
Abstract
Sequencing of the human genome is nearing completion and biologists, molecular biologists, and bioinformatics specialists have teamed up to develop global genomic technologies to help decipher the complex nature of pathophysiologic gene function. This review will focus on differential gene expression in ischemic stroke. It will discuss inheritance in the broader stroke population, how experimental models of spontaneous stroke might be applied to humans to identify chromosomal loci of increased risk and ischemic sensitivity, and also how the gene expression induced by stroke is related to the poststroke processes of brain injury, repair, and recovery. In addition, we discuss and summarise the literature of experimental stroke genomics and compare several approaches of differential gene expression analyzes. These include a comparison of representational difference analysis we have provided using an experimental stroke model that is representative of stroke evolution observed most often in man, and a summary of available data on stroke differential gene expression. Issues regarding validation of potential genes as stroke targets, the verification of message translation to protein products, the relevance of the expression of neuroprotective and neurodestructive genes and their specific timings, and the emerging problems of handling novel genes that may be discovered during differential gene expression analyses will also be addressed.
Keywords
ISCHEMIC STROKE
Genomics technology
As efforts to complete sequencing of the human genome are nearing conclusion, there is an increased interest in the application of genomic approaches to aid in the discovery, development, and rationale of the use of drugs. As databases of differential gene expression have expanded, so has the expectation of identifying novel drug targets for disease intervention. Indeed, significant work has already been performed to understand gene expression changes in the ischemic heart (Stanton et al., 2000) and the ischemic brain (Barone and Feuerstein, 1999; Soriano et al., 2000).
Early epidemiologic studies of the 1970s provided initial evidence for a genetic influence in stroke. The Framingham study was one of the first studies to suggest that a positive parental history of stroke contributed significantly to the risk of the offspring (Kannel et al., 1970). Thirty years later stroke remains an area of substantial unmet medical need. The complexity of stroke undoubtedly reflects the heterogeneity of the human stroke population, the contribution of monogenic and polygenic disorders to this disease process, and the interactions of these with a multitude of environmental factors.
This review will briefly focus on the genetics of risk and sensitivity to ischemic stroke. It will discuss how genetic history relates to the broader stroke population and will provide a detailed discussion of the stroke genomics literature. This review will describe how preclinical models of spontaneous stroke can be applied to humans to identify the chromosomal loci of risk, and how the changes in gene expression associated with stroke are associated with poststroke brain injury, resolution of brain injury, and brain recovery processes. In addition, it will provide detailed discussion of several differential gene expression analysis techniques. This will include a detailed comparison of the emerging technology of representational difference analysis and its application to a stroke model that has been well-characterized and represents the type of stroke in evolution most often observed in humans. Issues will also be addressed regarding validation of potential stroke targets, the relevance of the expression of neuroprotective and neurodestructive genes and their specific timings, and the emerging problems with handling novel and unknown genes that may be discovered during analysis of differential gene expression.
Preponderance and risk
Stroke is the third largest cause of death in the U.S., ranking only behind heart disease and all forms of cancer. It is the leading cause of disability in the U.S. and has the highest disease burden cost. Ischemic strokes comprise approximately 80% of all strokes. No medical treatment is approved for the treatment of ischemic stroke other than tPA, a thrombolytic factor, which has to be administered within 3 hours after stroke. Only 1% to 2% of stroke patients meet the criteria for treatment with this thrombolytic agent. Aspirin and anticoagulants (where embolic phenomena are documented) are used as preventative therapy. Estimates indicate that there are approximately 775,000 stroke cases per year in the U.S., with approximately 4 million surviving, but at an increased risk of a secondary cardiovascular event. In the U.S., stroke is costly, with an annual health care cost of $30 to $50 billion. Estimates indicate that stroke is responsible for half of all patients hospitalized for acute neurologic disease (Stephenson, 1998; Fisher and Bogousslavsky, 1998).
Stroke risk factors include both genetic and environmental factors. Stroke risk factors that can be treated include high blood pressure, heart disease, cigarette smoking, transient ischemic attacks, and high red blood cell count. Risk factors for stroke that cannot be changed include increased age, gender (men have approximately a 19% greater chance of stroke than women), race (blacks have a greater risk of death and disability from stroke), diabetes mellitus, previous stroke, and a family history of strokes. Other controllable risk factors are secondary risk factors for stroke that contribute to heart disease, including high blood low-density lipoproteins (LDL)-cholesterol and lipids, physical inactivity, and obesity (Pancioli et al., 1998).
GENOTYPING IN STROKE
Genetics of increased stroke risk
The strongest evidence for a genetic risk to stroke is derived from twin studies. Proband concordance rates have long been used to identify the heritability of a trait or disorder. The concept of concordance is that for a disorder of genetic predisposition, the rate will be higher for monozygotic twins than dizygotic twins. Aside from genetic influence, it is assumed that other factors, such as environmental exposure, will be approximately similar for both types of twins (Hrubec and Robinette, 1984). Brass et al. (1992) confirmed an elevated probandwise concordance rate for stroke risk in monozygotic twins over dizygotic twins (17.7% vs. 3.6%), confirming a genetic predisposition to stroke. Subsequent reassessment of this cohort of patients 6 years later reported risk attributable to genetic influence, but with an increase in the role of environmental factors (Brass et al., 1992). A more recent twin study by Carmelli et al. (1998) has refined cohort analysis to stroke risk by assessing individual stroke phenotypes that may be influenced by genetic factors. In this study, the phenotype of white matter hyperintensity volumes using magnetic resonance imaging (MRI) was applied and genetic factors accounted for 71% of the variation in this endpoint.
A large number of familial studies have verified that a history of paternal or maternal stroke is associated with occurrence of stroke in offspring, and that a positive paternal history of stroke was an independent prognostic predictor of stroke (Kiely et al., 1993; Jousilahti et al., 1997; Liao et al., 1997). For example, Welin et al. (1987) reported that, in a cohort of men studied since 1913, maternal history of stroke increased relative risk by threefold. Similarly, Khaw and Barrett-Connor (1986) reported that a positive family history of stroke in any first degree relative is an independent predictor of stroke mortality in women aged 50 to 79, but not in men. Moreover, this study also identified that a family history of stroke was an independent predictor of coronary heart disease in men aged 50 to 64 years, indicating that genetic risk factors for stroke may be shared with other cardiovascular disorders that have a high genetic component. Indeed, studies of the relative risk of other cerebrovascular diseases with less heterogeneous phenotypes have documented strong patterns of inheritance. Bromberg et al. (1995) found that subarachnoid hemorrhage occurs with a relative risk of 6.6 in first degree relatives compared with second degree relatives. Indeed, defining specific stroke subtypes may be key in elucidating the exact degree of genetic contribution to any particular phenotype. From twin studies, it appears that the extent to which genetic factors may contribute to stroke risk varies with age. These factors are caveats to the identification of therapeutic targets from candidate gene strategies, and one must remember that a candidate gene approach for inheritance of risk factors may only be relevant to a highly limited stroke subpopulation.
Simple strokelike diseases: single gene mutations
Identification of possible genetic determinants of stroke risk has been hampered by the lack of homologous patient populations. Mendelian disorders with specific strokelike phenotypes have been explored as genetic models of the more general population. These disorders include cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL) (Tournier-Lasserve et al., 1993), mitochondrial encephalopathy-lactic acidosis-and strokelike syndrome (MELAS) (Hirano and Pavlakis, 1994), Sneddon syndrome (Lossos et al., 1995), familial hemiplegic migraine (Joutel et al., 1993), and hereditary coagulopathies (Hassan and Markus, 2000). Although these subgroups contribute little to the overall prevalence of stroke, genes identified from them are hoped to highlight potential commonalties in the wider patient population. Studies on CADASIL and MELAS are examples of such approaches.
CADASIL was originally described by Sourander and Walinder (1977) as an inherited, autosomally-dominant dementia with multiple infarcts. Epidemiologically, CADASIL is limited to sporadic identification in Europe (Chabriat et al., 1995; Dichgans et al., 1998) and North America (Hedera and Friedland, 1997; Desmond et al., 1998). The principal symptoms of CADASIL are migraine with aura, ischemic stroke, and psychiatric symptoms including dementia (Viitanen and Kalimo, 2000). In these patients, T2-weighted MRI discloses small periventricular white matter hyperintensities often involving the internal capsule (Chabriat et al., 1998). The CADASIL gene, identified as Notch 3, is located at the chromosome loci 19p13.1 – 13.2 (Joutel et al., 1996; Dichgans et al., 1996). The Notch genes regulate the lin-12/sel-12 signalling pathway important in development, although the normal adult function of Notch genes remains unknown (Artavanis-Tsakonas et al., 1999). An interesting association of the Notch 3 gene with Alzheimer disease also has been discovered. Notch gene products interact with the presenilin-1 pathway as substrates for γ-secretase. This enzyme is known to have a key pathologic role in the production of Aβ peptide, although the modulatory role that Notch 3 may have in this disease process is undefined (Levitan and Greenwald, 1995; Viitanen and Kalimo, 2000). The Notch 3 gene encodes a transmembrane protein composed of 2,321 amino acids, presumed to have a receptor function and located primarily on smooth muscle cells (Viitanen and Kalimo, 2000). In CADASIL, approximately 90% of patients have missense mutations in extracellular domains of the protein product, whereas in approximately 70% of patients, the mutation is located within exons 3 and 4 (Joutel et al., 1997). All known mutations associated with CADASIL result in removal or addition of cysteine residues, and it is proposed that expression of these mutated Notch 3 proteins results in cerebral vascular smooth muscle dysfunction (Joutel and Tournier-Lasserve, 1998).
