
Abstract
Recently, the authors isolated a novel gene of the HSP110 family, ischemia responsive protein 94 kDa (irp94), and demonstrated the expression of this gene after transient forebrain ischemia. In the current study, the authors investigated the expression profiles of all HSP110 family members including hsp110/105 and osp94/apg-1, after transient forebrain ischemia using rat four-vessel occlusion model. Among three members of the HSP110 family, induction of hsp110/105 was the most prominent after ischemia. hsp110/105 mRNA expression was clearly enhanced from 4 to 24 hours after a 6-minute or longer ischemic period. First, hsp110/105 mRNA expression was induced in the dentate gyrus, and later in the pyramidal layer. HSP110/105 protein expression also was enhanced by a 6-minute or longer period of ischemia. Profiles of HSP110/105 expression after ischemia were similar to those of inducible HSP70. After transient forebrain ischemia for 10 minutes, HSP110/105 protein was induced in the dentate gyrus and the CA3 pyramidal layer, but not in the CA1 pyramidal neurons. However, 6 minutes of ischemia induced the HSP110/105 protein, as well as the HSP70 protein, in the CA1 region. CA1 pyramidal neurons expressing HSP110/105 acquired tolerance against subsequent severe ischemia. In conclusion, HSP110/105 showed the most prominent induction after ischemia among the three HSP110 gene family members. Colocalization of HSP110/105 and HSP70 in the CA1 neurons that acquired tolerance suggested that induced HSP110/105 might contribute to ischemic tolerance together with HSP70.
It has been reported that induction of several genes, including heat shock proteins (HSPs), is observed in the hippocampus, cerebral cortex, and other brain regions after transient forebrain ischemia (Nowak 1985, 1991; Kawagoe et al., 1993; Nishi et al., 1993; Kato et al., 1994; Lowenstein et al., 1994; Takeda et al., 1994; Xue et al., 1998; Yagita et al., 1999). HSP70 have been well studied regarding neuronal response against ischemic injury. HSP70 family proteins act as molecular chaperones under normal and stress conditions. Those proteins were induced by ischemia and are believed to support neuronal survival through refolding of the denatured protein (Sharp et al., 2000). It was reported that gene transfer of HSP72 into the brain (Yenari et al., 1998) and hsp70 overexpression in transgenic mice (Rajdev et al., 2000) causes resistance against ischemic brain injury.
Recently, the authors isolated an ischemia responsive protein of 94 kDa (irp94), a novel member of the HSP110 family, and demonstrated the expression of this gene after transient forebrain ischemia (Yagita et al., 1999). The HSP110 family is known to diverge from the HSP70 family, consisting of three members in mammals—HSP110/105, osmotic stress protein 94 (OSP94)/APG-1, and IRP94/APG-2 (Lee-Yoon et al., 1995; Yasuda et al., 1995; Kojima et al., 1996; Kaneko et al., 1997a, b ; Yagita et al., 1999). There are two members of HSP110/105 proteins—HSP105α and HSP105β (Yasuda et al., 1995). HSP105α is a constitutively expressed 105-kDa stress protein, whereas HSP105β is a 90-kDa stress protein specifically induced by heat shock at 42°C. HSP105β is an alternative spliced form of HSP105α lacking 44 amino acids in the region between the positions 530 and 573. HSP110/105 proteins were suggested to be associated with HSC70, a constitutive type of the HSP70 gene family (Hatayama et al., 1998, Yamagishi et al., 2000; Wang et al., 2000). These facts suggest that the HSP110 family correlates with the HSP70 family and also plays an important role for neuronal survival in the ischemic brain. However, the expression profile of the HSP110 family after cerebral ischemia has not been fully elucidated; furthermore, the involvement of HSP110 in the neuronal stress response remains unclear.
In the current study, gene induction of three HSP110 family members after ischemia was compared. Afterward, the authors investigated HSP110/105 expression after transient forebrain ischemia, because its gene induction after ischemia was the most predominant among the three members. Furthermore, the expression profile of HSP110/105, as well as HSP70, after acquisition of ischemic tolerance was investigated.
MATERIALS AND METHODS
Animal model
Adult, male Wistar rats weighing 350 to 500 g were used in the current study. The experimental protocol was approved by the Institutional Animal Care and Use Committee of Osaka University Graduate School of Medicine. Transient forebrain ischemia was induced by four-vessel occlusion and reperfusion as previously described (Pulsinelli et al., 1982; Yagita et al., 1999). Briefly, both vertebral arteries were cauterized through each alar foramen using an electrocautery needle under pentobarbital (45 mg/kg, intraperitoneally) anesthesia. The next day, rats were anesthetized with 3% halothane inhalation, and both common carotid arteries were occluded with miniature aneurysmal clips for various durations from 3 to 15 minutes. Among 238 rats that underwent surgery, 150 rats that showed complete and lasting loss of the righting reflex, pain reflex, and corneal reflex during 4-vessel occlusion were chosen for the ischemic rats. Ten ischemic rats among them died during the recirculation period. Rectal temperature was monitored continuously during ischemia and was maintained at 37°C to 37.5 °C with an overhead heating lamp for up to 1 hour after removal of the clips. Skull temperature also was monitored with a needle thermometer probe placed on the parietal area of the skull and maintained at 36.0°C during ischemia. Sham-operated animals went through the first day's procedure and were treated similarly to those subjected to ischemia, except for occlusion of the carotid arteries.
Histologic analysis
Fifty-six rats were used for histologic analysis of the hippocampus. Eight rats were recirculated after 3, 6, 10, and 15 minutes of ischemia. Eight sham-operated animals were used as controls. For induction of tolerance, 8 rats were subjected to 6 minutes of ischemia or sham-operation, and 2 days later they were subjected to 15 minutes of ischemia. Seven days after the last ischemia, animals were perfusion-fixed with 4% paraformaldehyde (PFA) under deep pentobarbital anesthesia. Brains were removed, dehydrated, and embedded in paraffin. Then, 5-μm-thick cross-sections at a coronal level 3.6 mm caudal to the bregma were stained with hematoxylin and eosin. The number of undamaged neurons from 3 defined fields, 400 μm in length, of the CA1 subsector of the dorsal hippocampus, the medial, the middle, and the lateral CA1 was calculated in both hemispheres as described previously (Shamloo and Wieloch, 1999). Results of the undamaged neurons are presented as a percentage of the sham-operated group value.
