
Abstract
Estradiol reduces brain injury from many diseases, including stroke and trauma. To investigate the molecular mechanisms of this protection, the effects of 17-β-estradiol on heat shock protein (HSP) expression were studied in normal male and female rats and in male gerbils after global ischemia. 17-β-Estradiol was given intraperitoneally (46 or 460 ng/kg, or 4.6 μg/kg) and Western blots performed for HSPs. 17-β-Estradiol increased hemeoxygenase-1, HSP25/27, and HSP70 in the brain of male and female rats. Six hours after the administration of 17-β-estradiol, hemeoxygenase-1 increased 3.9-fold (460 ng/kg) and 5.4-fold (4.6 μg/kg), HSP25/27 increased 2.1-fold (4.6 μg/kg), and Hsp70 increased 2.3-fold (460 ng/kg). Immunocytochemistry showed that hemeoxygenase-1, HSP25/27,and HSP70 induction was localized to cerebral arteries in male rats, possibly in vascular smooth muscle cells. 17-β-Estradiol was injected intraperitoneally 20 minutes before transient occlusion of both carotids in adult gerbils. Six hours after global cerebral ischemia, 17-β-estradiol (460 ng/kg) increased levels of hemeoxygenase-1 protein 2.4-fold compared with ischemia alone, and HSP25/27 levels increased 1.8-fold compared with ischemia alone. Hemeoxygenase-1 was induced in striatal oligodendrocytes and hippocampal neurons, and HSP25/27 levels increased in striatal astrocytes and hippocampal neurons. Finally, Western blot analysis confirmed that estrogen induced heat shock factor-1, providing a possible mechanism by which estrogen induces HSPs in brain and other tissues. The induction of HSPs may be an important mechanism for estrogen protection against cerebral ischemia and other types of injury.
Keywords
Epidemiologic studies associate postmenopausal estrogen use with a reduction in risk of Alzheimer and Parkinson disease, and reduced death from stroke and cardiovascular disease (Garcia-Segura et al., 2001; Green and Simpkins, 2000; Hurn and Macrae, 2000). Estrogen protects in several neuronal culture model systems, including serum deprivation, β-amyloid–induced toxicity, excitotoxicity, and oxygen-glucose deprivation. Estrogens attenuate neuronal death in rodent models of cerebral ischemia, traumatic injury, and Parkinson disease (Hurn and Macrae, 2000). Although estrogens are known to exert several direct and indirect effects on neurons and the brain, the cellular and molecular mechanisms of protection by this steroid are just beginning to be elucidated (Garcia-Segura et al., 2001; Green and Simpkins, 2000; Hurn and Macrae, 2000). One mechanism of protection may be that native estrogens or exogenous 17-β-estradiol act as direct vasodilators or through vasodilatory mediators during ischemia. Estrogen-mediated improvement in cerebral blood flow has been most clearly identified in models of global cerebral ischemia (Hurn and Macrae, 2000).
Increased expression of heat shock protein (HSP) 70, 27 (HSP27), and 32 (hemeoxygenase-1) in the brain has been extensively documented in association with a variety of insults, including ischemia, and may play a role in cell survival and recovery after injury (Rajdev et al., 2000; Sharp et al., 1999; Xanthoudakis and Nicholson, 2000). Estrogen has been shown to modulate HSPs in a variety of tissues. Estrogen increases heat-shock factor expression in endometrium (Yang et al., 1995), and activates HSF-1 and induces HSP72 in isolated cardiac myocytes (Knowlton and Sun, 2001). Estradiol increases HSP90 and HSP70 mRNA in myometrium and endometrium of nonpregnant ewes (Wu et al., 1996), and induces HSP27 in human endometrial glandular epithelium (Padwick et al., 1994), endothelial cells (Piotrowicz et al., 1995), and in MCF7 human breast cancer cells (Porter et al., 2001).
There are only a few isolated reports that estrogen might modulate HSP expression in the brain. Estrogen increases HSP90 protein in ventromedial hypothalamus, heat shock cognate 73 (Hsc73) in the pituitary and ventromedial hypothalamus, and induces HSP72, HSP27, and HSP90 in astrocytes in the arcuate nucleus and in the wall surrounding the third ventricle (Olazabal et al., 1992; Krebs et al., 1999; Mydlarski et al., 1995). The significance of the localized estrogen-related HSP induction is not known, nor is it known whether this localized induction relates to the protection afforded by estrogen.
In this study, we postulated that estradiol would induce HSPs in the brain because of reports that estrogen induces heat shock factors (HSFs) and binds HSP90 (Yang et al., 1995; Knowlton and Sun, 2001), both of which should induce HSPs. We found that 17-β-estradiol induced hemeoxygenase-1, HSP25/27, and HSP70 in brains of male and female rats, and that this induction occurred mainly in cerebral blood vessels in male rats. 17-β-Estradiol also increased hemeoxygenase-1 and HSP25/27 brain expression 6 hours after global cerebral ischemia in gerbils. Hemeoxygenase-1 was induced in striatal oligodendrocytes and hippocampal neurons, and HSP25 was induced in striatal astrocytes and hippocampal neurons. These results suggest that estrogen induction of HSPs may be one of the important mechanisms by which estrogen protects against ischemia.
MATERIALS AND METHODS
Animal studies
Male and female Sprague-Dawley rats (n = 40, 200–240 g) and adult male Mongolian gerbils (n = 12, 60–80 g) were used in this study. All procedures were approved by the University of Cincinnati Animal Care Committee in accordance with the National Institutes of Health Guide for Care and Use of Laboratory Animals. Water-soluble 17-β-estradiol (E4389; Sigma, St. Louis, MO, U.S.A.) was dissolved in 0.9% saline solution and administered intraperitoneally at a dose of 46 or 460 ng/kg or 4.6 μg/kg in male and female rats. Controls were injected with 0.9% saline. Positive controls were injected with kainic acid (10 mg/kg, intraperitoneally). After 6 or 24 hours, rats were anesthetized with isoflurane and euthanized for Western blot analysis (two animals for each group); or animals were anesthetized with ketamine (100 mg/kg) and xylazine (20 mg/kg) and perfused with 4% paraformaldehyde in 0.1 mol/L phosphate buffer (pH 7.4) for immunohistochemical analysis (two animals for each group).
Adult male Mongolian gerbils were anesthetized with 3% isoflurane in a mixture of 20% oxygen and 77% nitrogen. 17-β-Estradiol (460 ng/kg or 4.6 μg/kg) or saline (0.9%) vehicle was injected intraperitoneally 20 minutes before ischemia. To produce ischemia, both common carotid arteries were exposed and occluded with aneurysm clips for 5 minutes. The clips were then removed to restore cerebral blood flow. The rectal temperature was maintained at 36.5 to 37.5°C with a heating blanket until the animals recovered from surgery. After recovery, the animals were monitored for 2 hours to prevent hypothermia. At 6 or 24 hours after ischemia, the gerbils were anesthetized with isoflurane and euthanized before the brains were removed for Western blot analysis (two animals for each group), or were anesthetized with ketamine (100 mg/kg) and xylazine (20 mg/kg) and perfused with 4% paraformaldehyde before the brains were processed for immunohistochemical analysis (two animals for each group).