Whether abnormalities in Notch signaling impact on the broader stroke population is currently unknown, although the pathogenesis of CADASIL, characterized by progressive disruption of vascular endothelium, secondary fibrosis, and thrombosis is typical of some stroke subpopulations (Ruchoux and Maurage, 1998). Interestingly, anticoagulant therapy has been tried in CADASIL without positive results (Viitanen and Kalimo, 2000). More broadly, CADASIL also has close relations to Alzheimer disease, and signaling components of the presenilin pathway are shared with the Notch pathway (De Strooper et al., 1999). The presenilin-1 regulated γ-secretase cleaves both the Notch intracellular domain and β-amyloid precursor protein for subsequent translocation to the nucleus and binding to DNA (De Strooper et al., 1999). Therefore, although the pathology of CADASIL may bear similarity to stroke, the cell biology is also reminiscent of Alzheimer disease. Because vascular risk factors, or disease, or both, can impact vascular dementia and Alzheimer disease, these relations are intriguing (Kudo et al., 2000; Schmidt et al., 2000; Skoog, 2000).
Mitochondrial encephalomyelopathy-lactic acidosis-and strokelike episodes is characterized by migrainelike headache, nausea, seizures and strokelike episodes. Lesions are most commonly found in occipital and parietal regions, with high lactate levels found within lesions under proton nuclear magnetic resonance (Hassan and Markus, 2000). Patients typically have mutations of mitochondrial DNA for the tRNA-leu gene at an A-G transition mutation at nucleotide position 3243 (Ciafoaloni et al., 1992; Macmillan et al., 1993) and at a T-C transition at 3271 (Sakuta et al., 1993). Kovalenko et al. (1996) speculated that as mutations accumulate, a gradual mitochondrial dysfunction develops. It is unclear how widespread such mutations are in the broader stroke population. Indeed, cases of MELAS have been reported without a family history, suggesting that these point mutations may be spontaneous (Rastenyte et al., 1998). Pharmacologic interventions have reflected a unique nature of MELAS within stroke and cardiovascular disease subpopulations. Antithrombotic therapy has been used in MELAS patients for cardiac complications associated with left ventricular dysfunction (Kosinski et al., 1995).
CADASIL and MELAS demonstrate that several relatively rare “strokelike” mendelian syndromes can be used to explore potential genetic determinants of stroke. Parallel strategies have been adopted with similar success in other more complex multifactorial polygenic traits such as hypertension (Dominiczak et al., 2000). Genes such as 11β-hydroxylase in glucocorticoid remediable aldosteronism have been shown to mediate the hereditary hypertension in these patients (Lifton et al., 1992). However, as in stroke genetics, narrowing heterogeneity and studying single gene and mendelian disorders has been found to limit the application to the broader patient population.
Ischemic stroke: a disease having complex genetic associations
In common with many diseases, there are individuals with complex genetic profiles and with complex profiles of poststroke gene expression that can contribute to the risk of ischemic stroke and increased cerebral ischemic stroke sensitivity, respectively. Candidate gene studies in heterogeneous stroke populations negate issues of limited patient population by the a priori choice of a functionally relevant gene and its relation with a particular phenotype. This is often termed ‘association’ and is a statistical measure of the dependence of a particular phenotype (for example, ischemic stroke with the presence of a particular candidate gene/allele). Therefore, association can be positive (that is, has a significant statistical relationship/association between the gene of choice and phenotype) (see Table 1), or negative (that is, has no significant relationship/association between gene/allele and phenotype) (see Table 2). Candidate gene choice is frequently driven by accepted stroke risk factors (for example, hypertension, hemostasis, and abnormalities in lipid metabolism), and indeed, significant positive associations of numerous markers with ischemic stroke have been identified, including ApoE ε2 allele and D/D genotype of angiotensin converting enzyme-1 (Table 1). However, there are also numerous examples of negative (or lack of) associations of genes with ischemic stroke. For example, a negative association was identified for some hemostasis factors (for example, Factor V, Q506 polymorphism, and Factor VII R353 Q polymorphism) and some hypertension factors (for example, angiotensinogen and M235T polymorphism) (Table 2). Currently, it is difficult to identify clear patterns of candidate gene associations with ischemic stroke. Furthermore, the reproducibility of these gene expression associations in different patient populations (for example, different race or genetic backgrounds) is not known. The bewildering combination of possible outcomes for candidate gene association studies is emphasized by the genomic and phenotypic heterogeneity of the global stroke population. Studies are typically designed with case controls or by cohorts to enable close approximation of phenotype between affected and nonaffected individuals. Superimposed upon these levels of variation are issues in the timing of stroke onset, in the variability of environmental influences and penetrance (that is, not all individuals of a given genotype will express the phenotype). Finally, although the human genome project continues apace (Genome International Sequencing Consortium, 2001), identifying functionality of gene products lags considerably. Current estimates propose only approximately 10% of the human genome has been ascribed function (Hassan and Markus, 2000). Certainly a lot of work needs to be done in this area, and issues related to stroke genomics that include risk and the expression of genes underlying brain vulnerability and ischemic sensitivity must be considered.
Candidate genes with a positive association with ischemic stroke
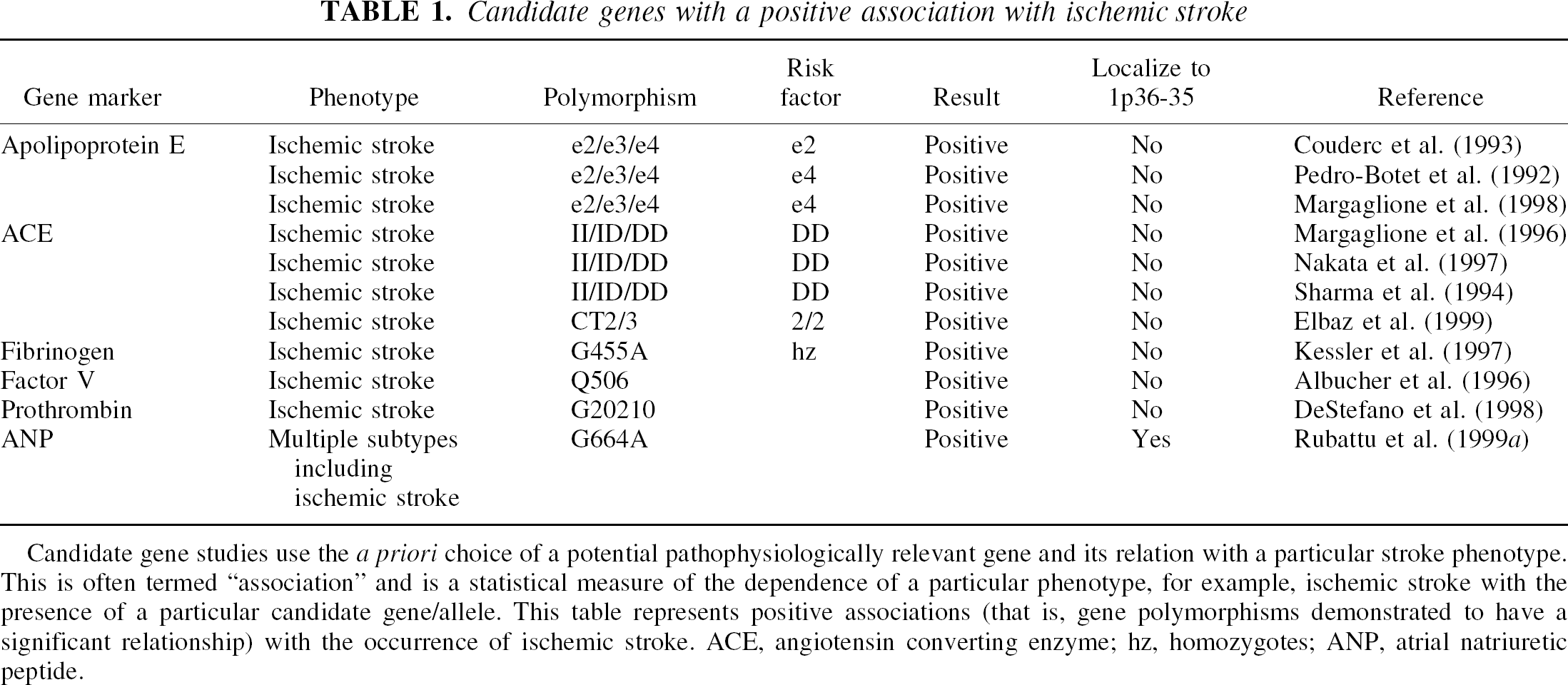
Candidate gene studies use the a priori choice of a potential pathophysiologically relevant gene and its relation with a particular stroke phenotype. This is often termed “association” and is a statistical measure of the dependence of a particular phenotype, for example, ischemic stroke with the presence of a particular candidate gene/allele. This table represents positive associations (that is, gene polymorphisms demonstrated to have a significant relationship) with the occurrence of ischemic stroke. ACE, angiotensin converting enzyme; hz, homozygotes; ANP, atrial natriuretic peptide.