cDNA probe
To obtain a rat-specific cDNA probe, the authors screened a rat hippocampal cDNA library (Stratagene, La Jolla, CA, U.S.A.) using polymerase chain reaction (PCR) products of hsp110/105 and osp94/apg-1 as probes, because cDNA sequences encoding hsp110/105 and osp94/apg-1 in the rat have not been published. mRNA was isolated from the C57BL/6 mouse hippocampus using an oligo-dT cellulose column with a mRNA purification kit (Pharmacia, San Diego, U.S.A.). Purified mRNA then was reverse-transcribed with SuperScriptII (GIBCO BRL, Gathersburg, U.S.A.) in the presence of a random hexamer for 60 minutes at 37°C. Obtained first-strand cDNA were subjected to 35 cycles of PCR with a setting of 94°C for 30 seconds, 62°C for 1 minute, 72°C for 1 minute, and 72°C for 5 minutes. Polymerase chain reaction primers were as follows: for mouse hsp105 (GenBank accession no. D67016), forward 5′-CAAGATTGCAGCGGACTTCA-3′ (corresponding to nucleotides 2238 to 2257) and reverse 5′-ACAGAGCCCTTCTCATTAGG-3′ (corresponding to nucleotides 2593 to 2612); and for mouse apg-1 (GenBank accession no. D49482): forward 5′-AACGATGCCGCTATGGAGAC-3′ (corresponding to nucleotides 1581 to 1600) and reverse 5′-CTGTAGAGGCTACTCTGGAT-3′ (corresponding to nucleotides 1809 to 1828). Amplified PCR products were subcloned into a pGEM-T vector (Promega, Madison, U.S.A.), and used as a probe for cDNA library screening. Sequencing of cloned fragments was performed with a Taq dye terminator cycle sequencing core kit (Applied Biosystems, Tokyo, Japan) and an ABI PRISM 310 Genetic Analyzer (Applied Biosystems). The cDNA library of the rat hippocampus constructed with λZAPII (Stratagene) was screened with the 32P-labeled cDNA probes. Positive clones were excised with a helper phage and recircularized to generate subclones in a pBluescriptIISK(-) phagemid vector (Stratagene). Obtained cDNA were used for Northern blotting as probes.
Northern blotting
Total RNA was isolated from the hippocampus of each rat (n = 4 for each group) and several organs using ISOGEN (Nippon Gene, Tokyo, Japan), according to the manufacturer's instructions. Ten microgram RNA samples from each group were electrophoresed on 1.0% formaldehyde-agarose gels and transferred onto GeneScreen membranes (DuPont NEN, Boston, MA, U.S.A.). Membranes were hybridized with the 32P-labeled cDNA probe. After washing at a final stringency of 0.1 × saline-sodium citrate buffer (SSC) at room temperature, membranes were autoradiographed. β-actin cDNA was used as an internal standard probe. Densitometric values for hsp110/105 mRNA were normalized to values for β-actin mRNA on the same lane.
Reverse transcription-polymerase chain reaction analysis
To examine whether hsp105β, alternative spliced isoform of hsp105α, is expressed after ischemia, the authors performed RT-PCR analysis using primers by which the deletion of 132 nucleotides would be detected. Forward and reverse PCR primers were 5′-GCACAGAAAGATGGAGAGAA-3′ (corresponding to nucleotides 1459–1478) and 5′-ACCAAGTTGGCTTCTACAGG-3′ (corresponding to nucletotides 1846–1865), respectively. First strand hippocampal cDNA from control and ischemic rats (24 hours after transient ischemia) were subjected to 30 cycles of PCR with a setting of 94°C for 30 seconds, 60°C for 1 minute, 72°C for 1 minute. The products were electrophoresed on a 2% agarose gel. Molecular size markers (100-base pair DNA Ladder; Life Technologies) were used for calibration of the sizes of the PCR product.
In situ hybridization
For in situ hybridization, 4 nonoperated control rats, 20 ischemic rats at 1, 4, 24, and 48 hours, and 4 days after transient forebrain ischemia for 10 minutes (4 rats per each postischemic period), and 10 sham-operated rats (2 rats per each postischemic period) were studied. Almost all procedures were performed as described previously (Yagita et al., 1999). Amplified rat cDNA using same primer set of mouse HSP105 was subcloned into a pGEM-T vector (Promega) for the in situ hybridization probe. The plasmid was linearized with NcoI and HincII, and then in vitro transcription and digoxigenin-labeling were performed to prepare sense and antisense RNA probes using a digoxigenin RNA labeling kit (Beohringer Mannheim, Mannheim, Germany). Frozen coronal sections including the hippocampus were treated with 1.0 mg/mL proteinase K at 37°C for 5 minutes, treated with 0.2 mol/L HCl for 20 minutes to quench endogenous alkaline phosphatase, and then acetylated with 0.1 mol/L triethanolamine-HCl, pH 8.0/0.25% acetic anhydride for 13 minutes at room temperature. After prehybridization for 20 minutes at 55°C with 50% formamide, 10% dextran sulfate, 1X Denhardt's solution, 20 mmol/L Tris-HCl, pH 8.0, 300 mmol/L NaCl, 0.2% sarcosine, and 0.02% transfer RNA, digoxigenin-labeled probes were hybridized to brain sections at 55°C for 16 hours. Brain sections were washed in 5 × SSC at room temperature for 20 minutes and in 50% formamide/2 × SSC at 65°C for 1 hour. An antidigoxigenin antibody conjugated to alkaline phosphatase (Boehringer Mannheim) was reacted at 1:500 dilution, and a color reaction was performed with 4-nitro blue tetrazolium chloride and 5-bromo-4-chloro-3-indoyl-phosphate.
Western blot
Hippocampi of each rat (n = 4 for each group) were homogenated with a lysis buffer containing 0.15 mol/L NaCl, 50 mmol/L Tris-HCl, and 5 mmol/L EDTA. Lysate was subjected to 7.5% sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred to a polyvinylidine difluoride sheet (Immobilon P; Millipore, Bedford, MA, U.S.A.). A blocking reaction was performed in 5% dry milk/PBS containing 0.05% Tween-20 (T-PBS). Membranes were incubated with a rabbit polyclonal anti-HSP110 antibody (1:5000, SPA-1103; Stressgen, Victoria, Canada), which was raised against 15 amino acids (670–684) of hamster HSP110, or a mouse monoclonal anti-HSP70 antibody (1:1000, C92F3A-5; Stressgen) at 4°C overnight. After washing the membrane with T-PBS, peroxidase-conjugated anti-mouse or anti-rabbit IgG (Amersham Pharmacia Biotechnology, Buckinghamshire, U.K.) were used as secondary antibodies. The ECL system (Amersham Pharmacia Biotechnology) was used for immunodetection.