Tissue preparation and Western blot analysis
Brains were homogenized in ristocetin-induced platelet agglutination buffer (9.1 mmol/L dibasic sodium phosphate, 1.7 mmol/L monobasic sodium phosphate, 150 mmol/L sodium chloride, 10 μl/mL Igepal (I-3021, Sigma) CA-630, 5 mg/mL sodium deoxycholate, 1 mg/mL sodium dodecyl sulfate, pH 7.4) with 0.01% PMSF, 3% aprotinin, and 0.02% sodium orthovanadate and centrifuged at 10,000 g for 10 minutes. The supernatant was removed and centrifuged again; the tissue was maintained at 4°C throughout. Protein concentrations were determined by the method of BCA (200 Protein Assay Kit) (PIERCE, Rockford, IL, U.S.A.), and gel samples prepared by adding sample buffer containing final concentrations of 50 mmol/L Tris (pH 6.7), 2% sodium dodecyl sulfate, 2% β-mercaptoethanol, and the marker bromphenol blue. Samples were boiled for 3 minutes, and 30 to 50 μg protein was loaded on 10% sodium dodecyl sulfate-polyacrylamide gels. Proteins were transferred to nitrocellulose membranes (Schleicher and Schuell, Keene, NH, U.S.A.) using standard electroblotting procedures. Nitrocellulose membranes were blocked with 10% nonfat dry milk. Blocking solutions were prepared in a buffer containing 100 mmol/L Tris-hydrochloride, 0.9% sodium chloride, and 0.05% Tween 20 (Tris-buffered saline-Tween (TBST), pH7.6).
Blots were incubated overnight at 4°C with antibodies against HSP70 (1:4000, 386032; Calbiochem, San Diego, CA, U.S.A.), hemeoxygenase-1 (1:1000, OSA-111; Stressgen, Victoria, BC, Canada), rodent HSP25 (1: 3500, SPA-801; Stressgen), HSP90 (1:5000, SPA-771; Stressgen), HSP60 (1:5000, SPA 807; Stressgen), HSP47 (1:500, SPA 470; Stressgen) and HSF1 (1:10000, Stressgen SPA-901), respectively. The rodent HSP25 is probably identical to the human HSP27 HSP and is referred to as HSP25/27 in this report. Immunoblots were washed in several changes of TBST at room temperature and then incubated with antimouse or antirabbit immunoglobulin linked to horseradish peroxidase (Santa Cruz Biotechnology, Santa Cruz, CA, U.S.A.). Immunoreactivity was detected with enhanced chemiluminescence (Amersham Pharmacia Biotech, Piscataway, NJ, U.S.A.) according to the manufacturer's recommended conditions. Blots were quantified using densitometric analyses with the MCID digital image analysis system (IMAGE Research, St. Catherines, Ontario, Canada). Western blots were performed in duplicate for two separate animals for each protein and each experimental condition.
Immunohistochemistry
Animals were anesthetized with ketamine (100 mg/kg) and xylazine (20 mg/kg) hydrochloride and perfused transcardially with 0.9% saline, followed by 4% paraformaldehyde in 0.1 mol/L phosphate buffer (pH 7.4). The brains were removed, postfixed for 6 hours in 4% paraformaldehyde phosphate buffer, and placed in 30% sucrose overnight. Fifty-micrometer coronal sections were cut on a sliding microtome and placed in 2% horse serum (or goat serum) and 0.2% Triton X-100/0.1% bovine serum albumin in phosphate buffer for 2 hours at room temperature, and then incubated for 2 hours at room temperature with mouse anti-HSP70 monoclonal antibody (1:1000, SPA-810; Stressgen), rabbit anti-HSP25 polyclonal antibody (1:1000, SPA-801; Stressgen), or rabbit antihemeoxygenase-1 polyclonal antibody (1:6000; gift from Ortiz De Montellano, University of California Davis) in 2% goat serum (or horse serum), 0.2% Triton X-100, and 0.1% bovine serum albumin in phosphate buffer. After three 10-minute phosphate buffer washes, the sections were incubated for 2 hours at room temperature with a biotinylated horse antimouse second antibody (1:200; Vector Labs) or goat antirabbit second antibody (1:400; Vector Labs), diluted in 2% goat serum (or horse serum), 0.2% Triton X-100, and 0.1% bovine serum albumin in phosphate buffer. After three 10-minute PB washes, the sections were incubated with ABC reagent (Vector Labs) for 2 hours. After three 10-minute phosphate buffer washes, the sections were incubated with a solution of 0.6 mg/mL diaminobenzidine and 0.05% hydrogen peroxide for 3 minutes. This incubation was terminated by three 10-minute phosphate buffer washes. Sections were mounted onto gelatin-coated slides and dried overnight before being covered with a cover slip. Representative sections from each animal were then photographed.
RESULTS
Hemeoxygenase-1 protein induction
The administration of 17-β-estradiol induced hemeoxygenase-1 at 6 hours. In female rats, 17-β-estradiol doses of 460 ng/kg (Fig. 1A, lane 7) and 4.6 μg/kg (Fig. 1A, lane 8) increased hemeoxygenase-1 protein 3.9-fold and 5.4-fold, respectively, compared with the 6-hour control (Fig. 1A, lane 2). There was no induction of hemeoxygenase-1 at 24 hours with any dose (Fig. 1A, lanes 3–5) compared with the 24-hour control (Fig. 1A, lane 1). In male rats, the highest 17-β-estradiol dose of 4.6 μg/kg increased hemeoxygenase-1 protein 3.7-fold at 6 hours (Fig. 1B, lane 8) compared with controls (Fig. 1B, lane 1). There was no induction of hemeoxygenase-1 at 6 hours with lower doses (Fig. 1B, lanes 6,7) and no induction of hemeoxygenase-1 at 24 hours after any dose (Fig. 1B, lanes 3–5). Six hours after global ischemia in gerbils, 17-β-estradiol (460 ng/kg administered 20 minutes before ischemia) increased hemeoxygenase-1 protein 2.4-fold (Fig. 1C, lane 2) compared with ischemia alone (Fig. 1C, lane 1). However, 24 hours after global ischemia, 17-β-estradiol (460 ng/kg) decreased hemeoxygenase-1 protein nearly fourfold (Fig. 1C, lane 4) compared with ischemia alone (Fig. 1C, lane 3).
