 0.05 was considered statistically significant.
RESULTS
Histopathology
Wild-type mice did not show any evidence of atherosclerosis (n = 3; not shown), whereas all ApoE−/− mice developed early changes consisting of scattered subendothelial foam cells in carotid arteries, and mild fatty streaks in the proximal aortic arch after 10 weeks of HFD starting at 4 weeks of age (n = 5; Supplementary Figure 1). None of the mice showed atherosclerotic luminal stenosis in cervical vessels at this age and HFD duration. In contrast, severe and often occlusive atherosclerotic plaques were found in the aorta and cervical arteries in 18-month-old ApoE−/− mice fed HFD for 6 months (n = 3). Intracranial vessels were free of any discernible atherosclerotic changes in all age groups and HFD durations studied (not shown).
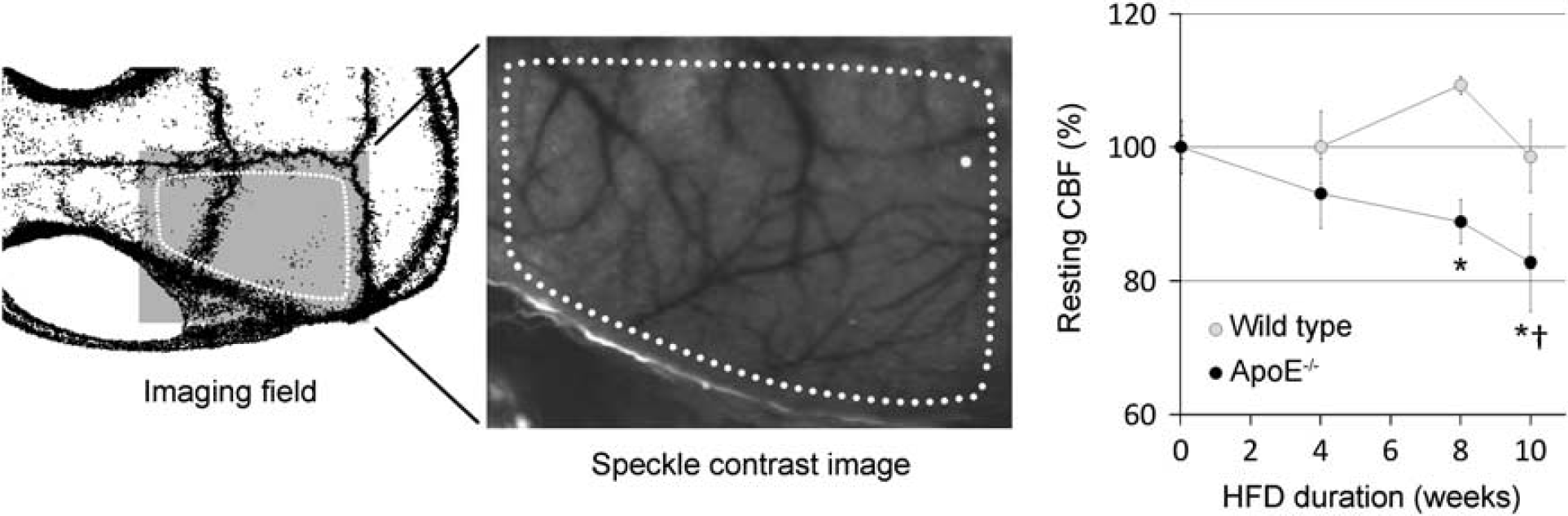
Resting cerebral perfusion. Laser speckle flowmetry provides a two-dimensional perfusion map of the entire dorsal cortex. Imaging field position over the right hemisphere (left) and a representative speckle contrast perfusion image (middle) are shown. Dotted lines show the region of interest for resting perfusion measurements in arbitrary units. Resting cerebral blood flow (CBF) progressively decreased after high-fat diet (HFD) in ApoE−/− mice, but not in the wild type. n = 9, 5, 8, 5 WT, and 6, 4, 9, 6 ApoE−/− at 0, 4, 8 and 10 week time points, respectively. ∗P < 0.05 vs. wild type (WT), †P < 0.05 vs. 0 weeks; two-way analysis of variance (ANOVA).
Systemic Physiology and Resting Cerebral Blood Flow
Arterial blood pressure was approximately 10% higher in ApoE−/− mice regardless of age and HFD duration, while body weights and arterial pH and blood gas values did not significantly differ from wild type (Supplementary Table 2). Despite higher blood pressures, absolute resting CBF was 20%–30% lower in ApoE−/− mice compared with wild type in cortex, striatum, and cerebellum suggesting increased cerebrovascular resistance (Supplementary Table 3). This was confirmed non-invasively by LSF, which showed a progressive reduction in resting CBF in ApoE−/− mice in relation to HFD duration (Figure 1). Wild-type mice did not show a consistent change in resting CBF even after 10 weeks of HFD.