Immunohistochemistry
Four nonoperated control rats, 12 ischemic rats at 4 hours, 24 hours, and 48 hours after transient forebrain ischemia for 10 minutes (4 rats per each postischemic period), and 6 sham-operated rats (2 rats per each postischemic period) were perfusion-fixed with chilled saline and subsequently with 4% PFA, as described earlier. Coronal sections (4-μm-thick), including the hippocampus, were subjected to immunohistochemistry for HSP105/110. Brain sections were incubated in the presence of a rabbit polyclonal anti-HSP105 antibody (1:50, N-187; SantaCruz), which was raised against amino acids in the region between 187 and 512 of mouse HSP105, at 4°C overnight, and after washing in Tris-HCl/NaCl (pH 7.6), a goat anti-rabbit IgG (Vector) was used as the secondary antibody for 1 hour at room temperature. Sections were washed in Tris-HCl/NaCl, and further incubated with a peroxidase-conjugated anti-peroxidase antibody (ICN Pharmaceuticals, Costa Mesa, U.S.A.) before incubation with diaminobenzidine and hydrogen peroxide. To compare the expression of HSP110/105 and HSP70 in the same hippocampus after preconditioning ischemia, 4 ischemic rats at 24 hours after transient forebrain ischemia for 6 minutes were perfusion-fixed with 4% PFA. Four percent PFA-fixed brain blocks containing the hippocampus were divided into two thinner coronal blocks. One was subjected to paraffin embedding for HSP110/105 immunohistochemistry, and another was sliced into 40-μm sections with a vibratome for HSP70 detection. A rabbit polyclonal anti-HSP105 antibody (SantaCruz, 1:50) and a mouse monoclonal anti-HSP70 (inducible type) antibody (StressGen, 1:200) were used for this procedure.
Statistical analysis
Data are presented as mean ± SD values. The neuronal cell number was assessed by analysis of variance followed by Scheffe's test (P < 0.05). The significance of the changes in the band density of Northern blots was determined with one-factor analysis of variance and Bonferroni–Dunn post hoc test.
RESULTS
Histopathologic outcome after ischemic pretreatment
Three minutes of ischemia did not cause any damage to the CA1 sector. Six minutes of ischemia did not cause damage to the CA1 sector in 5 of 8 rats, but caused scattered single cell necrosis in the CA1 sector in the remaining 3 rats. The number of intact neurons in the CA1 sector after 3 or 6 minutes of ischemia was almost the same as that of sham-operated rats (Fig. 1). Ischemia for 10 or 15 minutes caused approximately 70% to 90% cell damage in the CA1 sector, respectively (Fig. 1). The authors chose a 6-minute ischemic episode as the sublethal ischemic pretreatment and a 15-minute ischemia as the second ischemia. When sham-operated animals were exposed to 15 minutes of ischemia after a 2-day recovery period, only 27% ± 24% neuronal cells survived in the CA1 sector. In the pretreatment group, 71% ± 33% cells survived, and protection of pretreatment was significant (Fig. 1;P < 0.01). Therefore, 6 minutes of ischemia induced tolerance against subsequent lethal ischemia.
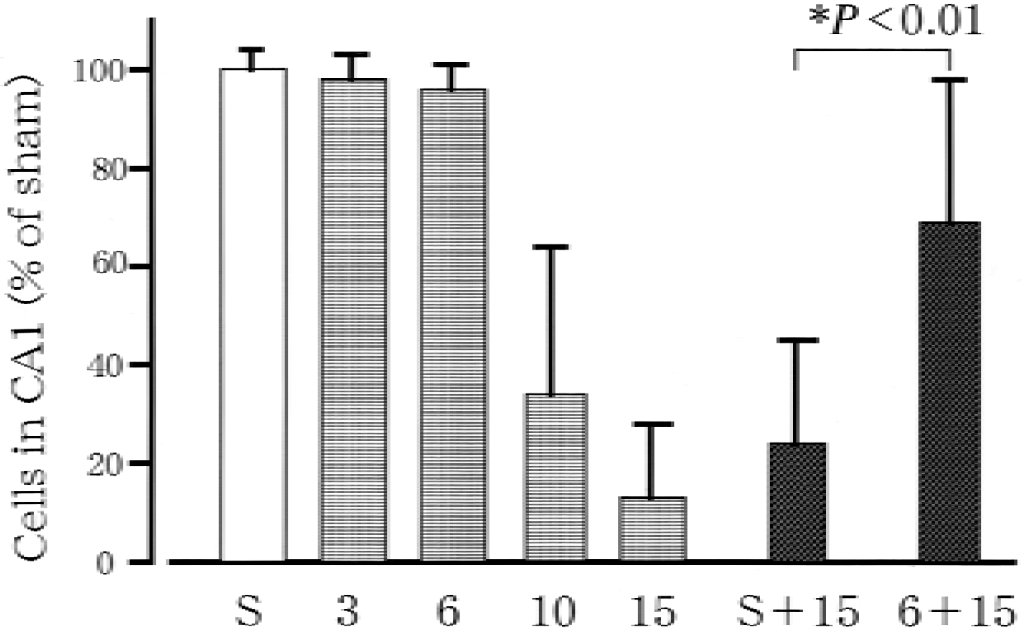
Number of neurons in the CA1 sector of the rat hippocampus, as a percentage of values of the sham-operated group (S). Animals were subjected to 3, 6, 10, or 15 minutes of ischemia. In the pretreatment group, animals were sham-operated or preconditioned with 6 minutes of ischemia followed by 2 days of recovery, and subsequently were subjected to 15 minutes of ischemia with S + 15 or 6 + 15, respectively. Data are mean ± SD. *Statistical difference between with and without pretreatment groups with P < 0.01, using analysis of variance followed by Scheffe's test.
Expression of the HSP110 family in normal tissues and hippocampus after transient forebrain ischemia
The authors obtained approximately 1.9 kbp osp94/apg-1 and approximately 2.8 kbp hsp110/105 cDNA from cDNA library screening (Fig. 2A). irp94/apg-2 cDNA was obtained in a previous study, and was expressed ubiquitously but more abundantly in the brain and testes (Yagita et al. 1999). hsp110/105 and osp94/apg-1 were primarily expressed in the brain and testes, respectively (Fig. 2B). Of these three genes (hsp110/105, osp94/apg-1 and irp94/apg-2), hsp110/105 was the most strongly induced by transient forebrain ischemia (Fig. 2C). osp94/apg-1 expression was less than the other two genes in the normal and ischemic hippocampus.