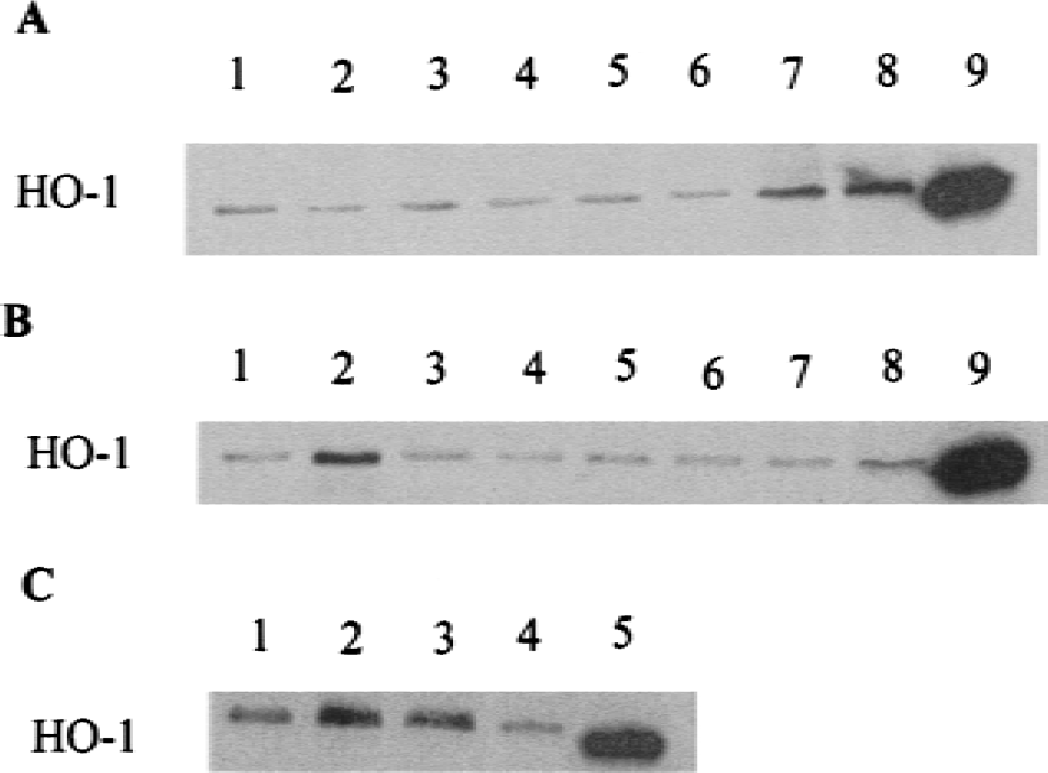
Western blots showing hemeoxygenase-1 protein from female rat brain
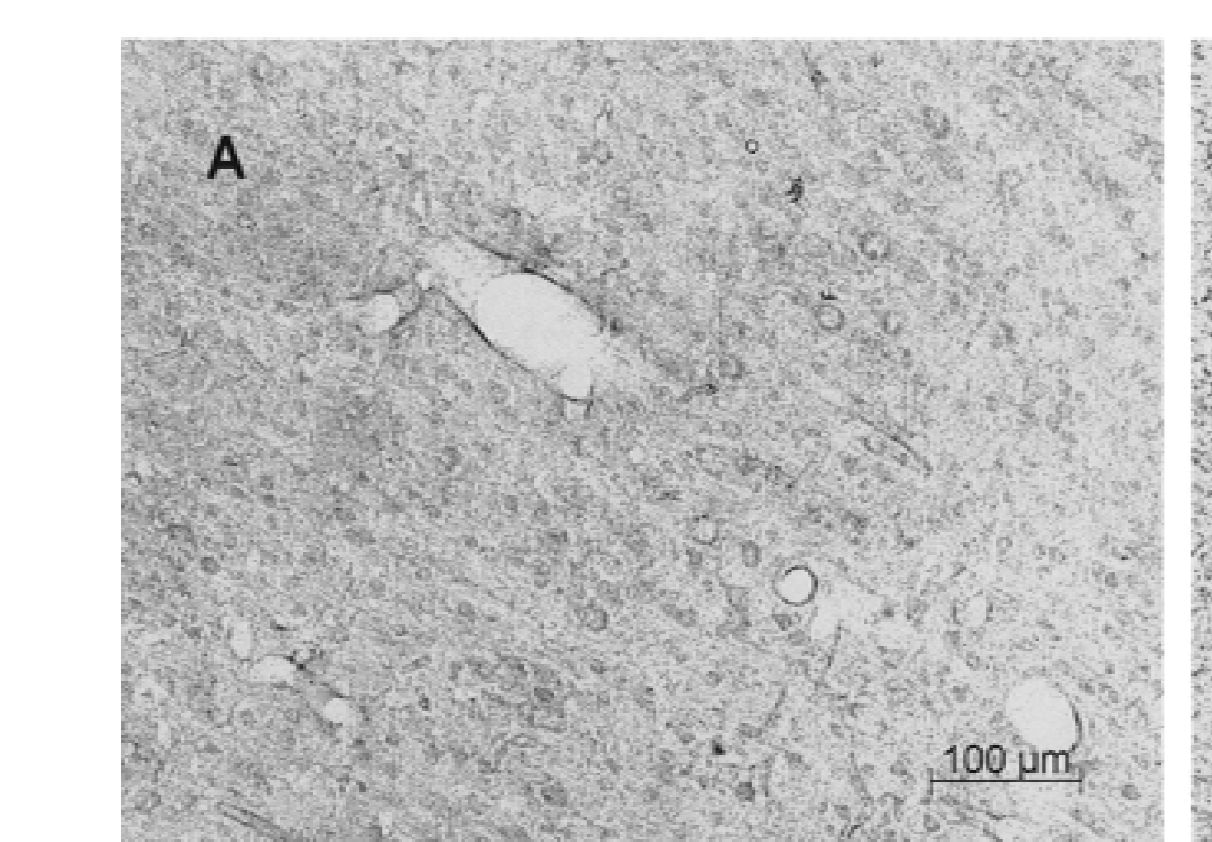
Immunohistochemical analysis of the normal adult rat brain showed that 17-β-estradiol (4.6 μg/kg) induced hemeoxygenase-1 in medium-sized vessels (most likely arteries) in the cerebral cortex (Fig. 2D), striatum (Fig. 2E), and hippocampus (Fig. 2F). There was no comparable staining of vessels in the control cortex (Fig. 2A), striatum (Fig. 2B), or hippocampus (Fig. 2C).
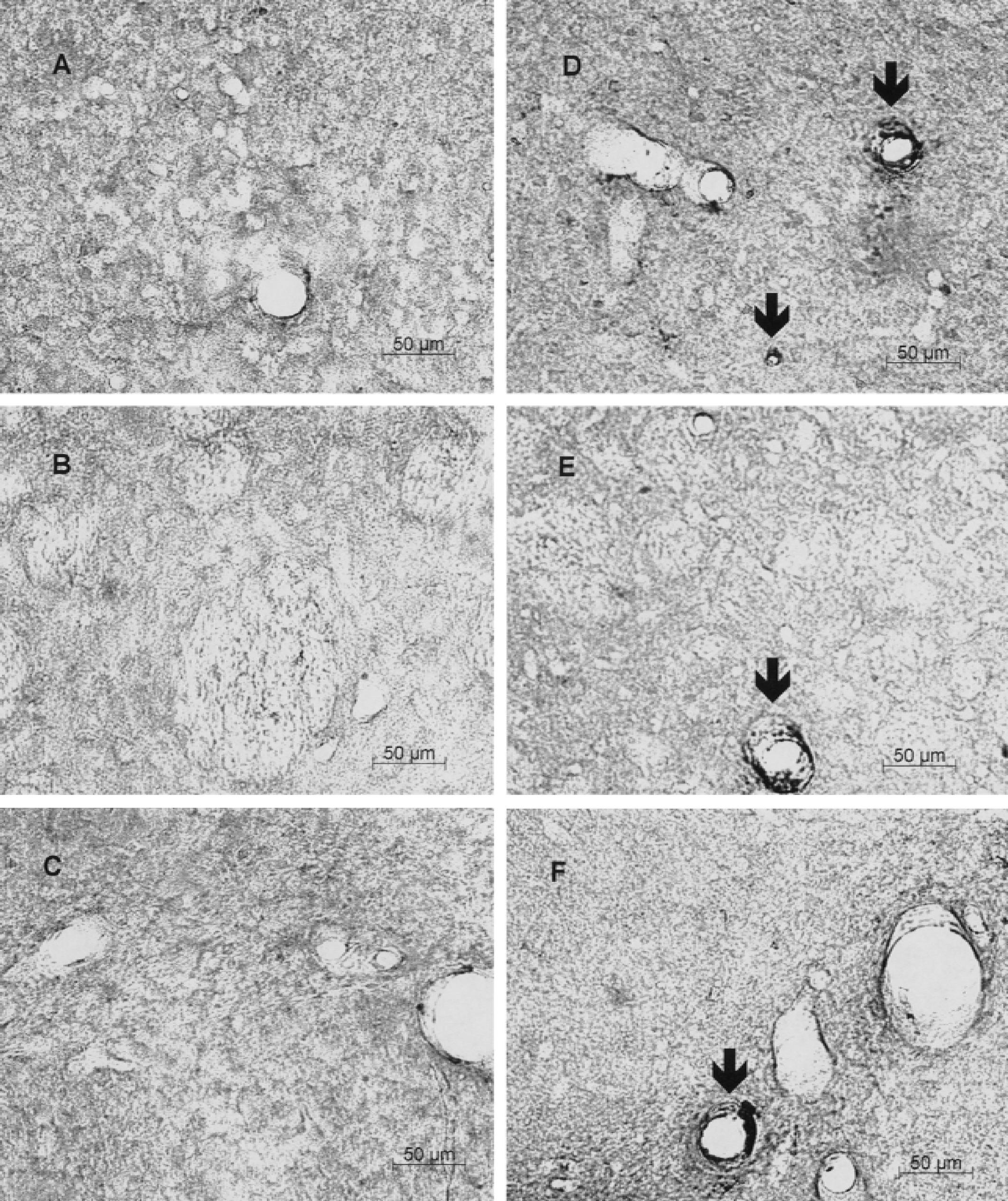
Hemeoxygenase-1 immunostaining in the cortex (
Six hours after brief global ischemia in gerbils, there was little induction in the cortex (not shown) or striatum (Fig. 3A) in these ischemic animals. After the administration of 17-β-estradiol (4.6 μg/kg) and 6 hours after global ischemia, hemeoxygenase-1 was induced in white matter tracts in the striatum (Fig. 3D) compared with ischemia alone (Figs. 3A). The staining in the white matter tracts appeared to be in cells with small cell bodies and fine processes, and probably represented oligodendrocytes (data not shown). 17-β-Estradiol (4.6 μg/kg) also induced hemeoxygenase-1 in CA1 (Fig. 3E), CA2 (Fig. 3F), and CA3 pyramidal neurons (data not shown), and to a lesser degree in dentate granule cell neurons of the hippocampus (data not shown) compared with vehicle-treated animals 6 hours after ischemia (Fig. 3B, C). The ischemia-induced staining in the lacunosom moleculare after ischemia alone was only slightly increased by estradiol (data not shown).