Candidate gene studies with negative association to ischemic stroke
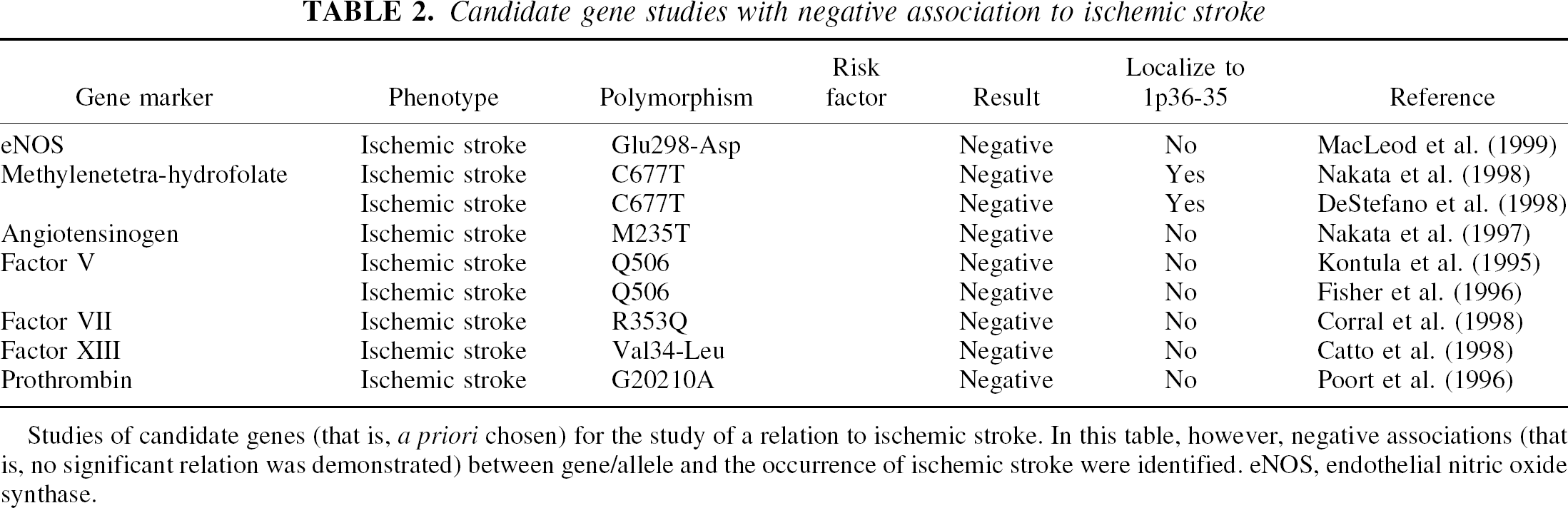
Studies of candidate genes (that is, a priori chosen) for the study of a relation to ischemic stroke. In this table, however, negative associations (that is, no significant relation was demonstrated) between gene/allele and the occurrence of ischemic stroke were identified. eNOS, endothelial nitric oxide synthase.
PRECLINICAL MODELS OF SPONTANEOUS STROKE
It is with these caveats in mind that studies have focused on animal models of spontaneous stroke, in which environmental and genetic variability can be controlled. Bioinformatic approaches using synteny can facilitate the matching of “stroke loci” found in stroke-prone rats to candidate genes on the human chromosome. Heterogeneity of risk factors and life events in humans has made it advantageous to study rodent models. Highly homogeneous populations of stroke-prone rats have been isolated from the incompletely inbred, spontaneously hypertensive rat (SHR) and then inbred further for this phenotype. Initial studies using the stroke-prone rat indicated that the degree of functional collateral blood flow after occlusion of the middle cerebral artery (MCAO) was inherited in an autosomally recessive manner (Coyle et al., 1984). The authors studied luminal diameters in vascular anastomoses between middle and anterior cerebral arteries and hypothesized that the collateral flow phenotype was determined by a single gene not directly linked to hypertension. Further genetic comparisons between strains were hampered by heterogeneity.
Narrowing the genotype by further crossing SHR rats with stroke-prone animals allowed cosegregation of genes defining various stroke phenotypes and for homogeneity of alleles for hypertension (Nagaoka et al., 1976). Two separate groups have used these inbred populations for identification of genes associated with manifestation of specific stroke phenotypes. Rubattu et al. (1996) performed a genome-wide screen in an F2 cross, obtained by mating stroke-prone and SHR rats, in which latency to stroke was used as a phenotype. They identified three major quantitative trait loci (QTLs)—STR1, STR2, and STR3. Of these, STR-2 and STR-3 conferred a protective effect against stroke in the presence of stroke-prone alleles, and STR-2 colocalized with the candidate gene encoding atrial natriuretic peptide (ANP) and brain natriuretic peptide (BNP). Furthermore, interactions between alleles from within STR1 and STR2 suggested that this phenotype was a reasonable model of the polygenicity of stroke in man. Follow-up sequencing to characterize ANP and BNP as candidates for stroke revealed point mutations in ANP and no differences in BNP. In vitro functional studies indicated less ANP promoter activation in endothelial cells from stroke-prone rats versus SHR, with significantly less ANP expression in the brain and no difference in BNP expression (Rubattu et al., 1999c). To determine the in vivo significance of the STR-2 lowered ANP promoter activation in stroke-prone animals compared with stroke-resistant animals, Rubattu et al. (1999b) performed a cosegregation analysis of stroke occurrence in SHR stroke-prone rats/SHR stroke-resistant F2 descendents and ANP expression. It was found that reduced expression of ANP did cosegregate with the appearance of “early” strokes in F2 animals; therefore, although lowered ANP expression may be part of the phenotype of the “protective” STR-2 QTL, it is unlikely that this is the primary protective mechanism in these animals. Parallel human studies of the role of ANP in cerebrovascular disease have confirmed that variation in the ANP gene may represent an independent risk factor for “cerebrovascular accidents” in humans (Rubattu et al., 1996) (Table 1) and may emphasize the utility of this cohort of animals as a model of ANP dysfunction in multiple subtypes of stroke.
Two other groups have used a modified model of the stroke-prone animal, using F2 hybrids derived from crossing the stroke-prone SHR with Wistar-Kyoto (WKY) rats (Ikeda et al., 1996; Jeffs et al., 1997). Ikeda et al. (1996) used brain weight poststroke as the phenotype for linkage analysis, after the discovery that F2 animals had higher levels of brain edema formation poststroke. Evidence of the linkage of phenotype to a gene on chromosome 4 was found that contributed to the severity of edema and was independent of blood pressure and STR3 identified by Rubattu et al. (1996). Jeffs et al. (1997) designed studies to identify the genetic component responsible for large infarct volumes in the stroke-prone rat in response to a focal ischemic insult. To do this, they performed a genome scan in an F2 cross, derived from the stroke-prone rat and the normotensive WKY rat. In contrast with Rubattu and coworkers, they were only able to identify one major QTL responsible for large infarct volumes. This QTL was located on rat chromosome 5, and like STR-2, it colocalized with ANP and BNP and was blood pressure independent. Unlike STR-2, this locus showed a much higher significance (lod 16.6) and accounted for 67% of phenotypic variance. Subsequent studies identified that infarct volumes in the F1 rats were approximately identical to those of the stroke-prone animals, suggesting a dominant mode of inheritance (Gratton et al., 1998).
Authors have argued over the significance of the overlap of STR2 identified from Rubattu et al. (1996) with the QTL identified by Jeffs et al. (1997) on chromosome 5. It is unclear how the two phenotypes studied—latency to stroke (that is, relative risk) (Rubattu et al., 1996) and size of infarct after occlusion (that is, sensitivity to focal ischemia) (Jeffs et al., 1997)—should physiologically relate to each other. This may only become apparent when individual genes can be cosegregated with each phenotype. Currently, altered ANP expression seems to play a role in the phenotype described by Rubattu et al. (1996), but has been excluded from a role in the colony used by Jeffs et al. (1997; Brosnan et al., 1999).
What can be concluded from each of these stroke-prone rat models? Certainly, each represents a unique and valid “model” of stroke for the study of inheritance and for a role of candidate genes in particular stroke phenotypes. Neither colony represents a definitive model of human stroke, although progress has been made with titrating identified candidate genes in these stroke-prone colonies to the human population (Rubattu et al., 1999a). One such research strategy we have used is the analysis of genomic synteny between the rat and human genome. This bioinformatic approach seeks to align regions of homology using evolutionary conserved markers and has been applied with some success in relating animal models to human genetics of other disease paradigms, for example, noninsulin-dependent diabetes (Ktorza et al., 1997). Relating identified loci from stroke-prone animals to the human genome offers a strategy for potential identification of candidate genes. For example, the STR2 region of rat chromosome 5 shows well-conserved gene order and synteny with the human chromosome region 1p35–36. The high level of synteny between these regions makes this region ideal for rat–human comparative analysis. Sequence tagged sites localized to this region have been identified and mapped to human transcript clusters. As many as 132 transcripts have been identified in this region. The main candidates are listed in Table 3.
1p36-p35 postional candidates with a biologic rationale in stroke

Identification of human candidate genes that are syntenic to the STR2 region of rat chromosome 5 identified by Rubattu et al. (1996) in a cohort of SHR-stroke prone animals. STR2 shows well-conserved gene order and synteny with the human chromosome region 1p35–36 (between D1S503-D1S2667). The high level of synteny between these regions makes this region ideal for rat–human comparative analysis. Sequence tagged sites localized to this region have been identified and mapped to human transcript clusters. Many transcripts, specifically 132 of them, have been identified in this region, with the main candidates listed above. KO, knockout; PDGF, platelet-derived growth factor; SHR, spontaneously hypertensive rats.
Interestingly, only a few candidate genes identified at 1p35–36 have been examined in association studies. ANP recently has been assessed for association with multiple subtypes of stroke (Rubattu et al., 1999a). The polymorphism G664A, responsible for a valine–methionine substitution in pro-ANP peptide was found to be positively associated with the occurrence of stroke (Table 1). In contrast, methylenetetrahydrofolate, another marker located at 1p35–36, was negatively associated with occurrence of stroke (Table 2). Further studies may elucidate the predictability of markers of 1p35–36 and association with stroke (Table 3).
In contrast, rat–human synteny in the regions of the rat STR-1 and STR-3 loci are not well conserved, as several disruptions of synteny appear to have been introduced during evolution. It may be difficult to determine exact regions of synteny between these rat loci and human chromosomal loci, and thus it may be difficult to extrapolate the candidate genes from rat to human. Human chromosomal regions syntenic with STR1 span regions of two human chromosomes, around 16p11 and 19q13. Human synteny with the STR3 region also appears to be disrupted, with regions of synteny mapping telomerically to opposite arms of chromosome 7 (7p21 & 7q35). Of course, this is a problem of animal modeling of human diseases in general and is not restricted only to ischemic stroke.