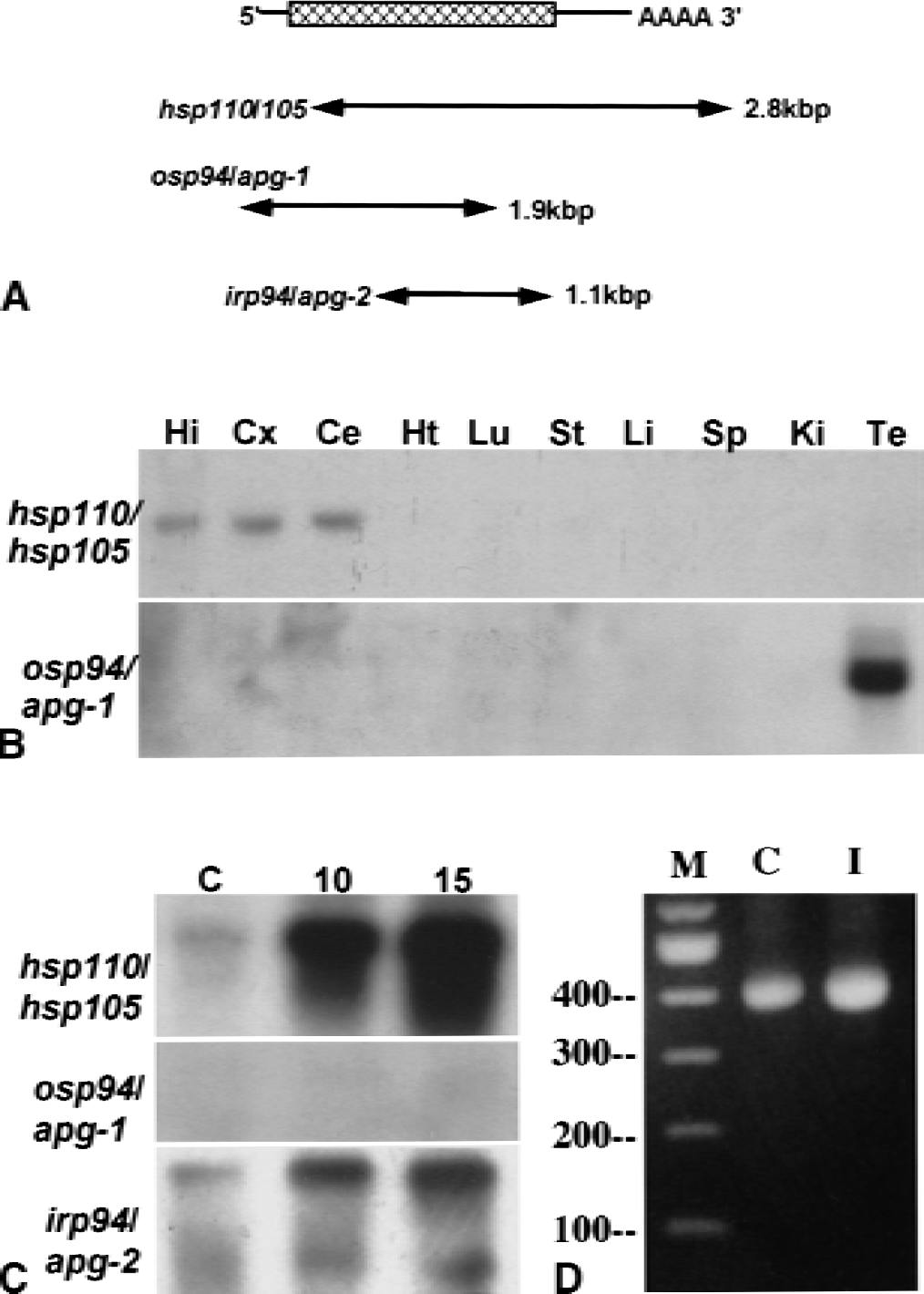
Tissue distribution and inducibility by transient forebrain ischemia of HSP110 family genes.
In RT-PCR analysis using primers by which the deletion of 132 nucleotides in hsp105β could be detected, only a single PCR band of approximately 400 bp was detected in cDNA prepared from control and ischemic rat hippocampus (Fig. 2D). An expected PCR band of 274 bp from hsp105β was not detected in either sample (Fig. 2D).
Temporal profiles of hsp110/105 mRNA expression after transient forebrain ischemia
Because hsp110/105 mRNA was abundant in the brain and strongly induced by ischemia, the expression profile of this gene after ischemia was investigated. In Northern blot analysis (Fig. 3), hsp110/105 mRNA was induced by a 6-minute (P = 0.12) or longer period of ischemia (P < 0.05), and its expression was clearly enhanced from 4 to 24 hours after ischemia. Amplified rat hsp105 cDNA was subcloned as the in situ hybridization probe. It was verified that the sequence of the cDNA was 92.8% matched to the corresponding part of mouse HSP105.