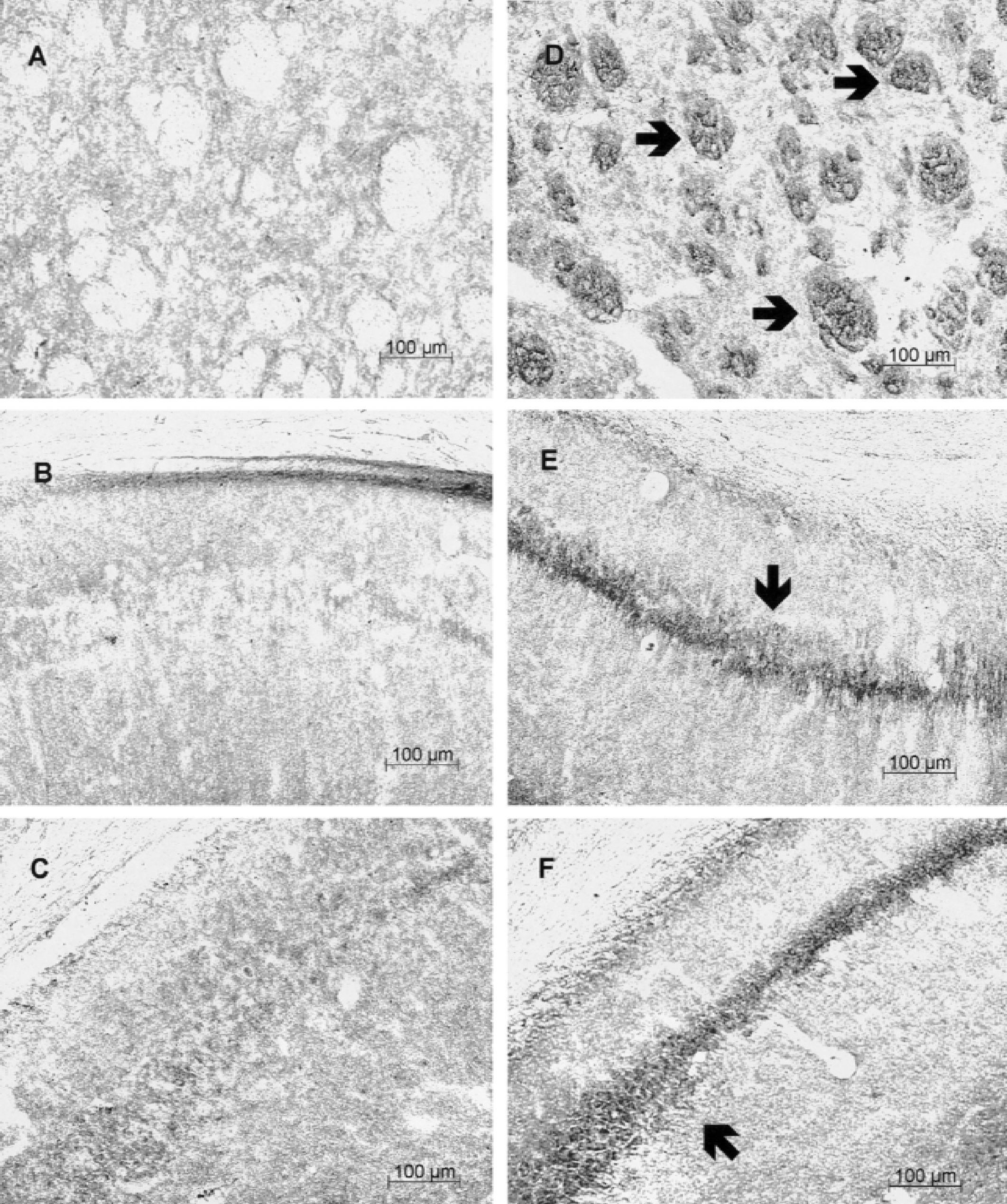
Hemeoxygenase-1 immunohistochemistry of ischemic gerbil brain
HSP25 induction
17-β-Estradiol also induced HSP25 in the rat brain (Figs. 4–6). Administration of 17-β-estradiol at doses of 460ng/kg (Fig. 4A, lane 7) and 4.6 μg/kg (Fig. 4A, lane 8) induced HSP25/27 approximately 1.5-fold and 2.1-fold, respectively, compared with the 6-hour control (Fig. 4A, lane 1). At 24 hours after the administration of 460 ng/kg 17-β-estradiol, HSP25/27 levels increased 2.1-fold (Fig. 4A, lane 4) compared with the 24-hour control (Fig. 4A, lane 2). In male rats, 17-β-estradiol only minimally increased HSP25/27 at the higher doses of 460ng/kg and 4.6 μg/kg at 24 hours (Fig. 4B, lanes 4,5) compared with 6 hours (Fig. 4B, lanes 7,8) and vehicle control (Fig. 4A, lane 1). Six hours after global ischemia in the gerbil, 17-β-estradiol (460 ng/kg) increased HSP25/27 levels 1.8-fold (Fig. 4C, lane 2) compared with ischemia alone (Fig. 4C, lane 1), whereas there was little difference between estrogen-treated (Fig. 4C, lane 4) and ischemia-alone (Fig. 4C, lane 3) groups 24 hours after ischemia.