Physiologic Cerebral Blood Flow Responses
Physiologic reflexes were studied at 16 ± 1 weeks of age after 10 ± 1 weeks of HFD in wild type, and at 14 ± 1 weeks of age after 11 ± 1 weeks of HFD in ApoE−/− mice. Hypercapnia (5% CO2 inhalation) caused a rapid increase in CBF throughout the entire hemisphere, reaching a peak within 4 minutes (Figure 2). Peak hypercapnic hyperemia was attenuated in ApoE−/− mice by approximately 30% compared with wild-type mice. The attenuation was even more marked when normalized to arterial pCO2 increase (hypercapnic index 2.9 ± 0.4 vs. 1.7 ± 0.3% CBF increase for each mm Hg pCO2 increase, in wild type and ApoE−/− mice, n = 11 and 7, respectively; P < 0.05). Autoregulation of CBF was assessed during progressive stepwise blood pressure reduction by controlled exsanguination (Figure 3). In wild-type mice (n = 11), reduced perfusion pressure was counterbalanced by a corresponding drop in cerebrovascular resistance (CVR), so that CBF did not significantly decrease from baseline until a blood pressure of 40 mm Hg was reached (i.e., lower limit of autoregulation). In ApoE−/− mice (n = 7), CVR remained higher during progressive hypotension. Consequently, CBF started to diminish at higher blood pressure levels and remained below wild-type mice throughout the tested blood pressure range, indicating impaired autoregulation. The lower limit of autoregulation was 60 mm Hg in ApoE−/− mice. Functional activation of the cortex, tested by manual stimulation of the entire whisker pad, evoked a regional hyperemic response that reached 28 ± 3% of baseline within the contralateral barrel cortex (n = 11; Figure 4). This functional hyperemia was blunted in ApoE−/− mice by 64% (10% ± 3%; n = 7; P < 0.05), suggesting impaired neurovascular coupling.
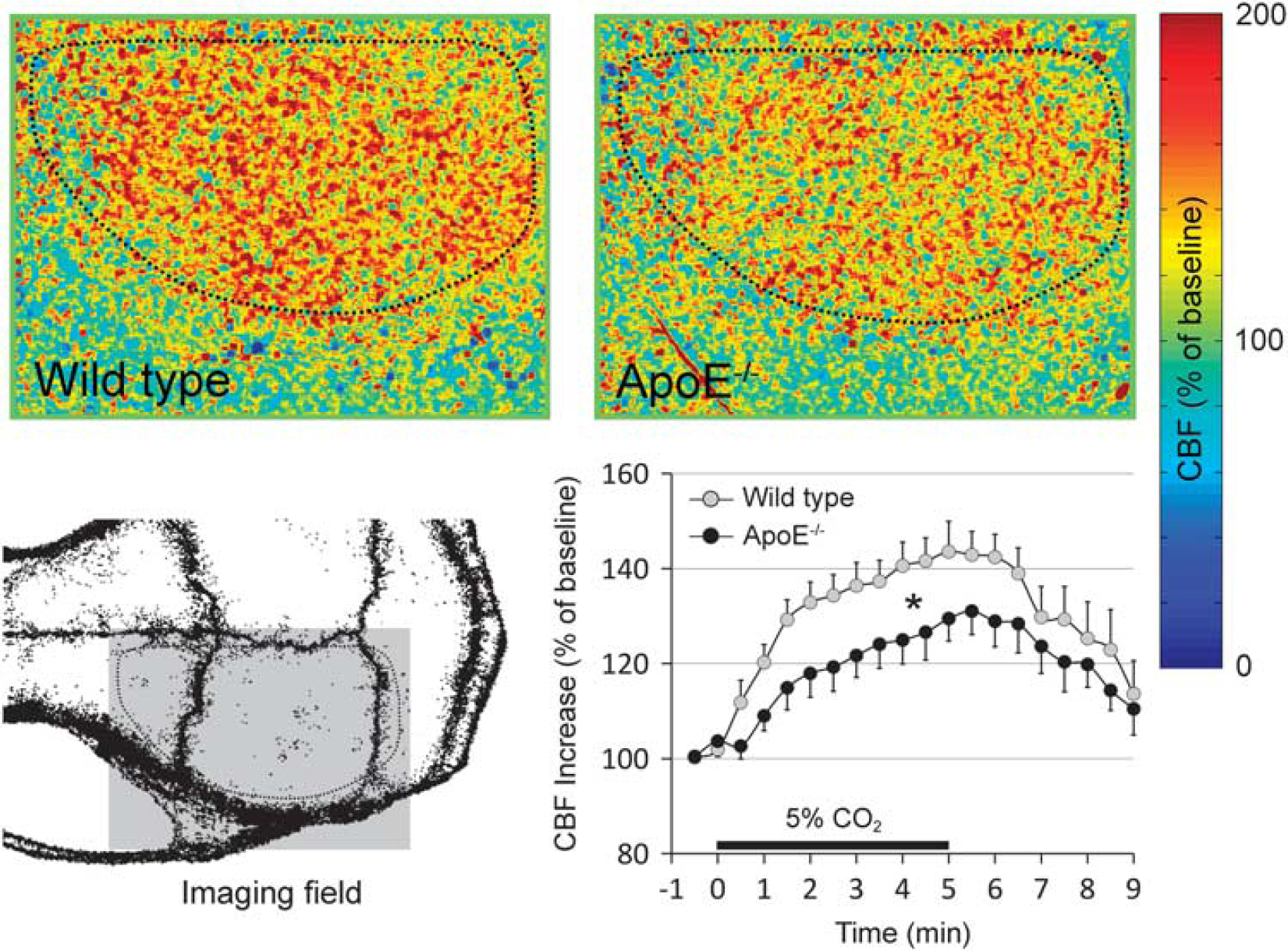
Hypercapnic hyperemia. Representative laser speckle perfusion images through intact skull show hypercapnic hyperemia (5% CO2 inhalation) throughout the dorsal cortex in wild type and ApoE−/− mice (upper row). Color bar indicates cerebral blood flow (CBF) relative to baseline. Imaging field position is shown on the lower left. Dotted outline indicates the cortical region of interest to quantify the time course of hypercapnic CBF changes (lower right). Both the rate of CBF increase and the peak hyperemia were attenuated in ApoE−/− mice during hypercapnia (n = 11 and 7, wild type and ApoE−/− mice, respectively; ∗P < 0.05, two-way analysis of variance (ANOVA) for repeated measures).