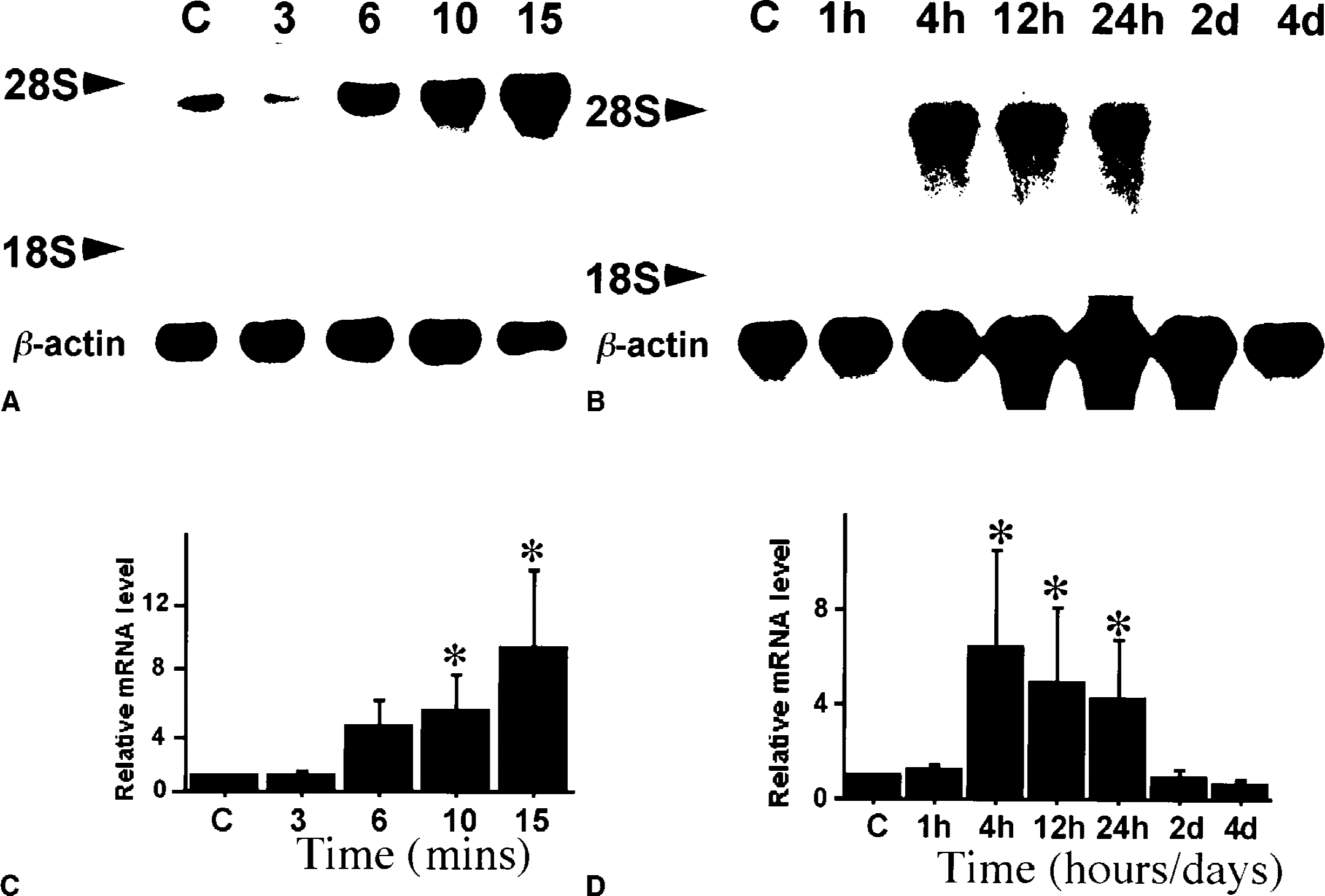
Expression of hsp110/105 mRNA in the hippocampus after transient forebrain ischemia. Four hours after recirculation, hsp110/105 mRNA tended to be induced by 6 minutes of ischemia (P = 0.12), and was significantly (P < 0.05) induced by longer periods of ischemia
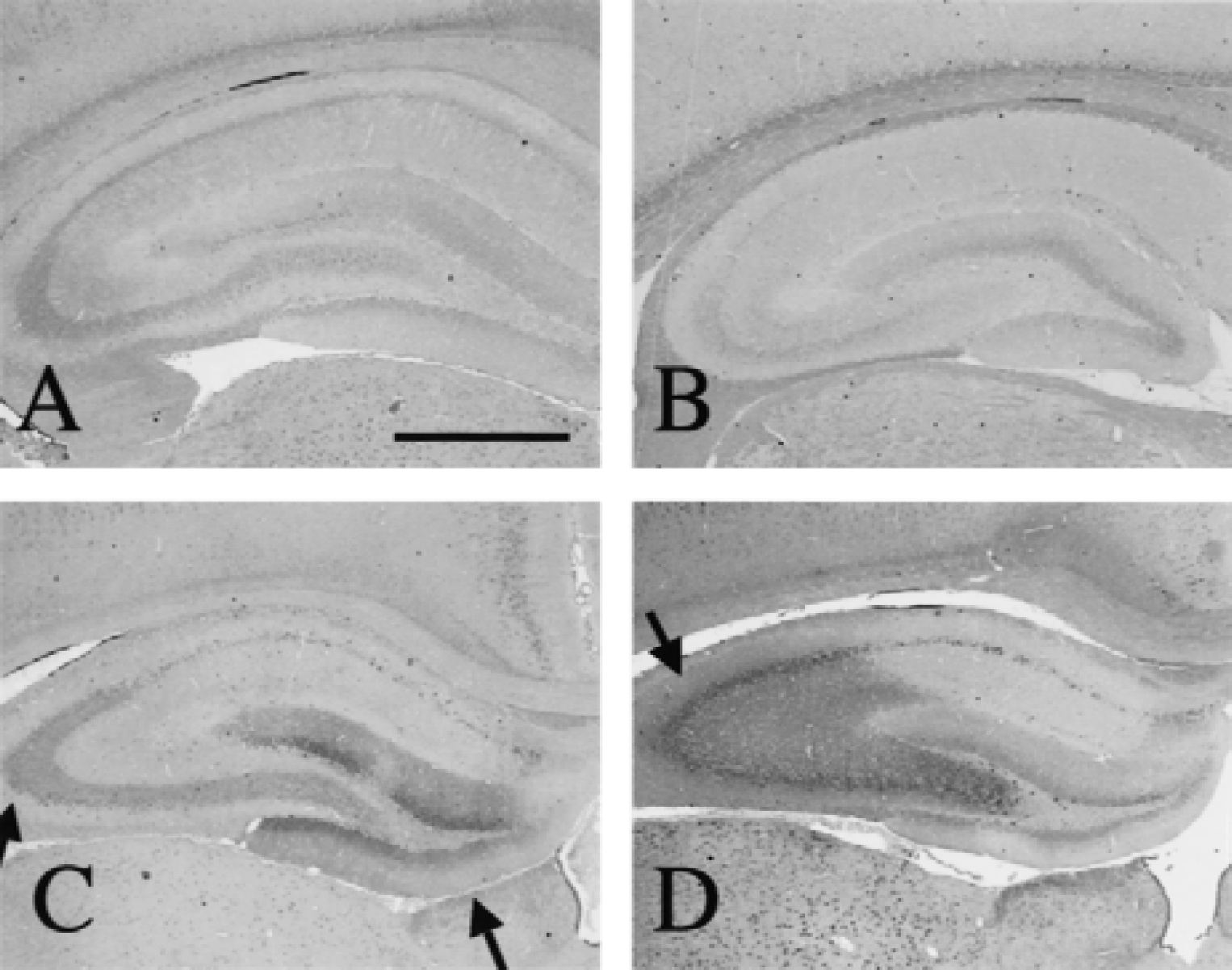
In in situ hybridization (Fig. 4), hsp110/105 mRNA expression was induced in the dentate gyrus as early as 1 hour later, and thereafter in the pyramidal layer (Table 1). Twenty-four hours after 10-minute ischemia, the signal density in the dentate gyrus decreased to the control level. In the CA3 region, up-regulation of signal density continued for at least 2 days after ischemia; however, the hsp110/105 mRNA signal disappeared in the CA1 region because of neuronal death 48 hours later.