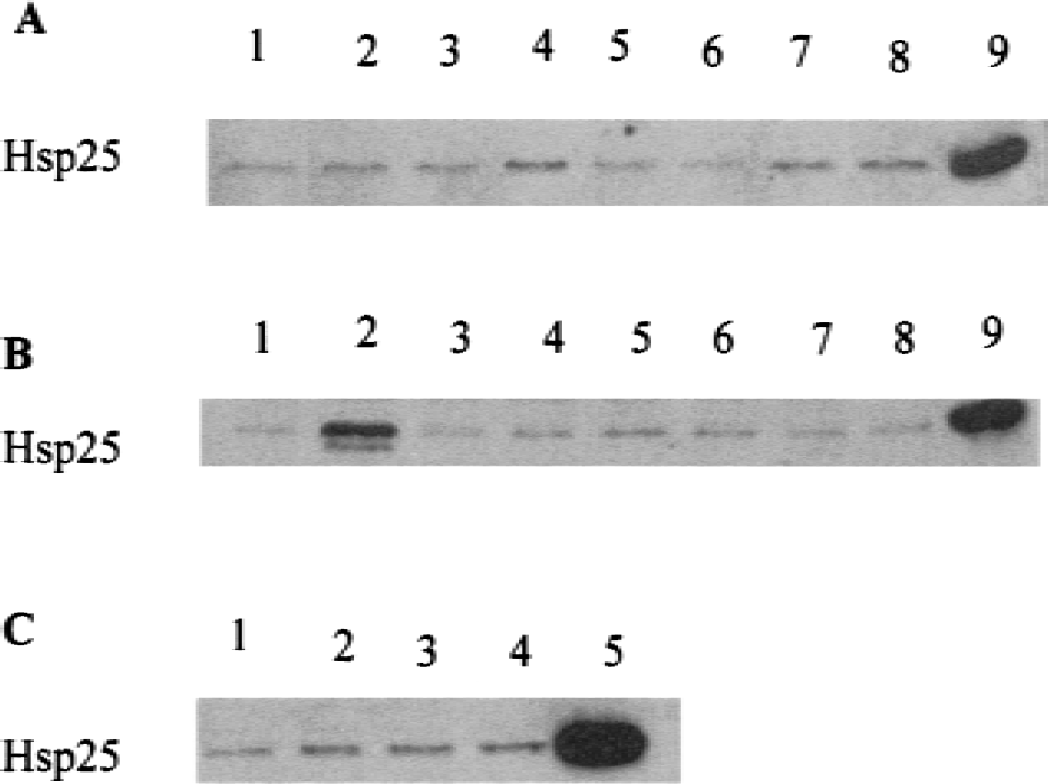
Western blots showing heat shock protein (HSP) 25/27 from the female
Immunocytochemical analysis showed similar, scattered HSP25/27 glial staining in brains of animals that received saline (Figs. 5A to 5C) and 17-β-estradiol (Figs. 5D to 5F). However, 6 hours after administration of 4.6 μg/kg 17-β-estradiol, there was marked upregulation of HSP25/27 in cerebral blood vessels (Figs. 5D to 5F) compared with saline-injected rats (Figs. 5A to 5C). Compared with vehicle-injected animals that had slight vascular staining (Figs. 5A to 5C), staining was localized to medium-sized vessels (probably arteries) in the cortex (Fig. 5D), striatum (Fig. 5E), hippocampus (Fig. 5F), and throughout other brain regions (data not shown). Six hours after global ischemia in gerbils, the HSP25/27 protein was induced in small arteries in many brain regions, including the striatum (Fig. 6A). There was also some HSP25/27 staining of neurons in hippocampal CA regions (Fig. 6B) and dentate gyrus (Fig. 6C). Six hours after global ischemia in gerbils pretreated with 17-β-estradiol (4.6 μg/kg), HSP25/27 was markedly induced in striatum astrocytes (arrow heads, Fig. 6D). HSP25/27 staining also increased, to some degree, in CA1 pyramidal neurons (Fig. 6E) and dentate gyrus granule cell neurons and hilar neurons (Fig. 6F) compared with ischemia alone (Figs. 6B and 6C).
Heat shock protein (HSP) 25/27 immunostaining in the cortex (
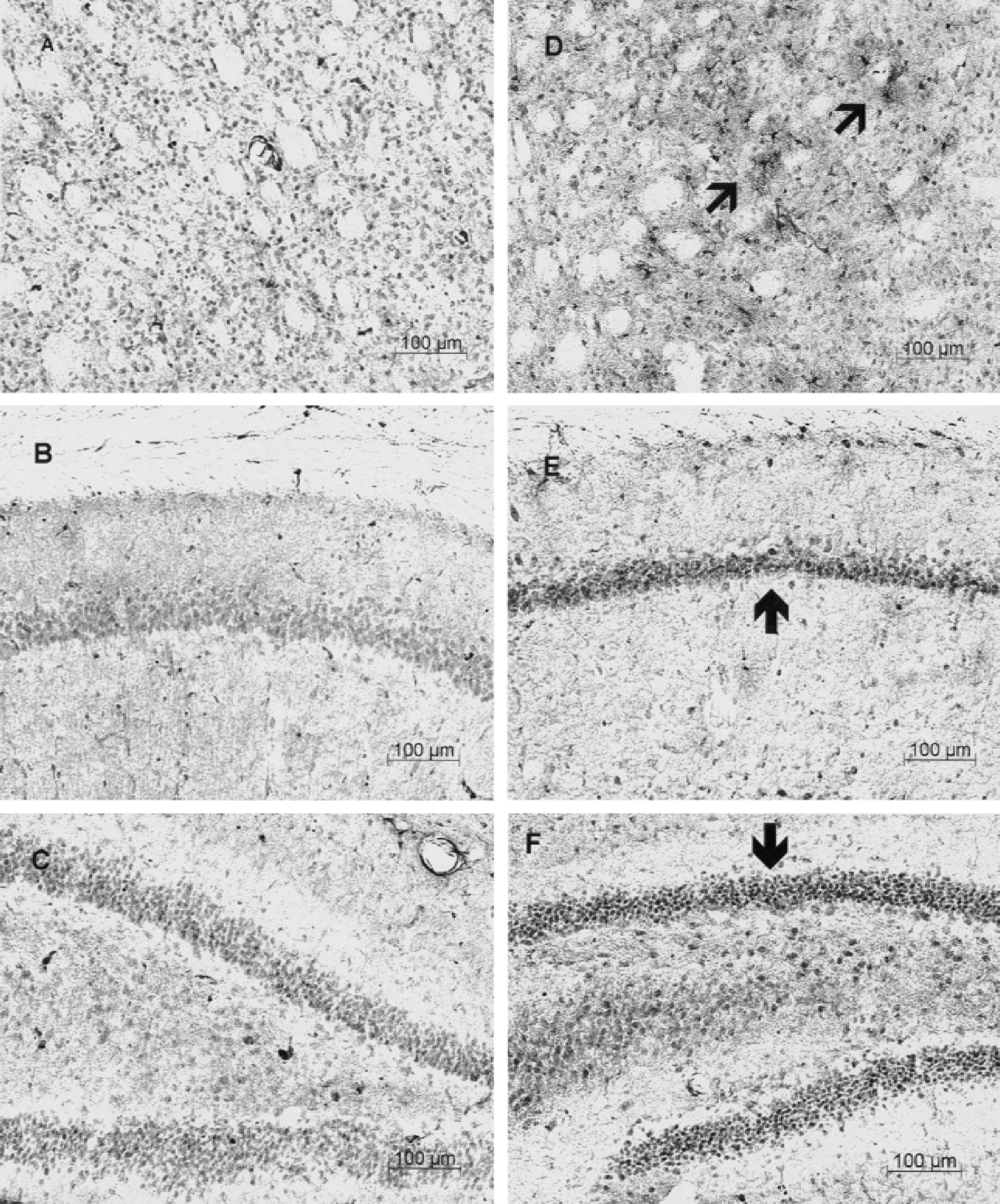
Heat shock protein (HSP) 25/27 immunohistochemistry of the ischemic gerbil brain
Heat shock protein 70 induction
The antibody used to detect HSP70 on Western blots stained two bands (Fig. 7A and 7B, lane 7). The lower band represents the constitutive Hsc70 protein that showed little change 6 (Figs. 7A and 7B, lanes 6–8) or 24 hours (Figs. 7A,B, lanes 3–5) after administration of varying doses of estrogen in female (Fig. 7A) or male rats (Fig. 7B). The upper band represents the inducible HSP70 (Figs. 7A and 7B, lane 9). Six hours after the administration of 17-β-estradiol, the 460-ng/kg dose induced HSP70 levels 2.3-fold in female rats (Fig. 7A, lane 7) compared with controls (Fig. 7A, lane 1); and the 460-ng/kg (Fig. 7B, lane 7) and 4.6-μg/kg (Fig. 7B, lane 8) doses of estradiol induced HSP70 levels 3.2-fold and twofold, respectively, in male rats compared with controls (Fig. 7B, lane 1). The levels of HSP70 decreased to control levels by 24 hours after estradiol in female (Fig. 7A, lanes 3–5) and male rats (Fig. 7B, lanes 3–5).