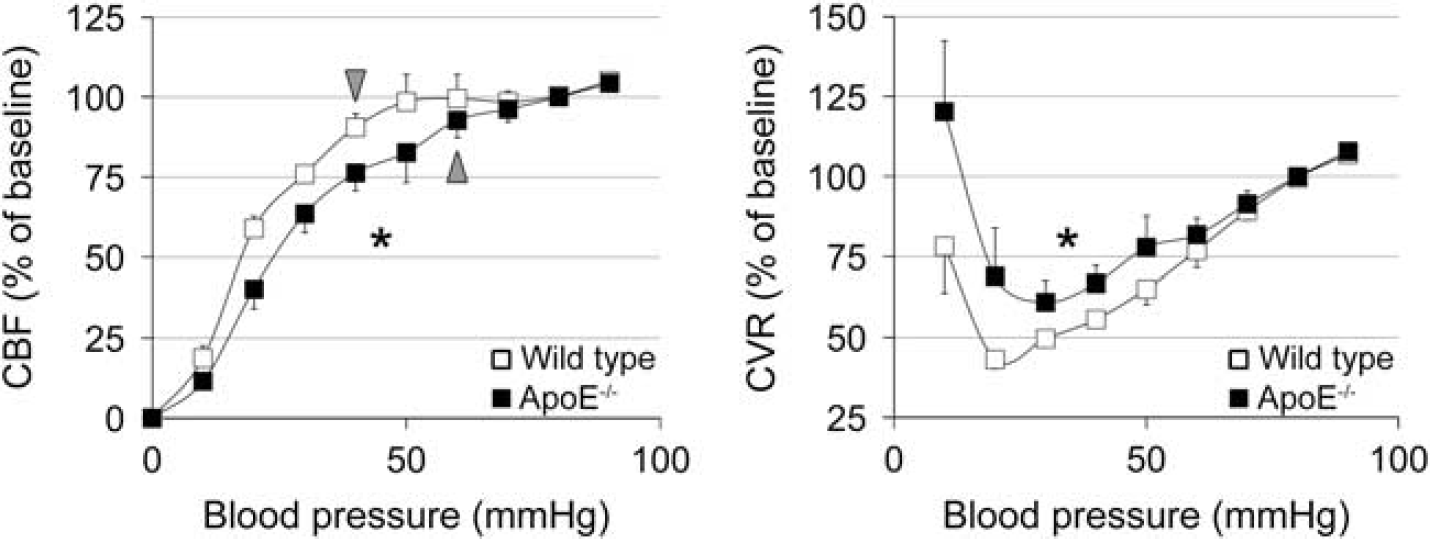
Autoregulation of cerebral blood flow. The lower limit of cerebral blood flow (CBF) autoregulation was tested by stepwise reduction in blood pressure by controlled exsanguination. Both CBF and cerebrovascular resistance (CVR) autoregulation curves significantly differed between wild type and ApoE−/− mice (n = 11 and 7, respectively; ∗P < 0.05, two-way analysis of variance (ANOVA) for repeated measures). The lower limit of autoregulation was 40 mm Hg in wild type compared with 60 mm Hg in ApoE−/− mice (arrowheads), defined as the blood pressure level below which CBF was statistically significantly lower than the baseline CBF at 80 mm Hg blood pressure (see Methods). Moreover, both the lowest level of CVR attained during hypotension (i.e., maximal vasodilation), and the associated blood pressure level were higher in ApoE−/− mice compared with wild type. CVR change was calculated as 100 × (BP/BP0)/(CBF/CBF0), where BP0 and CBF0 are baseline values (i.e., at 80 mm Hg blood pressure).