In situ hybridization of hsp110/105 mRNA after transient forebrain ischemia. hsp110/105 mRNA expression was low in the control
Frequency of up-regulation of hsp105/110 mRNA and HSP105/110 protein expression in each area of the hippocampus
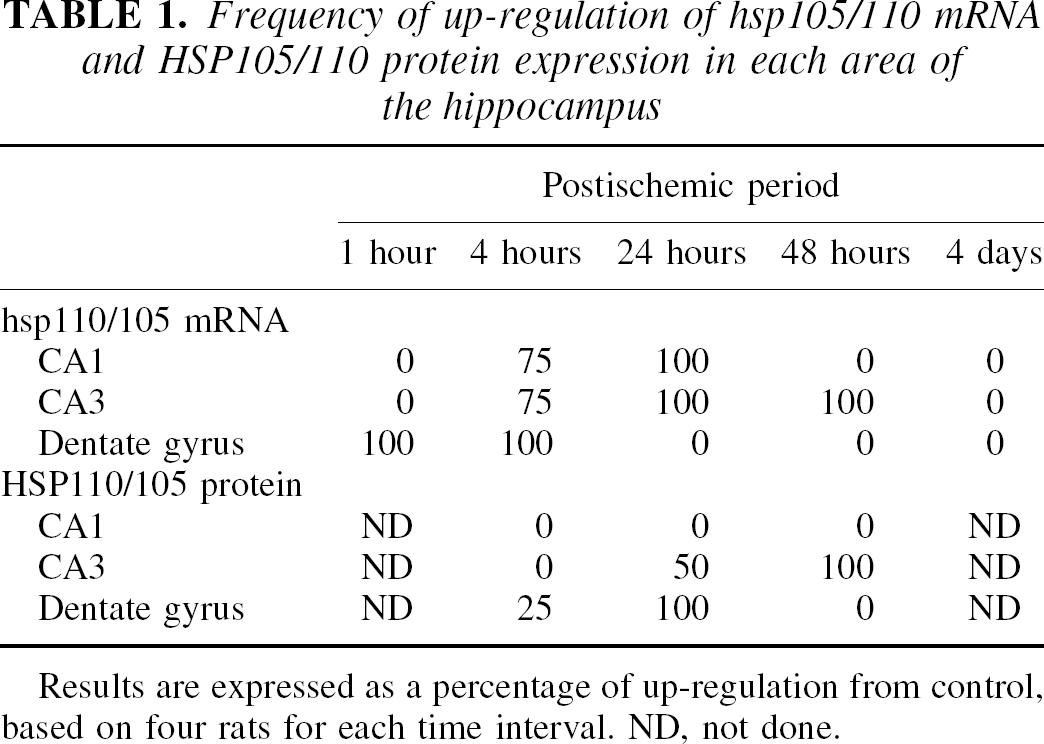
Results are expressed as a percentage of up-regulation from control, based on four rats for each time interval. ND, not done.
HSP110/105 protein expression after transient forebrain ischemia
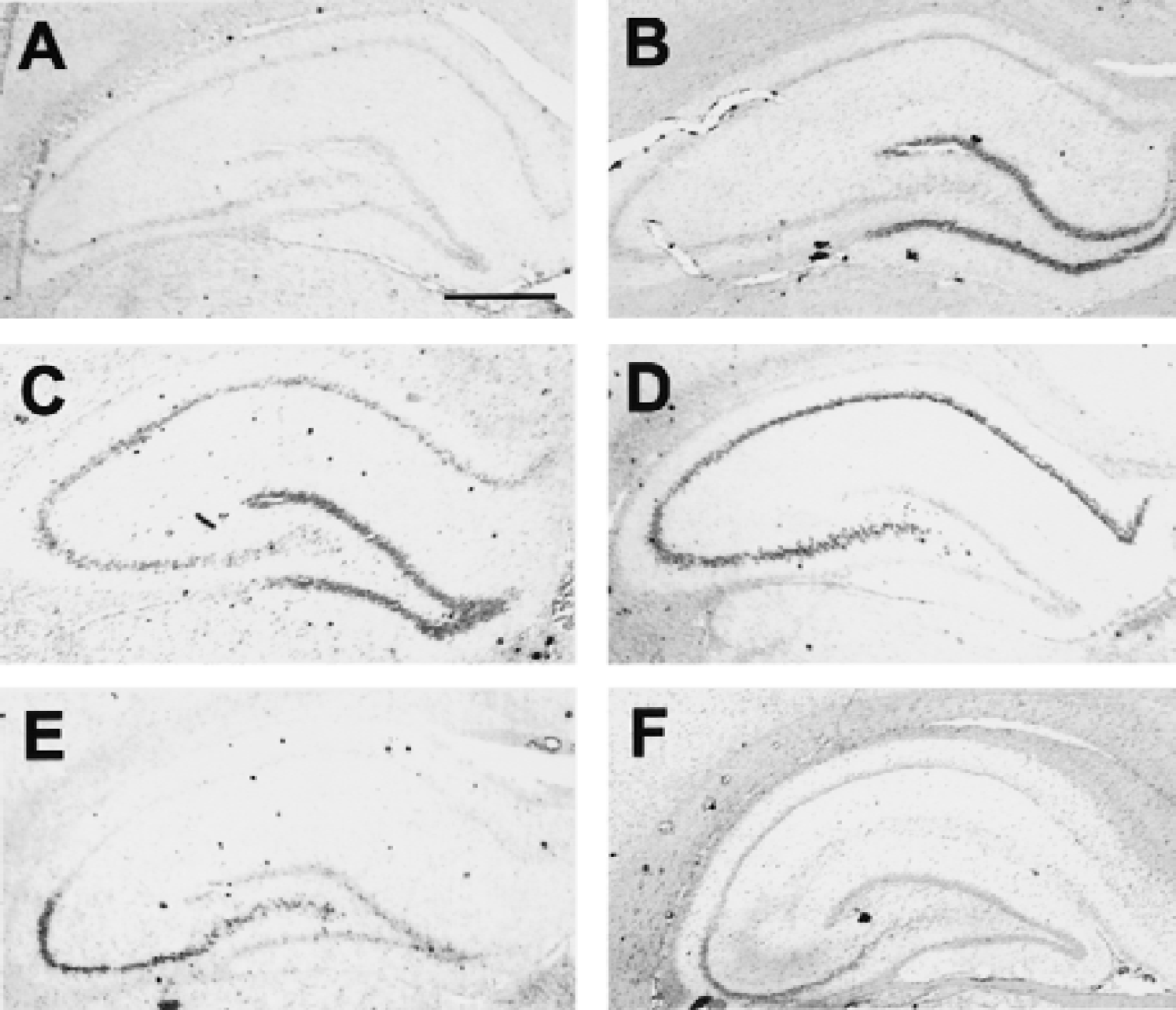
HSP110/105 was expressed in the control hippocampus, whereas HSP70 was not detected. In the ischemic hippocampus, HSP110/105 expression was enhanced by a 6-minute or longer ischemic period (Fig. 5A and 5C), similar to HSP70. Although the antibody against HSP110/105 recognizes both HSP105α and HSP105β, Western blot revealed a single band of 105-kDa HSP105α, and there was little expression of a 90-kDa HSP105β protein. Both HSP110/105 and HSP70 induction reached a peak 24 hours after 10 minutes of ischemia, and decreased after 48 hours (Fig. 5B). Because not only signal changes, but also neuronal death, could influence the results of Western blotting of the whole hippocampus, the authors examined HSP110/105 expression changes in each region after a 10-minute ischemia with immunohistochemistry (Fig. 6). In the control hippocampus (Fig. 6A), HSP110/105 was expressed only at a low level. The level of HSP110/105 expression was still low 4 hours after ischemia (Fig. 6B); however, HSP110/105 was markedly induced in the dentate gyrus and the CA3 region 24 hours after ischemia (Fig. 6C). After 48 hours of ischemia (Fig. 6D), HSP110/105 expression was prominent in the CA3 region, whereas it decreased in the dentate gyrus. Although hsp110/105 mRNA was induced in the CA1 region (Fig. 4D), HSP110/105 protein induction was hardly observed after ischemia. Frequency of up-regulation of HSP105 protein was summarized in Table 1.