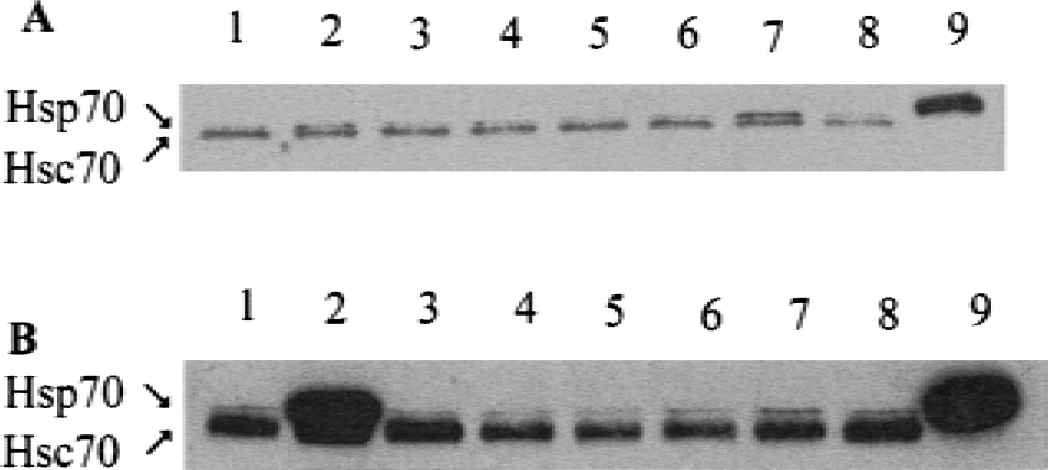
Western blots showing heat shock protein (HSP) 70 (upper band) and Hsc70 protein (lower band) from the female
Immunocytochemical analysis revealed that 17-β-estradiol (4.6 μg/kg) induced HSP70 mainly in small arteries in the cortex (Fig. 8D), striatum (Fig. 8E), hippocampus (Fig. 8F) compared with no vascular staining in vehicle-injected controls (Figs. 8A–C). The immunocytochemistry is thought to show HSP70 induction because only HSP70 and not Hsc70 was induced on the Western blots.
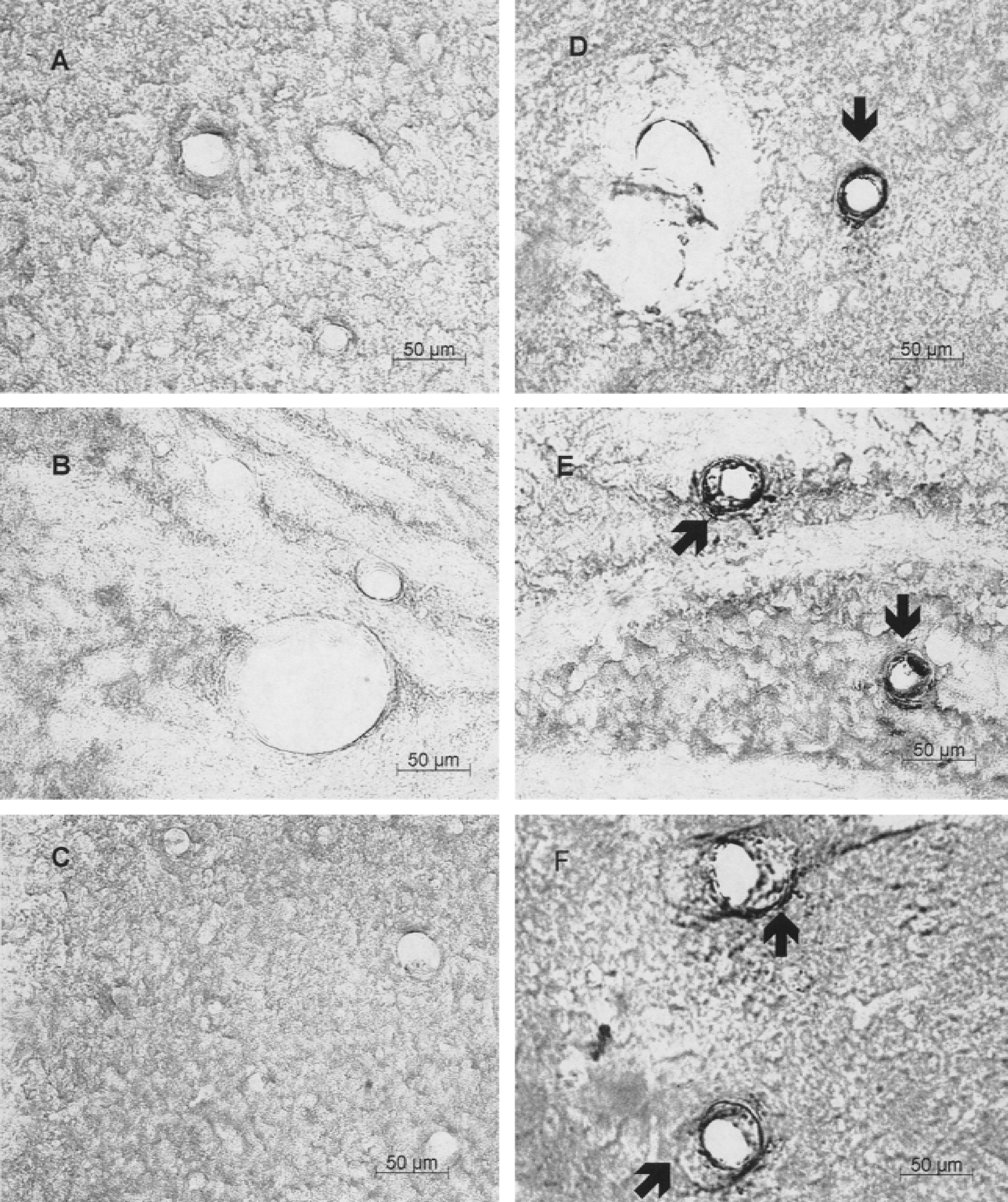
HSP70 immunostaining in cortex (
Heat shock protein 90, 47, and 60
Administration of 46 ng/kg, 460 ng/kg, and 4.6 μg/kg 17-β-estradiol did not affect HSP90 (Fig. 9A, lanes 6–8), HSP47 (Fig. 9B, lanes 6–8), or HSP60 (Fig. 9C, lanes 6–8) 6 hours after administration compared with 6-hour controls (Figs. 9A to 9C, lane 1). Similarly, 17-β estradiol did not affect HSP90 (Fig. 9A, lanes 3–5), HSP47 (Fig. 9B, lanes 3–5), or HSP60 (Fig. 9C, lanes 3–5) 24 hours after administration compared with 24-hour controls (Figs. 9A to 9C, lane 2).