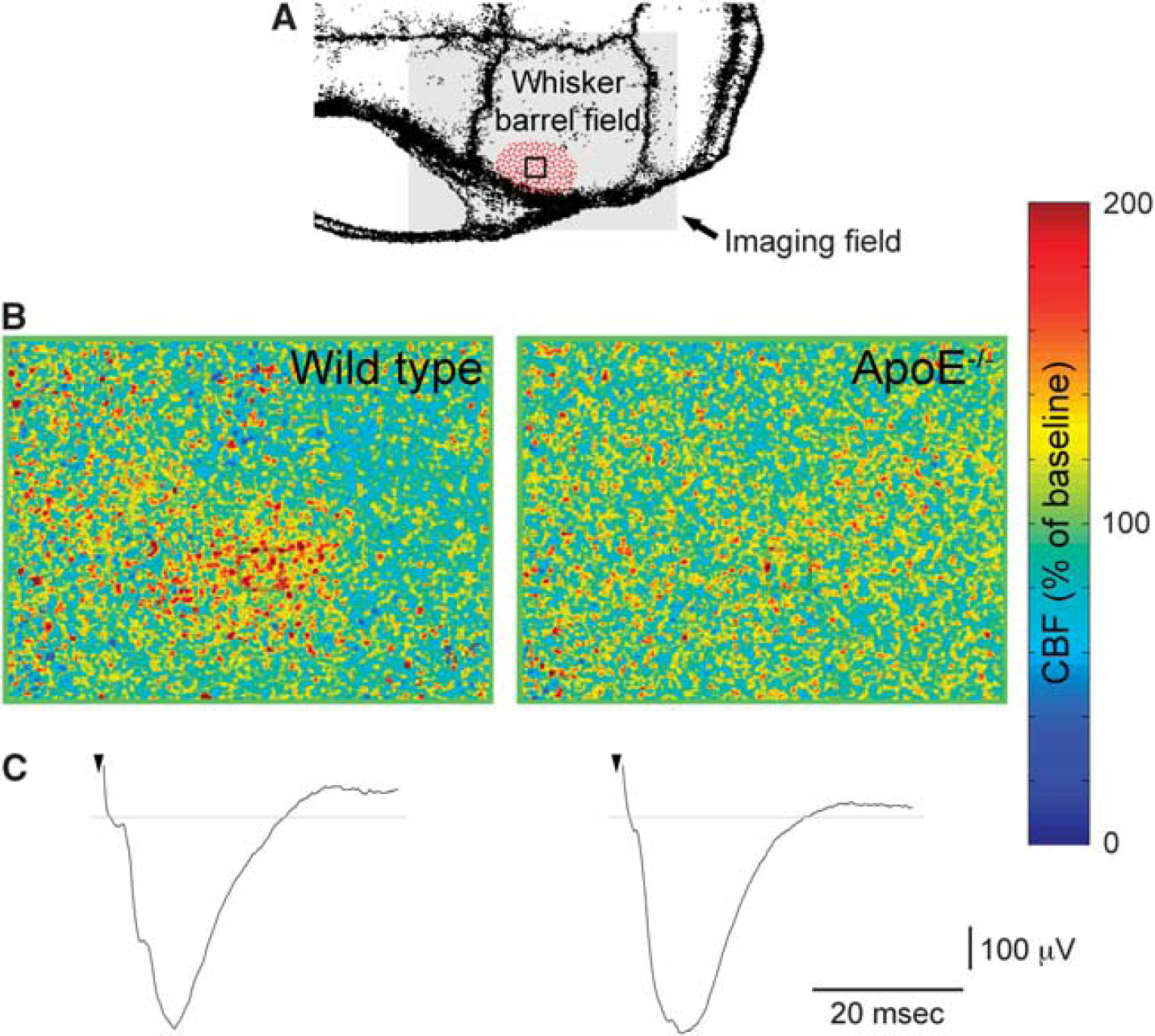
Functional neurovascular coupling in whisker barrel cortex. (
ApoE−/− mice reportedly develop progressive neurodegenerative changes that are histologically evident after 3–4 months of age. 17 Although we studied mice at an early age when neurodegenerative changes are histologically not evident, subtle changes can still interfere with cortical activation and secondarily diminish the associated functional hyperemia. To eliminate this potential confounder, we studied somatosensory-evoked field potentials in the whisker barrel cortex in a separate cohort, and found them comparable in ApoE−/− and wild-type mice (n = 6 and 9, respectively; Figure 4, Supplementary Table 4), suggesting that the attenuation of functional hyperemia was not secondary to impaired cortical activation.
Perfusion Defects after dMCAO
We examined the dynamic perfusion deficit during acute stroke using laser speckle imaging non-invasively through intact skull at various ages and HFD durations in separate cohorts (Figure 5). Distal MCAO abruptly reduced regional CBF over the dorsolateral cortex in all groups (Figure 5A). Penumbral perfusion reduction (square ROI) was more severe in ApoE−/− mice compared with wild type after 8 and 10 but not 4 weeks of HFD (Figure 5A, graphs). The difference persisted throughout the 60 minutes dMCAO. In the core, CBF was reduced to less than 20% and did not differ between the strains (P > 0.05; not shown). The CBF profile across the ischemic territory confirmed more severe perfusion deficit in ApoE−/− mice in penumbra after HFD (Figure 5B). Indeed, residual CBF was lower in ApoE−/− mice throughout the hemisphere, except in the most severely ischemic core, and tended to worsen as a function of HFD duration. As a result, the area of perfusion defect was larger and progressively worsened during dMCAO in ApoE−/− mice compared with age- and diet-matched wild-type controls (Figure 5C). In wild-type mice, HFD duration did not have a significant impact on perfusion deficit after dMCAO. In addition, we found a significant direct relationship between the area of perfusion defect (residual CBF ≤ 20%) and plasma total cholesterol (r = 0.65, P = 0.0006), LDL (r = 0.56, P = 0.0041), VLDL (r = 0.45, P = 0.0292), and CM (r = 0.67, P = 0.0003) fractions, when all data from WT and ApoE KO on 8 weeks of HFD were pooled (Spearman correlation). The inverse relationship between the perfusion defect and HDL levels did not reach statistical significance (r = 0.22, P = 0.3). These conclusions persisted when a residual CBF threshold of ≤30% was used to calculate the area of perfusion defect (not shown).