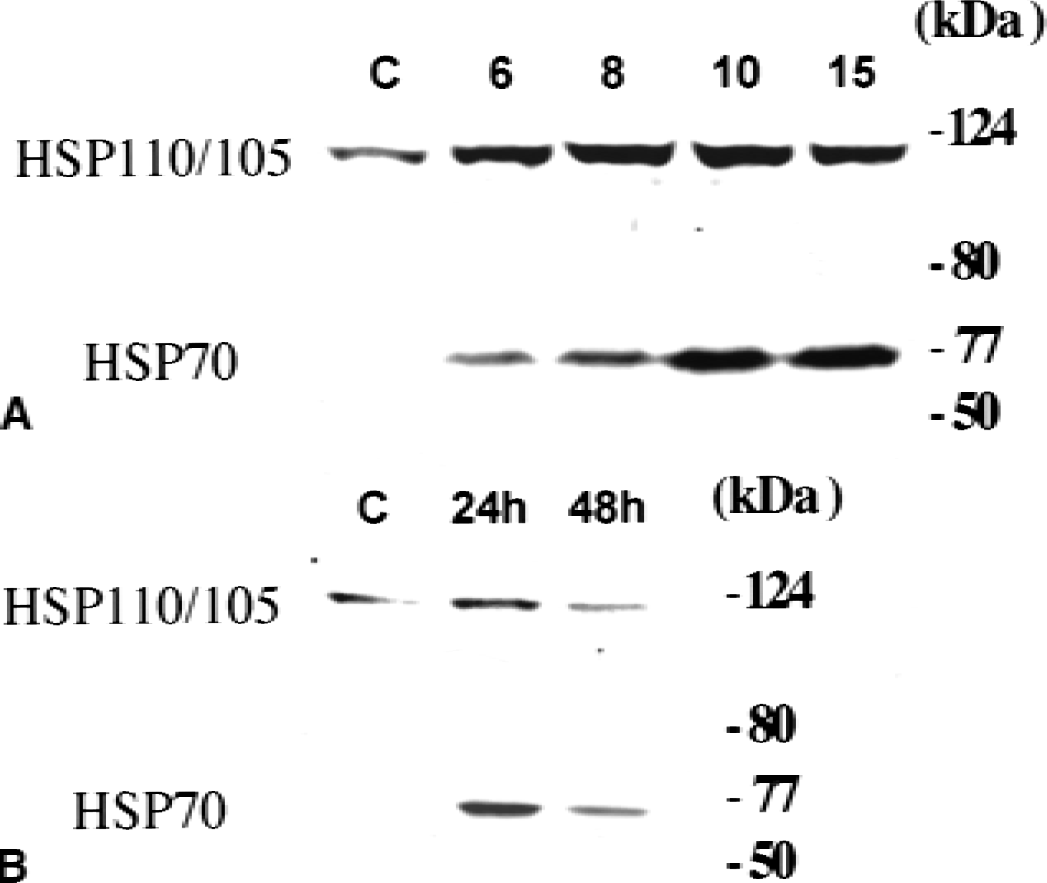
Western blot analysis of HSP110/105 in the hippocampus after transient forebrain ischemia. HSP110/105 protein expression also was enhanced by 6-minute or longer periods of ischemia
Immunohistochemical analysis of HSP110/105 expression after transient forebrain ischemia for 10 minutes. HSP110/105 protein expression was low in the control hippocampus
HSP110/105 and HSP70 expression and ischemic tolerance
To compare HSP110/105 and HSP70 induction in the hippocampus in the acquisition of ischemic tolerance, the authors examined immunoreactions for HSP110/105 and HSP70 in sections from the same hippocampus 2 days after 6 minutes of ischemia, when CA1 neurons acquired tolerance against subsequent severe ischemia (Fig. 7). HSP110/105 was clearly induced in the CA1 and CA3 sectors (Fig. 7A) and localized in the cytoplasm around the nucleus and dendrites (Fig. 7B). HSP70 also was strongly induced in the whole pyramidal cell layer, including CA1 neurons (Fig. 7C) and also was localized in the cytoplasm and dendrites (Fig. 7D).