GENE EXPRESSION IN THE EVOLUTION OF POSTSTROKE BRAIN INJURY
Cerebral ischemia is a powerful stimulus for the de novo expression and up-regulation of numerous gene systems (Barone and Feuerstein, 1999; Koistinaho and Hokfelt, 1997). In terms of isolation of gene candidates for a neuroprotection strategy, interpretation of expression changes has proven difficult. The multitude of animal models of ischemia with varying genetic heterogeneity and infarct pathophysiology is also complicated by spatial and temporal variations that has largely confounded interpretation (Sharp et al., 2000; Iadecola and Ross, 1997). Furthermore, assays of differential expression have varying sensitivity to the relative fold increase or decrease in mRNA expression. As a result, “fishing” exercises will often result in “catches” of differential gene expression that vary depending on the assay used.
With this bewildering array of complexity as a caveat, the following section addresses that assessment of animal model(s) that might be used, the appropriate assays available for differential gene expression analysis, the target confirmation methodology that is necessary after the identification and confirmation of a differentially expressed gene (that is, a “hit”), and ultimately, the functional assessment of these genes in the disease process. A hierarchical critical path that depicts the path from target identification to target confirmation and validation is depicted schematically in Fig. 1.
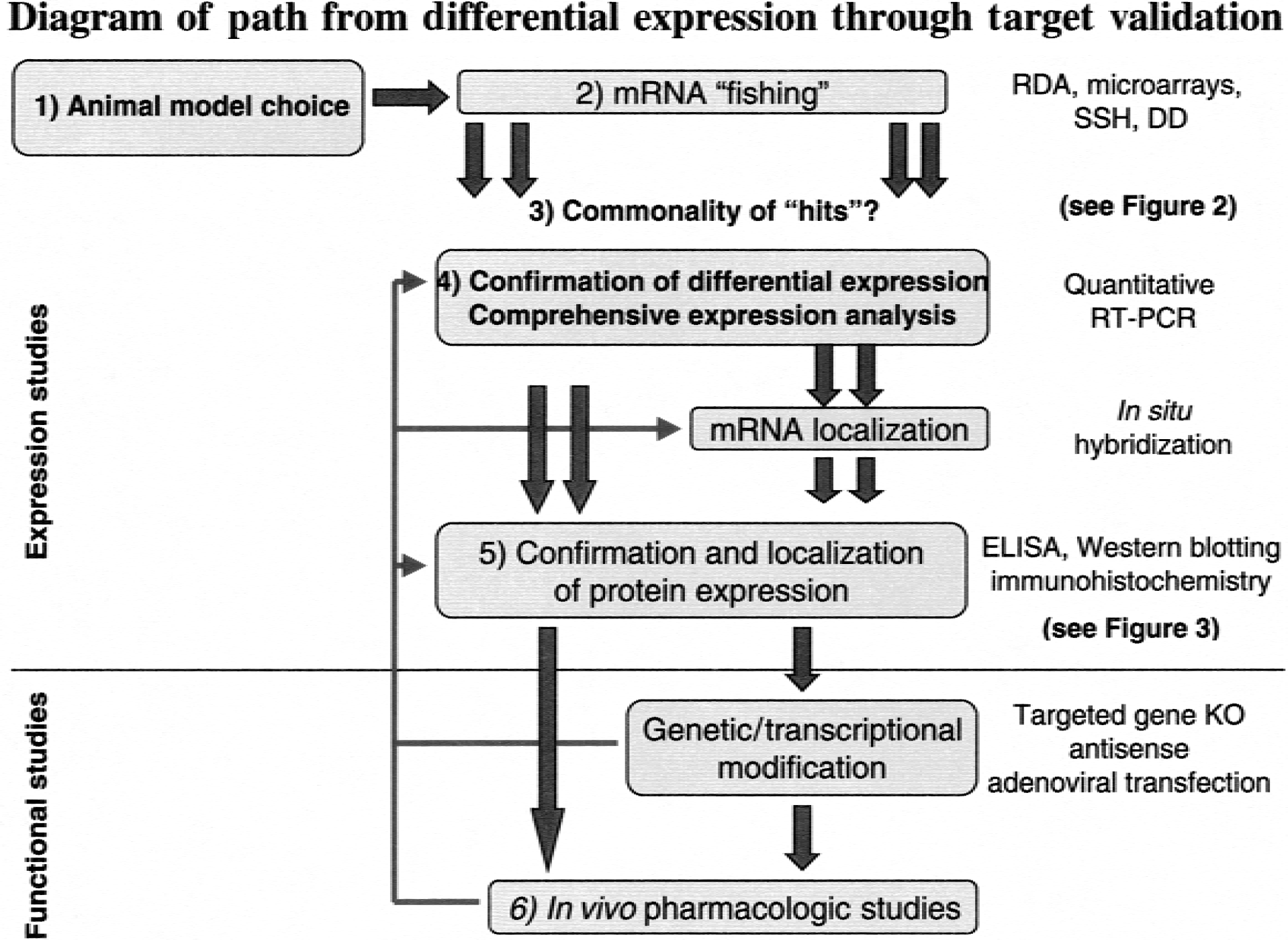
Hierarchical organization of target identification and confirmation. After selection of an animal model, appropriate to clinical subpopulation (1), broad mRNA fishing strategies are adopted using differential expression assays such as representational difference analysis (RDA), microarrays, subtractive hybridization (SSH), and differential display (DD). (2) Across these assays, commonalties in identified hits (3) are explored as a technique of prioritising subsequent confirmation studies (see Fig. 2). Comprehensive expression analysis using reverse transcription-polymerase chain reaction (RT-PCR) (4) allows confirmation of identified hits and fully quantified temporal profiling. Protein confirmation (5) by ELISA, Western blotting, and immunohistochemistry follows mRNA profiling to confirm translation. In an ischemic brain, pooling of mRNA and uncoupling of translation can occur (see Fig. 3). Finally, functional studies encompassing target gene knockout (KO), adenoviral transfection, and in vivo pharmacology (6) complete validation of a potential target gene.
ISCHEMIC STROKE MODELS: THE SEARCH FOR CLINICAL RELEVANCE
The failure of several putative neuroprotective agents in recent large multicentered clinical trials (Clark et al., 2000; Lees et al., 2000) has led to critical reexamination of the predictability of many preclinical models of ischemia (Feinklestein et al., 1999). Heterogeneity in the human stroke population and the multitude of well-defined animal models of ischemia have led to attempts to refine model choice as related to patient subgroups (DeGraba and Pettigrew, 2000; Parsons et al., 2000). In an effort to stratify patient groups that can be predicted using specific animal models, authors have focused on the use of MRI signatures, in particular, perfusion- (PWI) and diffusion-weighted imaging (DWI) mismatches. Two main groups of acute stroke patients are identifiable, those with evolving infarcts in which lesion PWI > DWI, or those with a stabilized infarct in which PWI ≤ DWI (Baird and Warach, 1998; Albers, 1999). Such PWI and DWI assessments have been proposed to correlate to the extent of salvageable tissue, with approximately 70% of patients exhibiting PWI lesions > DWI at 6 hours poststroke, and approximately 50% of patients exhibiting this mismatch at 24 hours poststroke (Albers, 1999).
Applying such imaging paradigms to animal models of focal ischemia should enable translation of preclinical pathophysiology into predictive outcomes in the appropriate patient population. Detailed comparisons of the development of PWI/DWI signatures between animal models of ischemia are difficult to establish because of the use of various rat strains, anesthetics, and modes of ischemia induction. However, broad comparisons are possible by exploring the development of DWI signal as a marker of lesion volume with respect to time. Therefore, data in certain animal models of focal stroke can show a delay in the development of DWI hyperintensity (that is, brain lesion size) that lags behind a perfusion deficit (that is, PWI changes associated with stroke and focal ischemia). This delay is attributable, in part, to the relative contribution of insufficient collateral flow and the periinfarct depolarizations that eventually significantly injure the poorly perfused, ischemic brain during infarct evolution (Hossman, 1996; Parsons et al., 2000).
Photothrombotic ischemia by the rose-bengal method produces a highly consistent focal infarct. Diffusion-weighted imaging lesion develops primarily during the first 24 hours, with an expanding volume of DWI deficit continuing over a subsequent 3 to 7 days (van Bruggen et al., 1992; De Ryck et al., 2000; Lee et al., 1996). The extensive thrombosis produced in this model in association with profound blood–brain barrier breakdown may limit the application of identified gene expression in this model to the clinic (that is, lesion development is rapid with little penumbra area to impact upon). Direct MCAO by proximal electrocoagulation of the MCA produces an expanding DWI lesion, with an initial marked expansion at 4 hours followed by a small increase from 4 to 24 hours (Gill et al., 1995). Modification of this occlusion to distal MCAO in SHR produces a rapidly evolving infarct with a near maximal lesion observed after 1 hour (Chandra et al., 1999).
Available embolic models of focal stroke using intraarterial injection of thrombin (Zhang et al., 1997), aged (Jiang et al., 2000) or fibrin-rich (Busch et al., 1998) clots have reported approximately similar expansion of DWI hyperintensity. For example, after thrombin injection, DWI hyperintensity is apparent 80 minutes postadministration, with the volume gradually expanding up to 24 hours (Zhang et al., 1997). Intraluminal suture occlusion produces a range of DWI lesion progression dependent on whether the filament is introduced through the common carotid artery (Koizumi et al., 1986) or the external carotid artery (Longa et al., 1989). Permanent occlusion through Koizumi suture MCAO produces a rapid evolution of DWI hyperintensity within minutes, followed by maximal expansion by 2 hours (Neumann-Haefelin et al., 2000; Li et al., 2000). In comparison, permanent MCAO (pMCAO) by the method of Longa et al. (1989) evokes an initial rapid expansion of DWI hyperintensity during the first 30 minutes followed by final infarct volume reached at 7 hours (Gyngell et al., 1995; Kohno et al., 1995a,b). A close inspection of this models identifies it as exhibiting a mismatch similar to that in humans, that is, representing the type of infarct in evolution that should provide information relevant to human stroke (Parsons et al., 2000). For this reason we have decided to use this model for our recent differential gene expression studies (Bates et al., 2001).