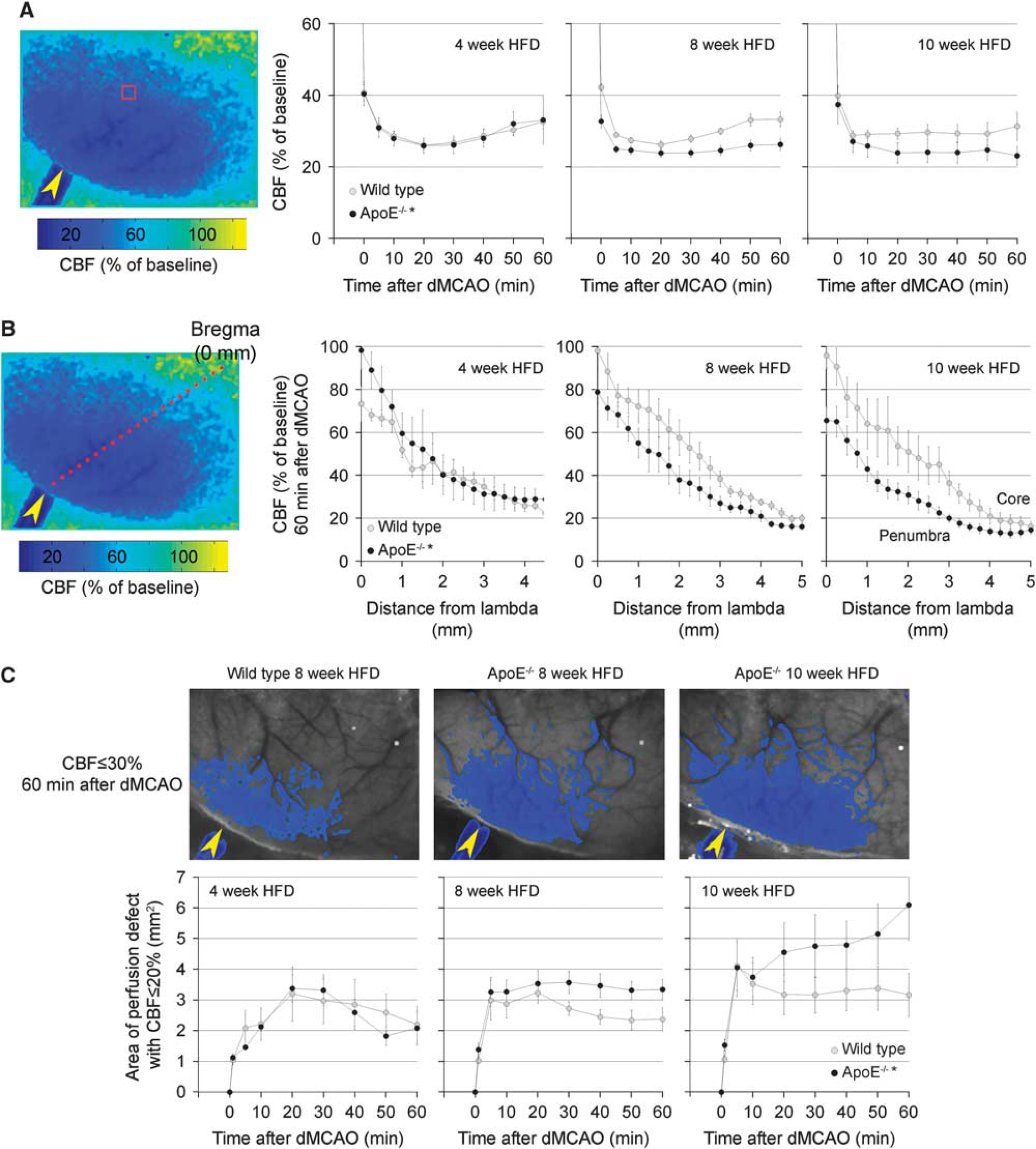
Perfusion defect during focal arterial occlusion. (
Infarct Volume
Consistent with worse perfusion deficits, infarct volumes were significantly larger in ApoE−/− mice compared with wild type after 8 but not 4 weeks of HFD when measured 48 hours after 1 hour transient dMCAO (14.5 ± 2.3 vs. 14.0 ± 2.9 mm3 after 4 weeks, and 11.0 ± 3.9 vs. 21.4 ± 3.2 mm3 after 8 weeks HFD in wild type and ApoE−/−, respectively; P < 0.05 after 8 weeks; Figure 6). Infarcts in wild type tended to be smaller after 8 weeks of HFD compared with 4 weeks. The difference was evident in all anteroposterior slice levels.