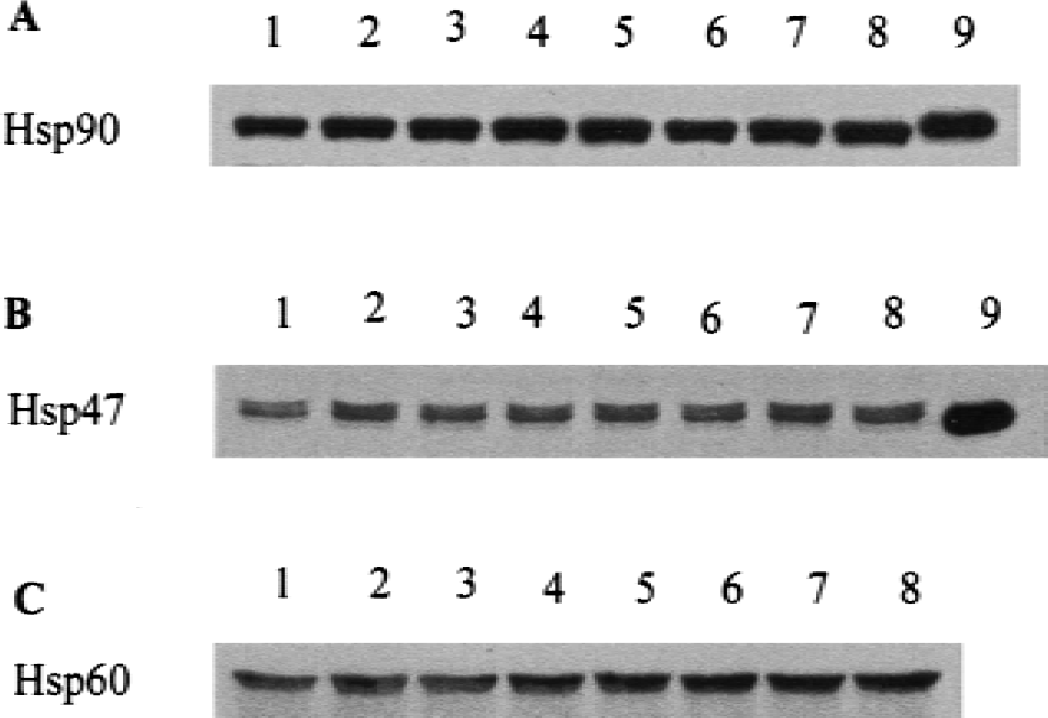
Western blots showing HSP90 protein
It should be emphasized that the changes of hemeoxygenase-1, HSP25/27, and HSP70 on Western blots of brains of estrogen-treated rats were generally modest. This finding appears to be due to dilution of the HSP induction, mainly in vessels with the HSPs in other cellular compartments that are not changing. During the interpretation of these HSP90, HSP47, and HSP60 data, it should be considered that changes in an underrepresented cell type will not be detected on the Western blots.
Heat shock factor-1 protein induction
The last study explored one possible mechanism by which estrogen might induce HSPs in the brain. We postulated that if HSF had an estrogen response element in its promoter, then estrogen could induce HSF. Administration of 17-β-estradiol (460 ng/kg) to nonischemic male rats increased levels of HSF1 protein 1.6-fold (Fig. 10A, lane 3) compared with controls (Fig. 10A, lane 1) at 6 hours, and at 24 hours increased HSF1 protein levels 1.5-fold (Fig. 10A, lane 4) compared with controls (Fig. 10A, lane 2). Six hours after global ischemia, 17-β-estradiol (460 ng/kg) increased HSF1 protein levels 1.6-fold (Fig. 10B, lane 2) compared with ischemia alone (Fig. 10B, lane 1). At 24 hours after global ischemia, 17-β-estradiol increased levels of HSF1 protein 2.1-fold (Fig. 10B, lane 4) compared with ischemia alone (Fig. 10B, lane 3).
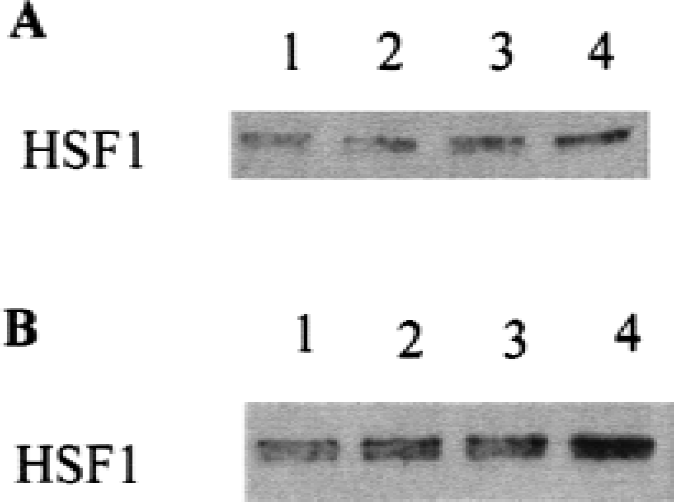
Heat-shock Factor1 (HSF1) Western blots of female rat brain
DISCUSSION
The results of this study show that estradiol induces hemeoxygenase-1, HSP25/27, and HSP70 in the brain 6 to 24 hours after acute administration. Estradiol induction of these HSPs occurs mostly in arteries in the normal brain and in arteries, glia, and neurons in the ischemic brain. Estrogen induction of heat-shock factors provides at least one mechanism by which estrogen induces HSPs in the brain and in other tissues. Finally, it is proposed that acute neuroprotection afforded by estrogen may be due at least in part to the induction or facilitation of the HSP response.
The present study provides the first evidence that 17-β-estradiol induces hemeoxygenase-1 protein in the normal and ischemic brain. Hemeoxygenase-1 was induced in blood vessels of nonischemic rats and in oligodendrocytes and hippocampal neurons of ischemic rats. The hemeoxygenase-1 gene is induced by various stimuli. Oxidative stress activates mitogen-activated kinases, mitogen-activated protein kinases that induces Nrf2, which induces hemeoxygenase-1 through an AP1 site (Elbirt and Bonkovsky, 1999; Immenschuh and Ramadori, 2000). Because estrogen activates mitogen-activated protein kinases (Green and Simpkins, 2000), it may induce hemeoxygenase-1 in part through this pathway. Nitric oxide is a potent hemeoxygenase-1 inducer (Bouton and Demple, 2000; Chen and Maines, 2000). Estradiol induces the association between HSP90 and endothelial nitric oxide synthase, the activation of endothelial nitric oxide synthase, and the release of nitric oxide (Russell et al., 2000). The estrogen receptor is localized to the caveola, where it directly interacts with the p85 subunit of PI3-kinase to activate the PI3-kinase-Akt endothelial nitric oxide synthase pathway (Mendelsohn, 2000). Cyclic adenosine monophosphate and cyclic guanosine monophosphate can activate protein kinase A and protein kinase G and increase hemeoxygenase-1 transcription (Immenschuh and Ramadori, 2000). Estrogen increases cyclic adenosine monophosphate accumulation and phosphorylation of cyclic adenosine monophosphate response element binding protein (Green and Simpkins, 2000). Therefore, estrogen likely induces hemeoxygenase-1 through multiple pathways, though the nitric oxide synthase and cyclic guanosine monophosphate pathways may be particularly important because estrogen induction of hemeoxygenase-1 appears to occur mainly in smooth muscle cells around cerebral arteries and arterioles.
Hemeoxygenase-1 is cytoprotective in various cell culture and animal models. Hemeoxygenase-1 gene induction is postulated to be an adaptive cellular defense mechanism against increases in intracellular heme (Lee et al., 1996; Yang et al., 1999). Hemeoxygenase-1 expression in vascular endothelial cells protects against oxidative stress-induced apoptosis (Foresti et al., 1999). Hemeoxygenase-1 and the products of heme catabolism attenuate the arterial response to injury and ensure vascular wall remodeling (Tulis et al., 2001). Neurons that over express hemeoxygenase-1 are resistant to glutamate and hydrogen peroxide-mediated cell death (Chen et al., 2000). Hemeoxygenase-1 overexpression in transgenic mice protects the brain from permanent focal ischemia (Panahian et al., 1999). The induction of hemeoxygenase protein protects neurons in cortex and striatum against transient forebrain ischemia (Takizawa et al., 1998). In this study, estrogen induction of hemeoxygenase-1 in arteries, glia, and neurons likely protects these cells from ischemic injury and provides a new molecular mechanism by which estrogen may protect the brain and heart against ischemia.
Estradiol also increased the levels of HSP25/27 in blood vessels of normal brain and in glia and neurons of ischemic brain. This finding is consistent with those of previous studies that 17-β-estradiol treatment of MCF7 human breast cancer cells increases HSP27 messenger RNA levels twofold for up to 24 hours (Porter et al., 2001). Estrogen increased HSP27 levels in the human endometrial glandular epithelium (Padwick et al., 1994), and estradiol treatment of endothelial cells resulted in an increase in both bovine and human HSP27 (Piotrowicz et al., 1995). These data strongly support our results.