METHODOLOGIES FOR DIFFERENTIAL GENE EXPRESSION IN FOCAL STROKE
The detection of genes that are differentially expressed because of stroke can be identified using the simpler (that is, well established and straightforward) techniques such as Northern blotting, reverse transcription-polymerase chain reaction (RT-PCR), in situ hybridization, and so on. These techniques involve the selection and study of a specific gene of interest (that is, based on previous data that provides a biological rationale for study in stroke or another specific disease). However, several more complex screening techniques are now available that can identify groups of differentially expressed genes, both known and unknown. These complex screening techniques include subtractive libraries/subtractive hybridisation, differential hybridisation, serial analysis of gene expression, representational differential analysis (RDA), and differential display (Feuerstein et al., 1996). For example, the identification of differentially expressed genes in stroke has used the simpler and complex techniques of differential display analysis, subtractive hybridization, DNA microarrays, and representational difference analysis. A complete summary of stroke-associated gene expression by all techniques is listed in Table 4. Variations in assay and threshold of detection often result in the isolation of gene sets that differ according to assay selection. Therefore, to ensure maximum confidence in the detection of adaptive up-regulation of gene expression, a pragmatic approach should be adopted. For example, commonalties in identified genes and pathways should be identified across several differential expression assays, or independent cross-validation of a gene's up-regulation should be emphasized, or both, rather than relying on the results of a single differential expression technique. Table 4 provides a list of only those gene and message expressions that have been confirmed within or between laboratories using more than one expression detection technique. A brief summary of the more complex techniques is provided below.
Summary of differential gene expression changes identified to date by techniques that measure transcription differences after focal ischemia stroke
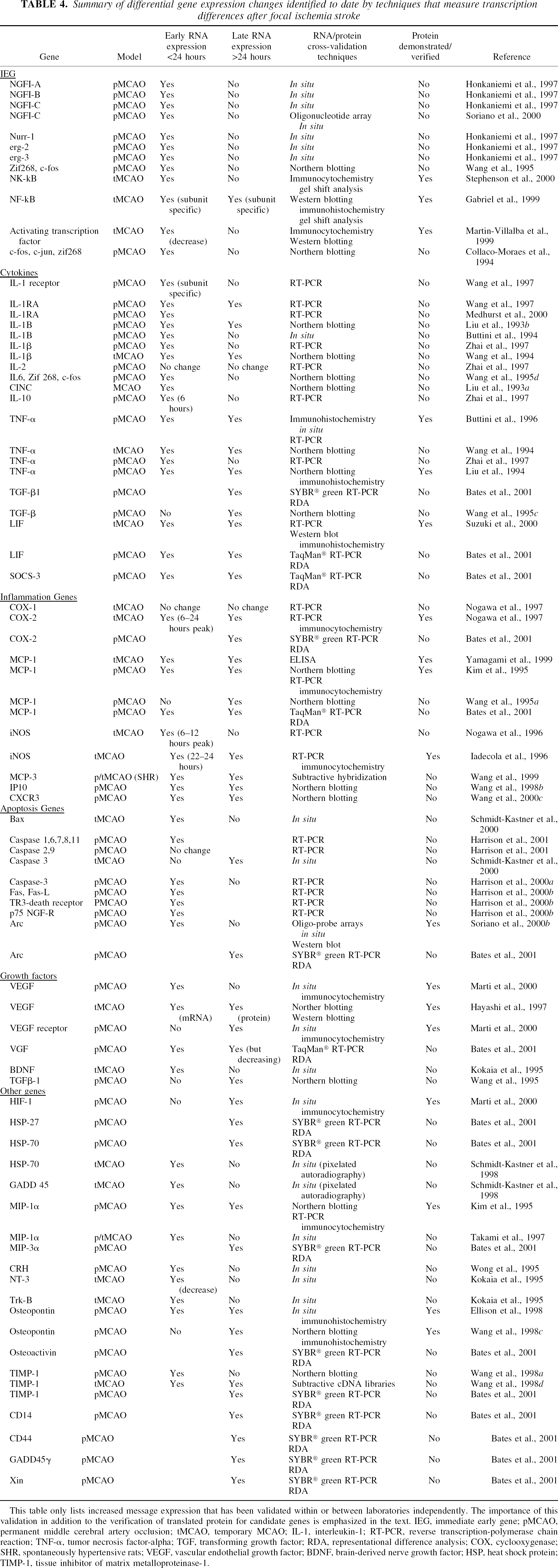
This table only lists increased message expression that has been validated within or between laboratories independently. The importance of this validation in addition to the verification of translated protein for candidate genes is emphasized in the text. IEG, immediate early gene; pMCAO, permanent middle cerebral artery occlusion; tMCAO, temporary MCAO; IL-1, interleukin-1; RT-PCR, reverse transcription-polymerase chain reaction; TNF-α, tumor necrosis factor-alpha; TGF, transforming growth factor; RDA, representational difference analysis; COX, cyclooxygenase; SHR, spontaneously hypertensive rats; VEGF, vascular endothelial growth factor; BDNF, brain-derived nerve growth factor; HSP, heat shock protein; TIMP-1, tissue inhibitor of matrix metalloproteinase-1.
Differential display
Differential display is a means of comparing all poly (A)+ mRNA between experimental and control populations. mRNA is converted into first strand cDNA through reverse transcription plus oligo(dT)-anchored primers followed by PCR with multiple sets of primers. The PCR products then are displayed with control and experimental samples side-by-side on high resolution denaturing gel, this way differential gene expression is immediately apparent. This technique has been applied with success in isolating differentially expressed products after experimental MCAO. Wang et al. (1995b), using mRNA differential display after rat focal MCAO, isolated a gene that encodes adrenomedullin, a member of the calcitonin gene-related peptide (CGRP) family. This was followed up by independent temporal studies using Northern analysis, which confirmed that expression of mRNA levels increased in the ischemic cortex at 3 hours and 6 hours post-MCAO. Levels remained elevated for up to 15 days post-MCAO. Immunohistochemical studies to confirm protein expression localized adrenomedullin to ischemic neuronal processes. In functional studies, synthetic adrenomedullin microinjected onto preconstricted rat pial arteries produced dose-dependent relaxation of these vessels. In addition, intracerebroventricular administration of adrenomedullin, before and after MCAO, increased the degree of focal ischemic injury.
Other groups also have used this technique in ischemia to identify differentially expressed mRNA such as serine protease inhibitors (SPI-3), zinc transporter gene (ZnT-1), and ADP-ribosylation factor like gene (ARF4L) in models of gerbil forebrain ischemia (Tsuda et al., 1996, 1997; Katayama et al., 1998). Proteosome expression was identified after rat photochemical ischemia (Keyvani et al., 2000). Transcription factor (SEF-2) and proteosome expression (p112) after rat global ischemia (Wigle et al., 1999) and chemokine identification (ST-38) after rat MCAO (Utans-Schneitz et al., 1998) also were demonstrated.
Differential display, although a labor-intensive technique, is very useful. For example, differential display is ideal for examining several RNA samples simultaneously and has been used extensively for temporal, dose-response, and multiple treatment studies. Also, although differential display is “semiquantitative,” only relatively small amounts of total RNA (approximately 15 μg) are required. However, as can be observed from the literature, problems with interpretation of data have been identified. High rates of false positives that can not be confirmed by RT-PCR or Northern blotting. Modifications to this approach have been used, such as subtracted differential display, which removes unregulated cDNA by mRNA subtraction before differential display (Wang and Uhl, 1998). Nevertheless, confidence in an isolated candidate gene can be improved by using other independent follow-up assays of gene expression, or other differential expression techniques in parallel, or both. Confirmation across several assays will aid the identification of false-positives and improve the confidence that a specific gene putatively up-regulated in ischemic stroke is significant.
Subtractive hybridization and suppression subtractive hybridization
Subtractive hybridization compares qualitative differences in gene expression between two experimental paradigms. This is usually achieved by hybridization of biotinylated “driver” cDNA to the mRNA pool from the “target” tissue. Duplexes of driver cDNA and target mRNA are then removed, resulting in a pool of target mRNA expressed only by the target tissue (Barr and Emanuel, 1990). Down-regulated mRNA are determined by performing the reaction in reverse. Modifications to the assay include suppression subtractive hybridization and RDA (see next subsection), where the PCR replaces physical subtraction methods to enrich for differentially expressed transcripts. Such modifications emphasize differential mRNA of both low and high abundance, rather than biasing selection of only highly expressed genes, as is the case with the more basic subtractive hybridization methodology.
Suppression subtractive hybridization has been successfully used to identify candidate genes with putative roles in experimental cerebral ischemia. Wang et al. (1999) identified the induced expression of a rat homologue to human monocyte chemotactic protein-3 (MCP-3) in ischemic brain. Independent Northern analysis identified increases in MCP-3 mRNA observed 12 hours postischemia, with 49-fold and 17-fold increases over control in permanent and temporary MCAO, respectively. Moreover, significant induction of MCP-3 in the ischemic cortex was sustained up to 5 days after ischemic injury. In other models, subtractive hybridization has been less widely used to identify candidate genes. This may be because of the difficulty of the subtraction approach, although false positives are less frequent. Nevertheless, Abe et al. (1996) have used subtractive hybridization to successfully identify a novel cDNA clone (pGSH3) expressed only after ischemia in the gerbil cortex. Basal cortical levels were found to be low, but 8 hours after a 10-minute transient forebrain ischemia the gene expression became prominent in the cerebral cortex. Analysis of DNA sequence revealed that the pGSH3 insert had a 91.3% homology with a 72-kd human heat shock protein (hsp70) gene.