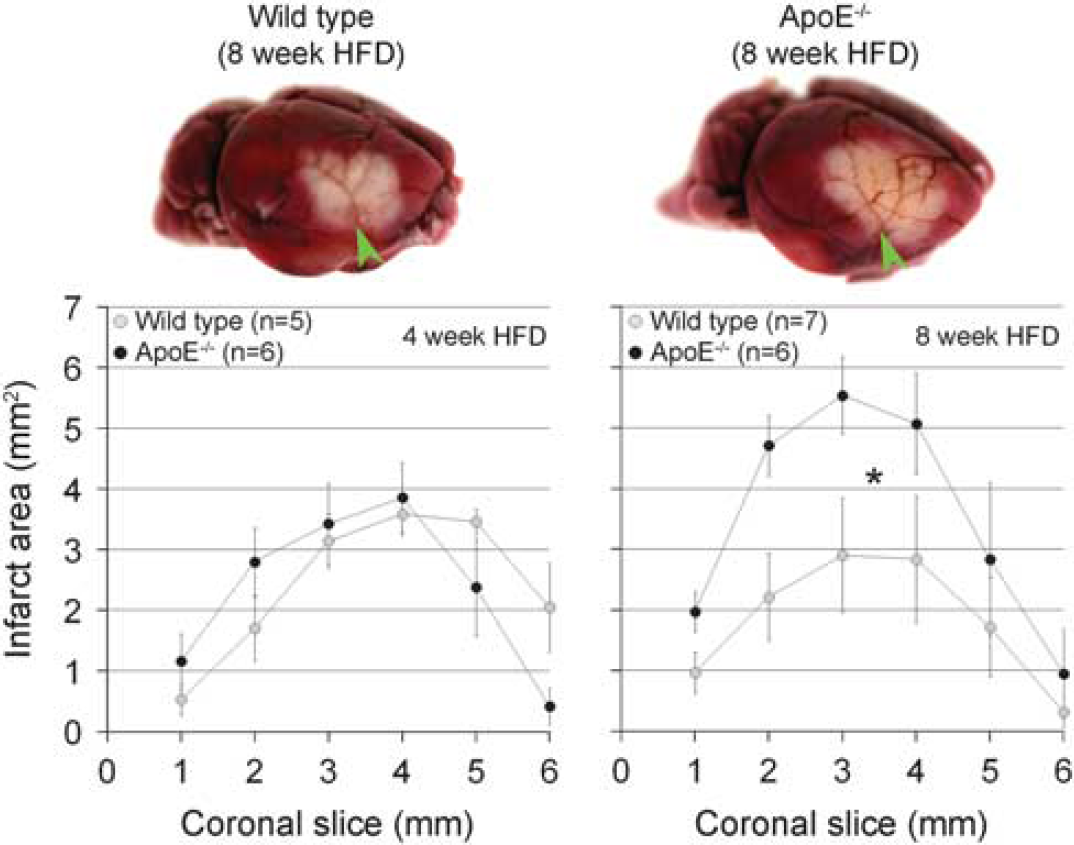
Infarct volume after transient focal arterial occlusion. Representative topical triphenyl-tetrazolium chloride (TTC) stained brains (upper row) from wild type and ApoE−/− mice show cortical infarct 48 hours after 1 hour transient distal middle cerebral artery occlusion (arrowheads). Infarct volumes, calculated by integrating the infarct area in 1 mm thick coronal slices, were almost doubled in ApoE−/− mice compared with wild type (WT) in a concentric fashion after 8 but not 4 weeks of high-fat diet (HFD) (lower row). Numbers of mice are shown on the graphs. ∗P < 0.05 vs. WT.
Viability Threshold
To test whether enlarged infarct volumes in ApoE−/− mice can be explained by hemodynamic factors alone, we calculated the viability threshold, defined as the CBF threshold below which infarction ensued (Figure 7). The CBF threshold for viability was significantly higher in ApoE−/− mice compared with wild type after 8 but not 4 weeks of HFD, suggesting that in addition to significantly worse CBF deficits, ApoE−/− brain tissue is also more sensitive to ischemia, and infarct ion ensues at milder hypoperfusion levels compared with wild type. Interestingly, 8 weeks of HFD appeared to decrease the viability threshold in wild-type mice, suggesting a neuroprotective effect.
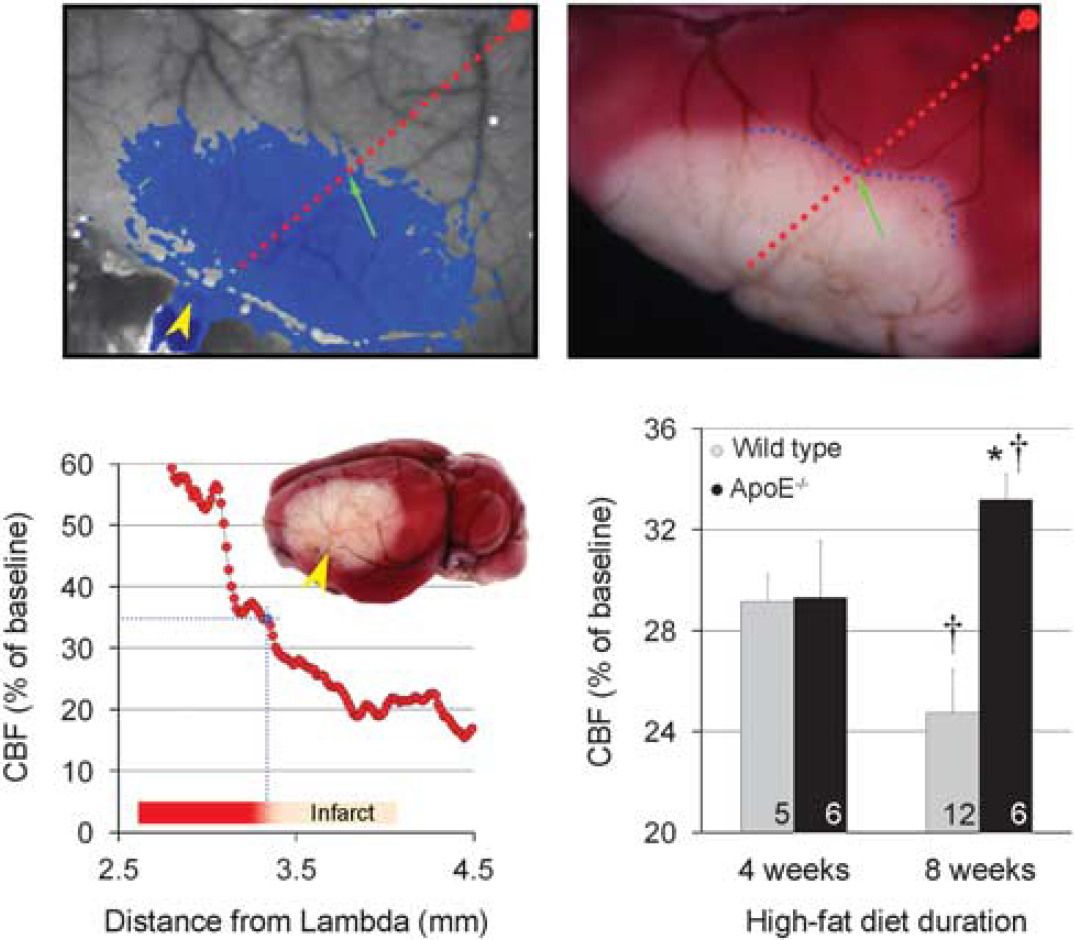
Perfusion threshold for viability. The laser speckle contrast image of a representative ApoE−/− mouse shows pixels with ≤35% residual cerebral blood flow (CBF) superimposed in blue at 60 minutes after distal middle cerebral artery occlusion (arrowhead) immediately before reperfusion (upper left). The triphenyl-tetrazolium chloride (TTC)-stained brain of the same animal shows the infarct 48 hours later (upper right). After spatially co-registering the two images using surface landmarks, a line profile (red dotted line) is drawn between lambda and the occluded middle cerebral artery branch, and CBF plotted along this profile as a function of distance from lambda using laser speckle images (lower left). The CBF level corresponding to the infarct edge (blue dotted line) is then located (green arrow) based on the distance of infarct edge from lambda on this profile. This value represents the CBF threshold for viability, below which the tissue infarcted (35% residual CBF in this representative mouse). When calculated in this way for each mouse, the viability threshold was significantly higher in ApoE−/− mice after 8 but not 4 weeks of high-fat diet (HFD) compared with age and diet-matched controls (lower right), suggesting that, in addition to suffering worse perfusion defects after distal middle cerebral artery occlusion (dMCAO), ApoE−/− brains require even higher CBF to survive. Numbers of mice in each group are shown on the bars. ∗P < 0.05 vs. wild type (WT); †P < 0.05 vs. 4 week HFD.