The mechanisms of estrogen induction of HSP25/27 are still being studied. Because the intron-exon structure of the mouse HSP25 gene is extremely similar to the human HSP27 gene, it is likely they represent the same gene in these two species and hence are referred to as the HSP25/27 gene herein. The promoter region of HSP25/27 contains putative transcription factor-binding elements, including two guanine–cytosine-rich Sp1-binding domains, two heat-shock elements, and an estrogen-responsive element half-site in direct proximity to the TATA box (Gaestel et al., 1993). The Sp1 and half-palindromic estrogen response element and formation of the Sp1/estrogen receptor complex are involved in estrogen-induced HSP27 gene expression (Porter et al., 1996). However, mutation of the estrogen response element halfsite does not result in loss of estrogen responsiveness, suggesting that estrogen inducibility is mediated through the Sp1-DNA motif. Moreover, Sp1 and estrogen receptor proteins physically interact to enhance Sp1-DNA binding that is estrogen dependent (Porter et al., 1997).
HSP27 reduces apoptotic and necrotic cell death (Mehlen et al., 1996; Samali and Cotter, 1996; Wagstaff et al., 1999). Estradiol rapidly activates p38β mitogen-activated protein kinase in endothelial cells, which activates the mitogen-activated protein kinase-activated protein kinase-2 and the phosphorylation of HSP27. Estradiol preserves the endothelial cells stress fiber formation and actin and membrane integrity in simulated ischemia, prevents hypoxia-induced apoptosis, and induces the migration of endothelial cells and the formation of primitive capillary tubes. These effects are reversed by the inhibition of p38β, by the expression of a dominant-negative mitogen-activated protein kinase-activated protein kinase-2 protein, or by the expression of a phosphorylation site mutant HSP27 (Razandi et al., 2000). Estrogen treatment increases SMER14 cell viability in the presence of staurosporine. Estrogen decreases annexin V staining and DNA fragmentation and increases HSP27 levels in SMER14 cells (Cooper et al., 2000). HSP27 expression in vascular smooth muscle cells induced by mild heat shock prevents inhibition of proliferation and induction of necrosis by heat shock (Champagne et al., 1999). HSP27 expression in sensory neurons promotes survival after axotomy or neurotrophin withdrawal (Lewis et al., 1999). Mild cerebral ischemia induces HSP27 that correlates with the onset of ischemic tolerance (Kato et al., 1994). The finding that estrogen induces HSP27 in blood vessels and glia and neurons in the brain may indicate an important mechanism for estrogen protection of cerebral ischemia.
Estrogen has been reported to induce HSP70 in a variety of tissues. Estradiol increased HSP70 messenger RNA in the myometrium and endometrium of nonpregnant ewes (Wu et al., 1996). Estradiol stimulated HSP70 expression in short-term cultures of steroid-responsive (T47-D) cells (Tang et al., 1995). Estrogen activated HSF1 and increased HSP72 in isolated cardiac myocytes (Knowlton and Sun, 2001). There have been few suggestions that estrogen could induce the inducible HSP70 protein in the brain. Estrogen and progesterone induce the constitutive HSP70 (Hsc73) in the pituitary and subregions of the ventromedial hypothalamus, as shown by Northern analysis and in situ hybridization (Krebs et al., 1999). Estrogen induced HSP72, HSP27, and HSP90, and subsequent granulation in astrocytes in estradiol receptor-rich brain regions including the arcuate nucleus and the walls of the third ventricle (Mydlarski et al., 1995). This is the first study to show that estrogen induces HSP70 in multiple brain regions and that the induction appears to occur mainly in blood vessels that are likely arteries and arterioles.
Estrogen induction of HSP70 most likely protects the brain. Overexpression of HSP70 in cultured cells protects against a variety of different injuries in vitro (Sharp et al., 1999; Yenari et al., 1999). Viral overexpression of HSP70 protects against ischemia and excitoxicity in vivo (Yenari et al., 1999). Overexpression of HSP70 in transgenic mice protects the mice against permanent middle cerebral artery occlusions (Rajdev et al., 2000). Estrogen induction of HSP70 in vessels might help to preserve blood flow or to decrease reperfusion-induced hemorrhages after spontaneous or tissue plasminogen activator-induced clot lysis.
In the rat, physiologic plasma 17-β-estradiol levels range between 10 and 30 pg/ml. In healthy Wistar male rats, plasma 17-β-estradiol levels reached 10 pg/ml after a 25-μg estradiol pellet was implanted under the skin for 7 to 10 days (Hurn and Macrae, 2000; Toung et al, 1998). Therefore, the 17-β-estradiol doses of 460 ng/kg (115 ng/rat) and 4.6 μg/kg used in this study to induce HSPs should be in the physiologic range. Estradiol in physiologic doses protects rats against cerebral ischemia. Importantly, the preservation of intraischemic cortical perfusion by estradiol was achieved by physiologic doses of estrogen and was lost at supraphysiologic doses (Hurn and Macrae, 2000). Induction of HSPs with low-dose estradiol may contribute to the neuroprotection produced by estrogen.
The mechanism by which estrogen can upregulate HSP32 (hemeoxygenase-1), HSP25/27, and HSP70 in cerebral arteries, glia, and neurons is of significant interest. Estrogen regulates HSF-1 and −2 at the messenger-RNA and protein levels in the endometrium (Yang et al., 1995). Estrogen and progesterone activate HSF-1 in adult male isolated cardiac myocytes, and this effect is followed by an increase in HSP72 protein (Knowlton and Sun, 2001). Transcriptional induction of HSP genes is generally mediated by binding of heat-shock transcription factor to the heat-shock element present in the promoters of heat-shock genes. In our study, estradiol increased HSF1 protein relative to controls at 6 and 24 hours. Moreover, estradiol induced hemeoxygenase-1 and HSP70 by 6 hours. Therefore, estrogen induction and activation of HSF1 expression could be one of the mechanisms that mediate estrogen induction of hemeoxygenase-1, HSP25/27, and HSP70 in vessels, glia, and neurons in the brain. Finally, pharmacologic doses of estrogen would bind HSP90, which would free HSFs from HSP90 (Zou et al., 1998). This HSP90 binding by estrogen could activate HSFs and HSP transcription just as the drug geldanamycin appears to bind HSP90 during periods of stress, free HSFs, and activate transcription of HSPs (Zou et al., 1998).
The estrogen induction of hemeoxygenase-1, HSP25/27, and HSP70 in blood vessels was one of the major surprise findings of this study. The HSP induction occurred mainly in medium-sized arteries to small arterioles, which would suggest that estrogen might induce HSPs in the smooth muscle cells or endothelial cells around arteries. This observation also suggests that estrogen likely induces HSPs in arteries throughout the body including coronary, pulmonary, and renal arteries. The estrogen receptors α and β are found in various regions of the rat brain (Pelletier, 2000; Commins and Yahr, 1985). In approximately 70% of the medial smooth muscle cells of the female rat aorta, tail artery, and uterine artery, nuclear immunoreactivity to estrogen receptor β was observed. In these vessels, endothelial cells also expressed estrogen receptor β. In the aorta and tail arteries, immunoreactivity for estrogen receptor α was observed in medial smooth muscle and endothelial cells of uterine vessels (Andersson et al., 2001). Why estrogen might target smooth muscle cells or endothelial cells around arteries is not clear. There could be an HSF unique to smooth muscle cells, a particular class of estrogen receptor in smooth muscle cells or endothelial cells that might signal heat-shock activation in smooth muscle cells.
In summary, 17-β-estradiol induces HSF1, hemeoxygenase-1 (HSP32), HSP25/27, and HSP70 in the normal and ischemic brain. Estrogen has an important influence on the expression of HSPs in normal blood vessels and under conditions of stress in most other cells. This effect may be an important molecular mechanism of estrogen protection in the brain and in other organs, including the heart.