Representational difference analysis (a form of subtractive hybridization)
Representational difference analysis is a relatively novel PCR-coupled, genome subtractive process (Hubank and Schatz, 1994; Lisitsyn et al., 1995) that until only very recently (Bates et al., 2001) had not been used to assay differential expression in models of cerebral ischemia. Representational difference analysis is conceptually similar to subtractive hybridization, but the unavailability of a commercially produced kit for RDA has meant that it has been less broadly exploited. Representational difference analysis was originally established to monitor differences in genomic DNA content between individuals, it was later modified to identify differences in gene expression (Hubank and Schatz, 1994; Lisitsyn et al., 1993).
The robust gene expression changes that characterize the MCAO model has also proved amenable to RDA, as we have recently been able to show (Bates et al., 2001). Subtracting ischemic cortex from rats 24 hours after pMCAO from similarly treated tissue from sham-operated animals, we were able to identify a number of candidate ischemia-regulated transcripts. Primary confirmation of the accumulation of these gene products in the ischemic cortex was confirmed using SYBR Green RT-PCR, followed by the more comprehensive time-course analysis using TaqMan RT-PCR in selected cases. Several genes identified through this approach previously had been reported to increase after MCAO, such as heat shock proteins (hsp27 and hsp70) and others (MCP-1, MIP3α, cyclooxygenase-2 (COX-2), TGF-β1, tissue inhibitor of matrix metalloproteinase-1 (TIMP-1), and Arc), and several were first identified to be MCAO-induced in this study (LIF, SOCS-3, VGF, CD44, CD14, CD81, osteoactivin, GADD45γ, and Xin) (Bates et al., 2001). These gene expressions and follow-up verifications of these and other differentially expressed genes are also listed, with appropriate references, in Table 4, and are discussed in more detail below.
Array technologies
All of the above strategies identify small numbers of differentially expressed genes. However, large numbers of DNA fragments (110 to 450bp) are produced in the process that need to be confirmed and frequently extended to full lengths to obtain gene identity. Although all of these technologies are useful for isolating candidate genes, they are of limited use in providing a broad characterization of the expression of large numbers of genes within a particular model.
Array-based technology allows such analysis. It provides for a full analysis of gene expression within a study including time-response profiling, drug treatment analysis, and so on. Whether using arrays of oligonucleotides (Lockhart et al., 1996; Lipshutz et al., 1999) or gene fragments (Schena et al., 1995), the technology allows parallel expression monitoring of several hundred genes at a time. The limitations and biases of the technique are obviously in the selection of genes to study on the array. Technology has been advancing rapidly in the area of cDNA array analysis and now short oligomers can be transferred to glass slides, allowing rapid development of important tools for genotyping and mRNA expression analysis (Young, 2000).
Soriano et al. (2000) pioneered this technique in the application of studying gene expression in the Tamura (1981) model of rat MCAO. The authors used an oligonucleotide probe array with 750 predetermined genes optimized for gene expression in rat bone and cartilage. The chip (ROEZ002; Hoffman-LaRoche Limited, Basel, Switerland) was used to monitor gene expression after 3 hours of permanent focal ischemia. To determine genes differentially expressed as a consequence of ischemia, the authors took tissue from the ipsilateral frontal and parietal cortices and compared the tissue with corresponding regions on the contralateral side. A significant change in transcription was defined as a twofold or greater increase or decrease in expression compared with the contralateral hemisphere. The authors describe a significant up-regulation of 24 genes in the parietal, frontal cortices, and striatum with particularly robust changes in c-fos, NGFI-A, NGFI-C, Krox20, NGFI-B, Nor-1, COX-2, and Arc.
The current study clearly demonstrates the utility of array technology for broad characterization of the regulation of genes during experimental cerebral ischemia. The use of arrays optimized for bone and cartilage genes unfortunately limited the usefulness to 15% of the total gene representation on the array. Nevertheless, key gene families such as the phosphatases (MKP-1 and MKP-3) and the chemokines (MCP-1 and MIP-1α) were represented and expression profiles agreed with previous findings (Wiessner et al., 1995; Gass et al., 1996; Kim et al., 1995). No change in housekeeping genes GAPDH and β-actin were found (ipsilateral vs. contralateral) at this 3-hour time point.
In our laboratories, we have further assessed the utility of microarrays. After MCAO, again using the Longa et al. (1989) technique and commercially available microarray grids from Affymax, we conducted differential gene expression analysis. Ipsilateral cortex samples were pooled from MCAO animals and sham-operated animals at 24 hours post-pMCAO. In common with Soriano et al. (2000), we identified several immediate early genes (IEGs) that were up-regulated, including c-fos (2.9-fold) and NGFI-A (2.9-fold). As reflected in later time points studied, fold induction of the IEGs was less than at 3 hours poststroke as studied by Soriano et al. (2000). Several heat shock proteins were found to be up-regulated, specifically HSP-27 (20.9-fold) and HSP-70 (9.8-fold), which were shown to be up-regulated after stroke by other methods that assess gene expression (Table 4). In addition, our microarray experiment revealed 74 genes/sequence candidates that are up-regulated in the MCAO model 24 hours postocclusion. Several candidate genes previously shown to be up-regulated or down-regulated in stroke were confirmed to be regulated at the level of transcription in this experiment. TIMP-1, Arc, osteopontin, glial fibrillary acidic protein (GFAP), VGF (NGF-induced), interferon-induced protein, calmodulin (and calmodulin binding proteins), and COX-2 have been shown to be up-regulated by us using microarrays, and in some cases by others using various techniques (see Table 4). Furthermore, many structural genes or their regulators (forms of actin and tubulin, vimentin, ARP2/3) and genes involved in basic cell metabolism (ribosomal proteins, polyA-binding protein, elongation factors, cysteine oxygen oxidoreductase, ribosomal RNA, mitochondrial cytochrome oxidase) were shown to be affected after stroke.
Interestingly, as documented by Soriano et al. (2000), differential expression of Arc also was found (2.6-fold). However, it should be noted that this was at 24 hours postocclusion. Using the Tamura et al. (1981) method of occlusion, Soriano and coworkers documented that Arc transcript levels decrease to basal levels by 24 hours. Given the putative role of Arc in mediation of cytoskeletal changes underlying neuronal plasticity (Fosnaugh et al., 1995), these findings emphasize temporal differences between models that may reflect different pathophysiologic processes (that is, evolving vs. terminal strokes as discussed earlier) (Parsons et al., 2000). Below we will discuss in more detail model to model differences, the importance of the poststroke timings of RNA sampling, different experimental paradigms in stroke that might help in the discovery of genes that have roles in brain protection or tolerance, and the specific studies that can be used to look for genes that might contribute to recovery and plasticity of the brain postinjury.
Between assay variation and increasing confidence in identified gene targets
In the preceding sections we have discussed many of the techniques available for detection of differential gene expression and some of the “within assay” issues associated with each technique. However, in generating and comparing gene expression data, there are several issues that warrant discussion, including the significant variability between techniques and the identification of false-positive and false-negative results. Assays of differential expression have an inherent variability dependent on assay methodology, sensitivity, and reaction efficiency. When exploring disease paradigms that are powerful stimulators of gene expression, such as cerebral ischemia, the tendency is to highlight vast gene sets that are up-regulated and differentially expressed. Given the large number of genes identified, it is difficult to confirm all differentially expressed genes and false-positive and false-negative differential gene expression becomes an issue. False-positives can be broadly defined as genes whose differential expression is not subsequently confirmed by an independent study (that is, by RT-PCR, Northern analysis, in situ hybridization, and so on). In contrast, false negatives are genes that are, in fact, differentially expressed, but are not detected as such by the complex assay used (for example, subtractive hybridisation, RDA, DNA microarray, and so on).
To manage these issues, we have used the strategy of using multiple assays of differential expression on the same RNA pool and then cross-validating all of the numerous differentially expressed products between assays. Confidence in particular products can then be increased by identifying commonalties in expression across assays. This approach is represented in Fig. 2, where targets are listed according to their detection across several assays of differential expression. Genes identified across all three assays (SSH, RDA, and gridding) are prioritized first, as they are associated with high levels of confidence. A table of descending confidence in hits can be constructed, which could include products such as TIMP-1, MCP-1, and hsp-27, prioritized first for subsequent levels of confirmation (for example, Taqman RT-PCR, protein expression). This technique for handling large numbers of “hits” circumvents (that is, avoids) issues of biasing the identification of differential expression to a single assay and also interassay variability. Subsequent analysis by Taqman RT-PCR confirmed that the robust “hits” (that is, differential gene expression identified across all assays) had particularly high fold increases in expression versus naïve rats (for example, MCP-1 were high at 603-fold), whereas other lower fold increases were also still robust “hits,” including HSP-70 at 20.9-fold, GFAP at 18.1-fold, and VGF at 7.3-fold versus naïve. Figure 2 also highlights that this strategy has been particularly useful for identifying false negatives (that is, products differentially expressed but not detected as such in assays). For example, in our studies, GFAP was identified to be up-regulated in stroke by gridding, whereas SOCS-3 was identified by subtractive hybridization, but by harnessing both techniques in a unified approach we were able to confirm (that is, using TaqMan RT-PCR) that both of these genes indeed did accumulate in the ischemic cortex after pMCAO. This “complimentary techniques” approach at target validation maximizes the coverage of differential gene expression by minimizing the losses caused by the technical vagaries of any single technique.