eNOS Phosphorylation
To explore the molecular basis for the cerebrovascular collateral dysfunction in ApoE−/− mice, we studied the expression and phosphorylation status of eNOS in homogenized brain tissue (Supplementary Figure 2). While total eNOS protein levels did not differ between ApoE−/− and wild-type mice after HFD, eNOS phosphorylation at the positive regulatory site Serine 1176 (S1176) was reduced in ApoE−/− mice, and phosphorylation at the negative regulatory site Threonine 494 increased compared with wild type after 8 weeks of HFD. ApoE−/− mice on regular diet also showed reduced S1176 phosphorylation albeit to a lesser extent, while Threonine 494 phosphorylation was unchanged.
DISCUSSION
Elevated stroke risk in hyperlipidemia is generally attributed to cervical or coronary atherosclerosis predisposing to atherothrombotic and cardioembolic strokes. However, our data show that even in the absence of atherosclerotic stenoses, both resting CBF and fundamental vasodilator responses to hypercapnia, functional cortical activation, and reduced perfusion pressure are diminished in hyperlipidemic ApoE−/− mice. Moreover, focal arterial occlusion results in larger perfusion defects, presumably because of impaired collateral flow. Taken together with elevated arterial blood pressure in ApoE−/− mice, detected in some but not all hyperlipidemic animal models,5,18 these data suggest significantly elevated resting CVR and severe vasodilator dysfunction in ApoE−/− mice. Such global CBF dysregulation may be a mechanism by which hyperlipidemia disrupts tissue homeostasis and predisposes to slowly progressive parenchymal neurodegeneration. For example, relative hypoperfusion at resting state and supply-demand mismatch during functional metabolic activation can lead to a chronic low-grade hypoxic–ischemic state, further exacerbated by impaired autoregulation during hypotensive transients. Moreover, resting hypoperfusion and impaired vasodilator function can compromise collateral flow, putting larger volumes of tissue at risk for infarction in hyperlipidemic patients upon stroke.
Previous work has shown diminished endothelium-dependent relaxations to pharmacologic agents in systemic (e.g., aorta) and cerebral arteries (e.g., carotid, basilar) in hyperlipidemic rabbits and ApoE−/− mice,3–5,7 although not all studies agree. 6 However, whether abnormal responses to pharmacologic agents translate into abnormal cerebrovascular physiology has been unclear. For example, resting CBF was reduced in rabbits on HFD, 19 but not in young ApoE−/− mice (8–10 weeks) on normal diet. 20 HFD attenuated hypercapnic hyperemia in cynomolgus monkeys and baboons,18,21 yet neither resting CBF nor autoregulation during hypotension appeared to be impaired in these models.