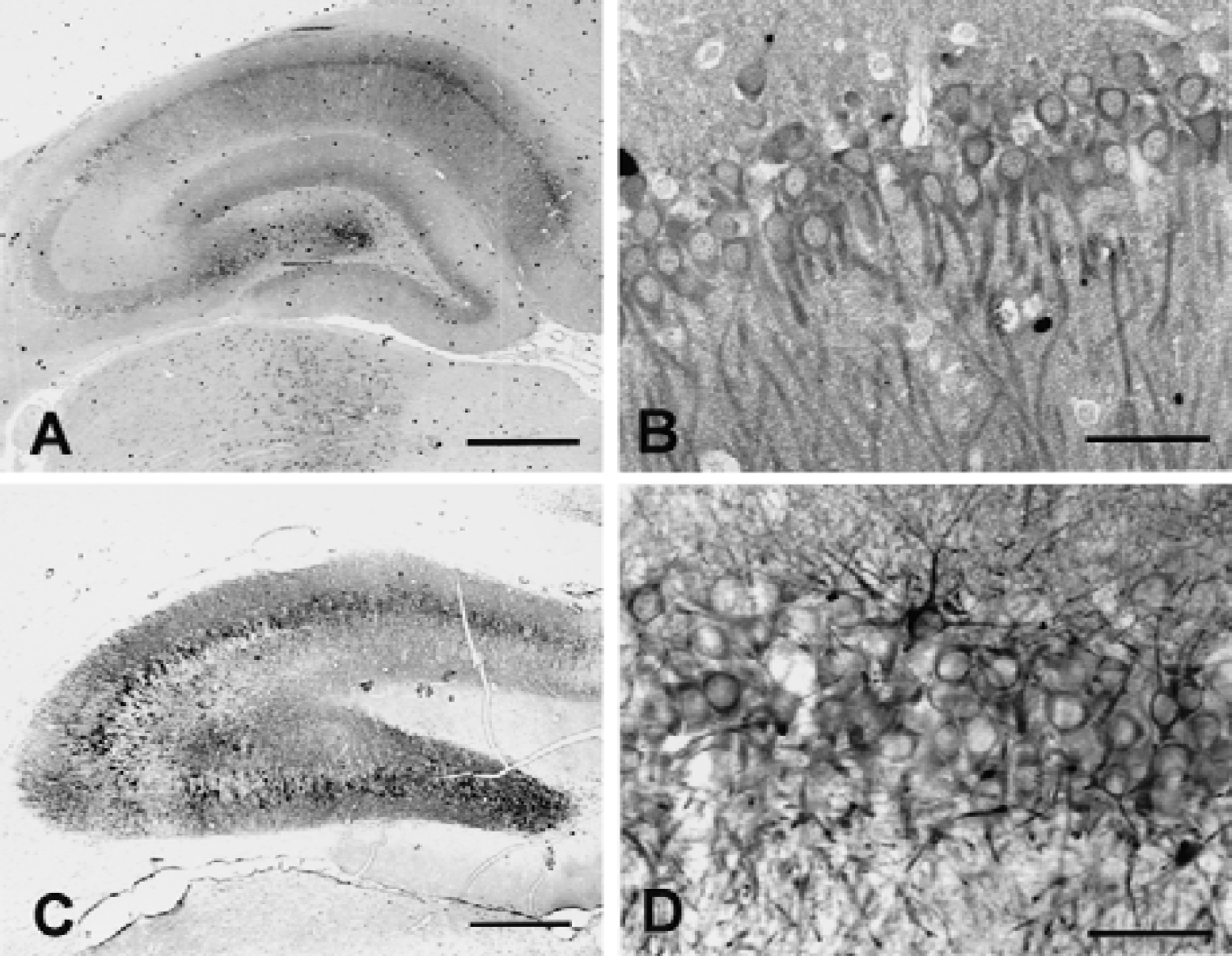
Comparison of HSP110/105 and HSP70 protein expression 24 hours after 6 minutes of transient forebrain ischemia. Six minutes of ischemia caused the hippocampus only ischemic stress without neuronal death in the CA1 region. HSP110/105 protein induction was stronger in the CA1 and a part of CA3 region than in the other regions of the hippocampus
DISCUSSION
In the current study, the authors demonstrated that hsp110/105 mRNA was abundantly expressed in the brain, and was most strongly induced by transient forebrain ischemia among the three HSP110 gene family members. Among two members of HSP110/105 proteins, hsp105α mRNA and HSP105α protein were expressed in the control hippocampus and after cerebral ischemia, whereas there was little hsp105β mRNA and HSP105β protein even after ischemia. This result led to the investigation of the expression profile of hsp110/105 mRNA and the HSP110/105 protein after transient forebrain ischemia. Northern and Western blotting analysis showed that 6-minute or longer periods of ischemia could induce hsp110/105 mRNA and HSP110/105 protein in hippocampal neurons. Then, in situ hybridization and immunohistochemistry showed that the induced hsp110/105 mRNA and HSP110/105 protein were localized in the dentate granule cells in the early phase after 10 minutes of transient forebrain ischemia. Subsequently, they were induced in the pyramidal layer, except for the CA1 region. In the CA1 region, hsp110/105 mRNA, but not the HSP110/105 protein, were induced. This profile was similar to HSP70 induction after transient forebrain ischemia (Vass et al., 1988; Nowak, 1991; Gonzalez et al., 1991; Simon et al., 1991; Aoki et al., 1993). It has been reported that HSP70 contributes to neuroprotection against cerebral ischemia (Yenari et al., 1998; Rajdev et al., 2000). In addition, HSP110/105 was suggested to be associated with HSC70, a constitutive expression molecule of the HSP70 family (Hatayama et al., 1998). The current results showed that induced HSP110/105 was colocalized with HSP70 in the CA1 region in the tolerant state after 6 minutes of sublethal ischemia. These findings suggested that HSP110/105 correlates to HSP70 and contributes to ischemic tolerance (Kitagawa et al., 1990, 1997; Kirino et al., 1991; Liu et al., 1992; Nishi et al., 1993; Glazier et al., 1994; Chen et al., 1996; Shamloo and Wieloch, 1999) or other stress responses to protect the neuron after ischemic stress. As acidic and basic isoforms of HSP105α and HSP105β have been reported recently (Ishihara et al., 2000), phosphorylation of HSP105α in the mechanism of ischemic tolerance may be examined in future studies.
It has been reported previously that the mRNA of HSP110 family genes was induced in the hippocampus after transient forebrain ischemia (Xue et al., 1998; Yagita et al., 1999). Xue et al. (1998) reported that apg-1/osp94 mRNA was induced in the hippocampus and cerebral cortex after transient forebrain ischemia. However, the authors did not observe any significant induction of osp94/apg-1 mRNA after 10 minutes of ischemia. This discrepancy may be because of the different severity of ischemia, because the previous study used a longer ischemic period than the current study. However, osp94/apg-1 mRNA was abundant in the testes, whereas only a small amount was expressed in the brain, as shown by the current data and previous studies (Kojima et al., 1996; Kaneko et al. 1997b). Therefore, osp94/apg-1 may be a testis-specific HSP110 family member and may play an important role mainly in the testes. In terms of the other member, irp94/apg-2, the authors demonstrated previously that 10 minutes, but not 6 minutes, of ischemia could induce this gene. Although constitutive expression of irp94/apg-2 in the brain (Kaneko et al., 1997a; Yagita et al., 1999) and inducibility by ischemia (Yagita et al., 1999) suggested some essential role in the ischemic brain of this gene, involvement of irp94/apg-2 in ischemic tolerance is unlikely.
The molecular mechanism of ischemic tolerance has been of great interest because it can be applied to the therapeutic strategy in ischemic brain injury. Stress proteins have been thought to be associated with ischemic tolerance through refolding of the denatured protein (Chen et al., 1996). However, it is still controversial whether HSP70 is the main molecule inducing ischemic tolerance because induction of tolerance was not always in parallel with that of HSP70 protein after preconditioning ischemia in both transient global and focal ischemia (Kirino et al., 1991; Chen et al., 1996). The fact that spreading depression induced tolerance without production of HSP70 (Kobayashi et al., 1995) also suggested that HSP70 was not necessary for tolerance. Thermotolerance induction was not blocked by inhibition of heat shock gene expression (Bader et al., 1992). These findings made it difficult to explain the induction of tolerance by only HSP70 expression. Although further study is necessary, HSP110/105 may play an important role in inducing tolerance by association with other stress proteins, such as HSP70 family members.
Footnotes
Acknowledgments:
The authors thank Ms. Y. Imaeda and Ms. R. Morimoto for secretarial assistance.