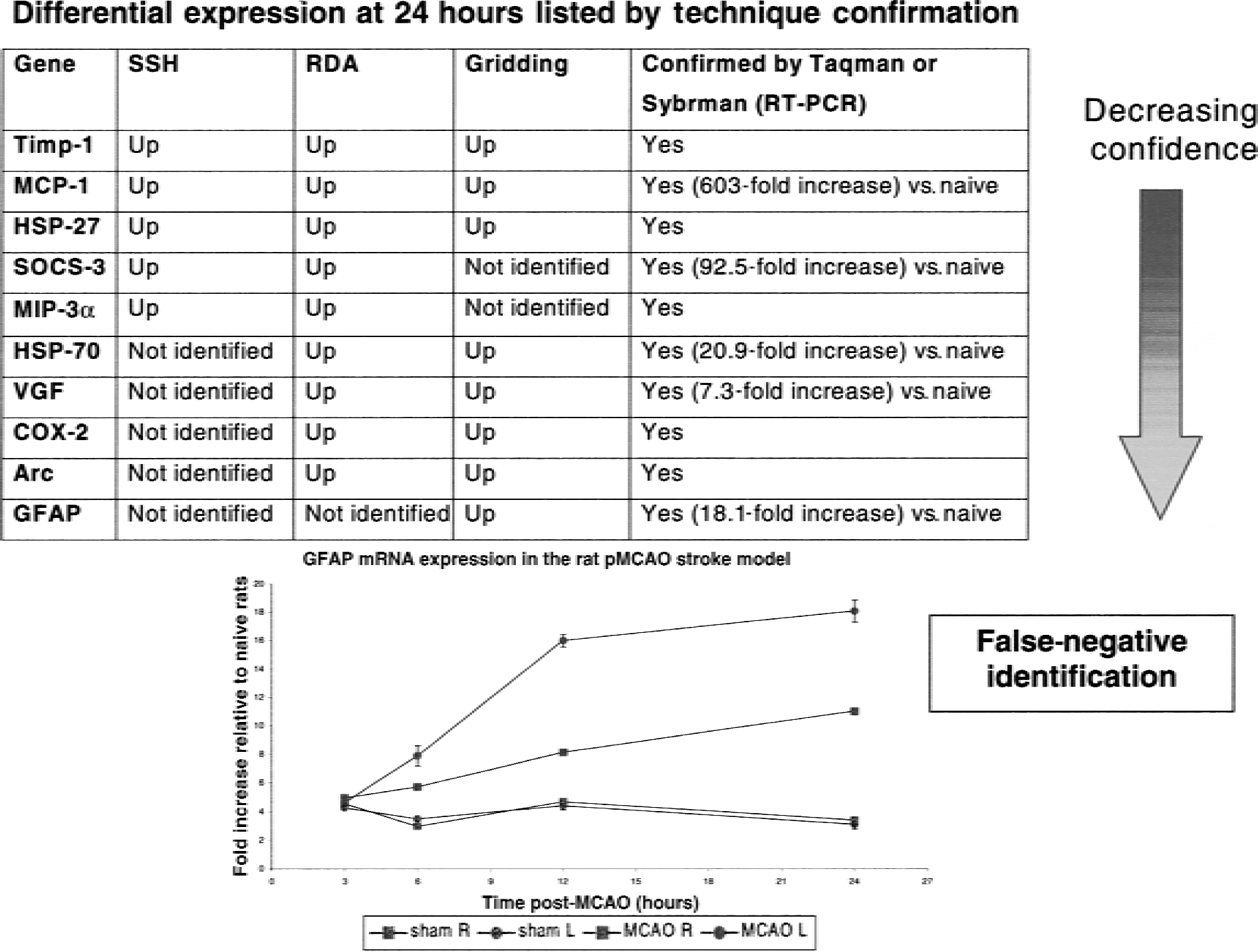
Example of prioritization of mRNA “fishing” hits before subsequent confirmation. Commonalities are identified across each assay of differential expression with greater levels of confidence assigned to “hits” highlighted with all techniques (subtractive hybridization (SSH), representational difference analysis (RDA), and gridding). This strategy is particularly useful for achieving the maximum breadth of fishing and highlighting false-negatives. In this example, glial fibrillary acidic protein (GFAP) was not identified by RDA or SSH, but microarray grids did highlight a differential expression. Subsequent analysis by reverse transcription-polymerase chain reaction (RT-PCR) demonstrated elevated expression in ipsilateral ischemic cortex (MCAO L) as compared with the contralateral control cortex (MCAO R) and with sham-operated cortices (SHAM L and R). HSP, heat shock protein; COX, cyclooxygenase; MCAO, middle cerebral artery occlusion.
Confirmation: an integral part of differential gene expression analysis
The techniques cited above for the identification of differentially expressed mRNA represent one of several starting points for an integrated approach to the study of gene expression in stroke models. All data derived by these methods require confirmation in an independent study to remove false positives, and this usually forms part of a broader analysis of expression of the gene that has been identified (Soriano et al., 2000; Wang et al., 1995b, 1998a, d ).
Traditional methods for analyzing gene expression include techniques such as Northern blotting, RNAse protection assay, in situ hybridization, and semiquantitative RT-PCR. All of these methodologies have been used on numerous occasions to study the expression of individual genes or small groups of genes in stroke models. However, the differential screening methodologies ideally generate large numbers of hits that require rapid confirmation in a higher throughput system.
Recently, real-time quantitative RT-PCR techniques have become available, such as the use of Taqman probes or SYBR green (Gibson et al., 1996), to monitor an accumulating PCR product in real time, hence allowing an accurate comparison of initial PCR template numbers. These assays can be performed in 96-or 384-well format and are highly amenable to the use of robots, reducing operator time and error. With these new techniques, it is possible to perform rapid confirmation of many differentially expressed genes simultaneously or to undertake a detailed expression analysis of a single hit (Medhurst et al., 2000). Taqman RT-PCR analysis has been extensively applied to the temporal profiling of caspase expression after MCAO in rats (Harrison et al., 2000c, 2001), and SYBR green has been used to confirm differentially expressed genes identified by RDA (Bates et al., 2001). Glial fibrillary acidic protein up-regulation after MCAO, identified as differential expression identified by microarray gridding, was confirmed by TaqMan RT-PCR (Fig. 2). Figure 2 shows the temporal profile of GFAP expression after MCAO (ipsilateral and contralateral cortices) and sham-operated animals (ipsilateral and contralateral cortices). In the ipsilateral cortex, GFAP expression increases over time to 24 hours at which time expression is 18.1-fold greater than in naïve animals.
Taqman methodology is a PCR-based technique that is more sensitive than other confirmatory technologies such as Northern blotting. Typically, Taqman RT-PCR uses approximately 50 ng total RNA per gene, whereas Northern blotting uses 10 to 20 μg. In addition, Taqman RT-PCR is as sensitive as in situ hybridization, with the added advantage of higher throughput. Perhaps most importantly for disease paradigms such as MCAO, where gene expression can exceed 600-fold over that observed in naïve animals (for example, MCP-1), Taqman PCR can quantitate gene expression over 5 to 6 orders of magnitude without multiple dilution series as necessitated by other assays (Medhurst et al., 2000). Clearly, PCR-based technologies such as Taqman RT-PCR and SYBR Green RT-PCR, although in their infancy in application to the study of cerebral ischemia (Bates et al., 2001; Harrison et al., 2000a, b , 2001), offer many advantages versus other assays for confirmation and expansion of data on differential gene expression.
These techniques are also of value in testing hypotheses about genes already known to be regulated in stroke models or where differential expression is suggested by other biologic evidence. The sensitivity of PCR-based methodologies suggests that sufficient RNA can be isolated from a single animal to allow the simultaneous assessment of several hundred genes. Thus, a large body of data can be gathered and the expression of many different genes can be compared in a single study without drawbacks such as variation between studies, operators, and cohorts of animals. The major drawback of high throughput quantitative RT-PCR is that although it allows for the rapid assessment of changes in gene expression at the level of mRNA, it is not able to provide information on the precise cellular localization of such changes. In the case of the Longa et al. (1989), stroke model that has been extensively studied in our laboratories, the cell-type and intracellular location(s) of changes in gene expression are likely to be invaluable. Neurones and oligodendrocytes die within the ischemic infarct (Bartus et al., 1995; Mandai et al., 1997), particularly after 12 hours of ischemia. Astrocytes and microglia are decreased in number in the core region of the lesion and proliferation of both of these cell types occurs in the marginal areas (Davies et al., 1998).
Polymorphonuclear leukocytes, and later macrophages, invade the lesion after approximately 12 hours and for days after (Clark et al., 1993; Davies et al., 1998; Kochanek and Hallenbeck, 1992). Changes in gene expression have to be understood in the context of the evolving cell types present at the time. Ultimately, this has to involve techniques, such as in situ hybridization and immunohistochemistry, which allow the localization of expression to be viewed in relation to the structure of the evolving lesion and the identification of the types of cells in which expression is occurring.
Confirmation and localization of protein expression: transcription to translation verification
Confirmation of protein expression and time course of translation is of primary importance. This is especially pertinent given the severe energy compromise in the ischemic brain. During this state, transcription and translation can become uncoupled because of the energetic demand of assembling functional protein. This will be temporally and spatially dependent, and has been extensively reviewed by Koistinaho and Hokfelt (1997) and Sharp et al. (2000).
Uncoupling of mRNA transcription and protein translation after pMCAO using the technique of Longa et al. (1989) is shown in Fig. 3. From previous and current data, it is apparent that both IL-1β (Liu et al., 1993b; Butinni et al., 1994; Wang et al., 1994) and IL-6 (Wang et al., 1995d) show an early increase in mRNA levels in ischemic versus sham-operated animals, 3 and 6 hours post-MCAO. However, protein determination by ELISA at 6 hours post-pMCAO fails to confirm a concomitant increase in IL-1β protein, although IL-6 protein levels are significantly elevated in ischemic animals at this time point, as expected. Issues associated with the sensitivity of protein detection assays must be considered for some proteins. However, from a family of related proteins, sampled from an identical cortical region, with an apparently similar mRNA expression profile over the first 6 hours post-pMCAO, completely opposing protein profiles have been obtained. An example of both message and protein up-regulation like that seen for IL-6 (that is, coupling of transcription and translation) can also be provided for tumor necrosis factor-α (TNF-α). TNF-α message and protein expression (that is, protein detected by the very sensitive immunohistochemical technique) can be detected at a similar and early time points poststroke (Butinni et al., 1996; Liu et al., 1994) (see Table 4). With the advent of the sequencing of the full human genome (Genome International Sequencing Consortium, 2001), we now find that only approximately 30,000 genes produce the much more copious and diverse number of proteins. Ultimately, it is proteins that are pivotal in cellular function; thus, proteomic analysis in addition to mRNA analysis points the way ahead.