Clinically, patients with familial hyperlipidemia have lower resting mean transcranial Doppler velocities and increased pulsatility index suggestive of increased downstream CVR. Furthermore, flow velocities acutely increase after apheresis suggesting a functional rather than anatomic (i.e., atheroscleroctic stenosis) restriction. 22 Mild-to-moderate hyperlipidemia, clinically more prevalent than severe genetic forms, also appears to disturb cerebrovascular physiology. In the prospective population-based Rotterdam study, elevated total cholesterol levels were associated with diminished CO2 reactivity index. 23 Moreover, acute reduction in LDL cholesterol, lipoprotein(a), and fibrinogen levels by heparin-mediated extracorporeal LDL precipitation increased the CO2 reactivity index by 60%, as determined by transcranial Doppler in patients with coronary artery disease and hyperlipidemia. 24 Conflicting data have also been reported, possibly reflecting heterogeneity in patient characteristics.25,26
Hyperlipidemic ApoE−/− mice do develop more severe tissue injury upon focal cerebral ischemia exacerbated by HFD. 27 However, whether enlarged infarcts in ApoE−/− mice are related to hyperlipidemic vascular dysfunction and worse perfusion defects has been unclear. In previous reports, CBF deficit was either not measured, or measured only in the ischemic core in younger ApoE−/− on regular diet and did not differ from the wild type.20,27–32 As a result, previous studies concluded that worse tissue outcome reflects non-vascular mechanisms. However, traditional methods to assess perfusion during focal arterial occlusion lack either spatial (e.g., laser Doppler) or temporal information (e.g., autoradiography). Therefore, whether hyperlipidemic vascular dysfunction can worsen the perfusion defect after focal arterial occlusion has not been adequately tested. Our data obtained using laser speckle flowmetry with high spatial and temporal resolution show significantly larger perfusion deficits as one mechanism for larger infarcts in ApoE−/− mice. The phenotype was progressive as a function of HFD duration.
Importantly, we did not find flow-limiting atherosclerotic lesions in ascending aorta and cervical arteries in ApoE−/− mice in the age groups studied, as previously reported.30,33 Moreover, neither the circle of Willis nor pial collaterals reportedly differed between ApoE−/− and wild-type mice, 20 eliminating a potential confounder, and further implicating vascular dysfunction as the mechanism for larger perfusion defects after dMCAO. Because hemodynamically significant atherosclerotic stenoses in cervical arteries can reduce distal perfusion pressure and diminish CBF responses, we chose to study mice at 14 weeks of age (i.e., 10 weeks of HFD), at a time when atherosclerotic changes were limited to subendothelial foam cells in the aortic root and arch, and major branches. In a previous study in ApoE−/− mice, endothelial dysfunction was shown after prolonged HFD (e.g., 6 months or more) 5 when potentially flow-limiting atherosclerotic stenoses were likely present in aortic arch and carotid arteries. Consistent with previous reports, we did not observe intracranial atherosclerotic changes even when advanced occlusive atherosclerotic plaques were present in aorta and its major branches in older mice. 5 Cerebral circulation appears to be relatively spared from atherosclerotic changes in human familial hyperlipidemia as well. 34
Notably, the increase in infarct volume in ApoE−/− mice appeared to be more than expected based on the increase in perfusion defect alone (Figures 5 and 6), suggesting that vascular dysfunction may not be the only mechanism of enlarged infarcts in the ApoE−/−. To test this we calculated the CBF threshold for tissue viability and indeed found that ApoE−/− brains required a higher level of CBF to survive compared with wild type (33% vs. 25% residual CBF, respectively), suggesting increased tissue sensitivity to ischemia. Increased sensitivity of ApoE−/− mice to global ischemia and excitotoxic injury has been reported, independent of vascular function.17,31 Mechanisms of neuronal sensitivity to ischemia in the ApoE−/− may relate to immunomodulatory and antioxidant effects of ApoE, or the primary neurodegenerative phenotype previously reported in the mutant, although the latter is not conspicuous in the age group we studied. The exact mechanism of neurodegeneration in ApoE−/− mice is unknown. Although ApoE plays a critical role in cytoskeletal stability and synaptic regeneration as a potential mechanism, chronic CBF dysregulation and loss of tissue homeostasis may also contribute to progressive neurodegeneration, as has been suggested for cerebral amyloid angiopathy and cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy. Interestingly, HFD for 8 weeks appeared to decrease the CBF threshold for viability in wild-type mice (i.e., reduced sensitivity to ischemia). These effects may be related to the known ketogenic properties of HFD exerting direct anti-excitatory and neuroprotective effects. 35 More work is needed to elucidate the mechanisms.
Mechanisms of hyperlipidemic vascular dysfunction are not completely understood. We detected reciprocal changes in eNOS phosphorylation at the positive and negative regulatory sites in ApoE−/− mice (i.e., reduced S1176 and increased T494 phosphorylation, respectively), exacerbated by HFD; consistent with previous reports total eNOS expression was not altered.3,36 Taken together with our previous study showing that eNOS phosphorylation at S1179 (bovine, corresponding to mouse S1176 and human S1177) regulates the CBF deficit after stroke, 37 eNOS dysfunction is a plausible mechanism for larger perfusion defects in the ApoE−/−, and can be pharmacologically targeted to improve ischemic tissue perfusion and outcome. 38
In summary, our data suggest that hyperlipidemia causes cerebrovascular dysfunction that predisposes to chronic supply–demand mismatch and compromises collateral perfusion in ischemic stroke, in the absence of hemodynamically significant, flow-limiting atherosclerotic stenoses in large arteries. As a result, hyperlipidemic patients may develop larger perfusion defects if and when they suffer cerebral arterial occlusions. More work is needed to determine the clinically relevant plasma lipid levels for vascular dysfunction and diminished collateral perfusion to become manifest in acute stroke.
DISCLOSURE/CONFLICT OF INTEREST
The authors declare no conflict of interest.
Supplementary Information accompanies the paper on the Journal of Cerebral Blood Flow & Metabolism website (http://www.nature.com/jcbfm)
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.