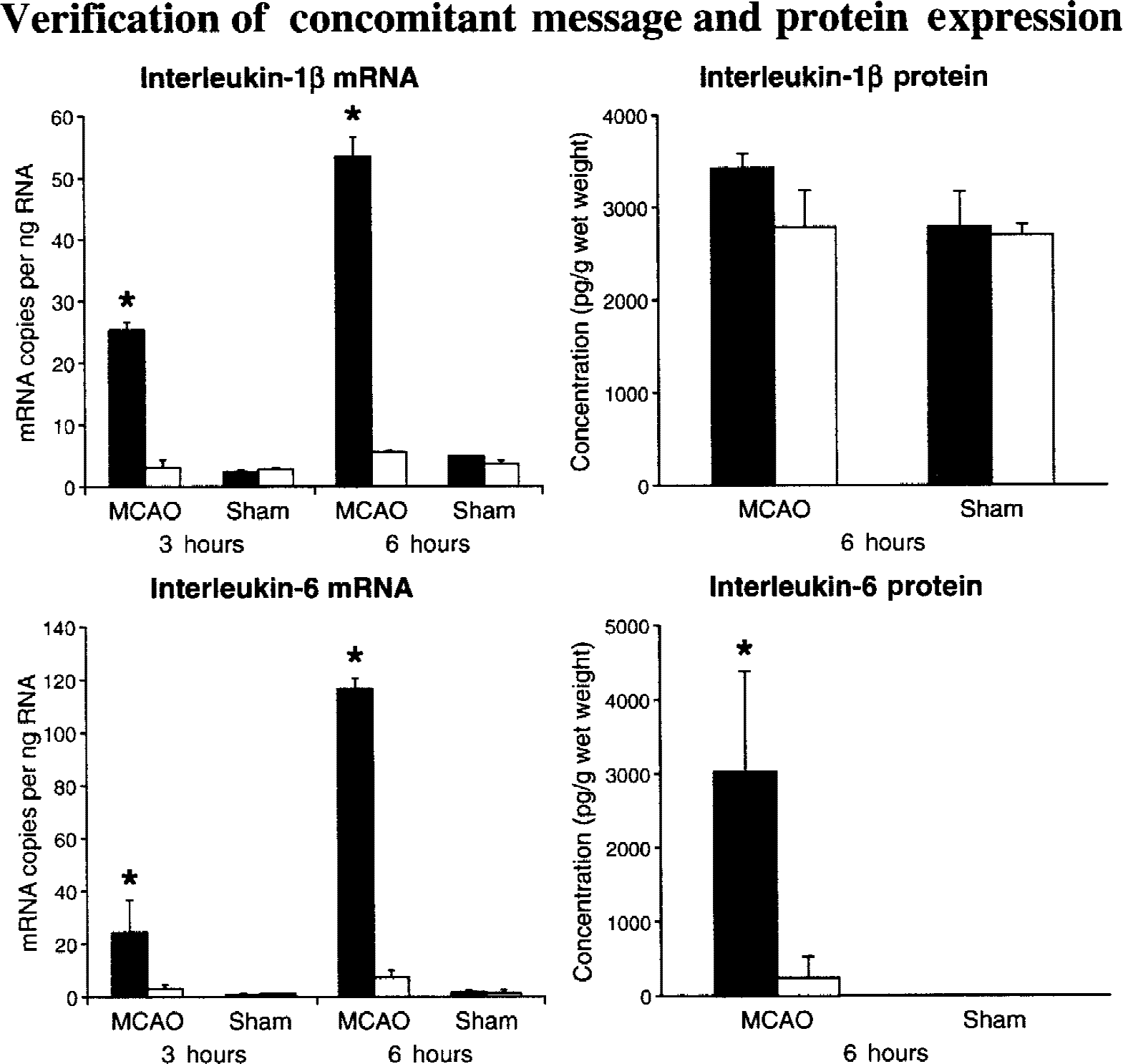
Uncoupling of transcription and translation can occur in stroke and disease, therefore, message expression information alone is not adequate. Confirmation of protein expression and time course of translation is of primary importance. During ischemic compromise, transcription and translation can become uncoupled because of the energetic demand of assembly of functional protein. After permanent occlusion of the middle cerebral artery (pMCAO) in normotensive Sprague-Dawley rats (by the “thread” method of Longa et al., 1989), TaqMan reverse transcription-polymerase chain reaction analysis of IL-1β and IL-6 mRNA shows an increase in mRNA levels in ischemic relative to sham-operated animals at 3 and 6 hours post-pMCAO. However, protein determination by ELISA at 6 hours post-pMCAO fails to confirm a concomitant increase in IL-1β protein (that is, not translated to protein within a similar time frame), although IL-6 translation was verified with measured protein levels being significantly elevated at 6 hours. Solid bars represent ischemic cortex mean ± SE. Open bars represent the contralateral control cortex mean ± SE. Bars represent the data from 5 to 6 rats. * P < 0.05, statistically significant difference as compared with the contralateral control cortex (analysis of variance, then Dunnett's test).
Functional studies, transgenic studies, and in vivo pharmacology
There are already many examples from the literature where transgenic animal studies, or pharmacologic studies, or both, have followed up or coincided with gene expression studies to demonstrate involvement of specific gene expression in focal stroke injury or protection. For example, COX-2–deficient mice exhibit reduced susceptibility to brain injury (Iadecola et al., 2001). Notably, IL-1ra was shown to be neuroprotective in brain injury (Relton and Rothwell, 1992) long before the altered expression of the IL-1 system in stroke was demonstrated (Wang et al., 1997). Also, IL-6 has been shown to be neuroprotective in stroke (Ali et al., 2000; Loddick et al., 1998). In addition, blocking thyrotropin-releasing hormone provides significant protection against ischemic brain damage and associated neurologic deficits (Borlongan et al., 1999; Yonemori et al., 2000). Finally, poststroke treatment with brain-derived nerve growth factor (BDNF) reduces brain injury to pMCAO (Yamashita et al., 1997). Genes for all of these proteins have been shown to be up-regulated in stroke models. These studies demonstrate the full circle and cross-validation of differential gene expression technology that is necessary in this research, and are directly relevant to the discussions on new directions and models provided below.
ADDITIONAL MODELS FOR PROBING DIFFERENTIAL GENE EXPRESSION
Neuroprotective and neurodestructive gene expression
As pointed out previously (Barone and Feuerstein, 1999), focal ischemia is a powerful stimulus to elicit responses in the brain in the form of multiple gene expression changes. Focal ischemia is a powerful reformatting and reprogramming stimulus for the brain. There are broad and robust gene expression responses that occur after focal stroke that are exhibited as temporal episodes or “waves” of expression of different groups of genes (Barone and Feuerstein, 1999). These “waves” are largely composed of increased expression of inflammatory cytokines, including IL-6 and IL-1ra. In addition, growth factors (for example, BDNF) that might be expected to play a neuroprotective role after stroke also increase in this time frame. The increased cytokine gene expression appears to drive leukocyte infiltration, a poststroke brain response to injury, and is associated with secondary brain injury and repair processes after stroke. Later waves of new gene expression include mediators that appear to be important in tissue remodeling (that is, resolution of ischemic tissue injury) and perhaps recovery of function. These issues are important in relation to the models suggested below for future differential gene expression analysis.
Preconditioning stress in brain tolerance strategies
A short ischemic event can result in subsequent ischemic tolerance, a resistance to severe ischemic tissue injury. This phenomenon, known as ischemic preconditioning, has been described in several organs, especially brain and heart, and may represent a fundamental protective response to injury after previous stress (Kitagawa et al., 1990, 1991; Yellon et al., 1998; Lawson and Downey, 1993). Ischemic preconditioning is a powerful inducer of ischemic brain tolerance to severe stroke as reflected by preservation of brain tissue and motor function for up to 7 days after the initial preconditioning stimulus (Barone et al., 1998). Ischemic tolerance is dependent on de novo protein synthesis at the preconditioned brain site, which contributes to the neuroprotection. These models of brain protection provide an opportunity to identify novel protective gene expression associated with the development of brain tolerance. It has been suggested that neurotrophic factors, stress proteins, and cytokines contribute to the tolerance response to ischemia and other forms of stress to the brain (Barone et al., 1998; Currie et al., 2000; Yanamoto et al., 2000; Chen et al., 1996; Chen and Simon, 1997; Wang et al., 2000a, b ). For example, ischemic tolerance is associated with increased expression of the neuroprotective protein, IL-1ra, and a reduced postischemic expression of the early response genes, c-fos and zif268. These brain tolerance models are amenable to the differential gene expression approaches described above. For example, Wang et al. (1998d) applied the suppressive subtractive hybridization methodology to discover genes responsible for ischemic tolerance after preconditioning. Using suppressive subtractive hybridization, TIMP-1 was identified. Northern analysis confirmed that TIMP-1 mRNA was significantly elevated at 24 hours and 2 days after preconditioning, which corresponded well to the onset of ischemic tolerance.
Later gene changes: strategies in brain recovery, plasticity, and recovery of function
Although initially neurologic functional deficits occur after stroke, there is a significant recovery of brain function that both occurs spontaneously and improves with training after stroke (Barone and Feuerstein, 1999; Rossini et al., 1999a, b ; Dobkin, 2000; Hunter et al., 2000). Sampling tissue during functional brain recovery (that is, at later time periods poststroke) might be expected to provide an opportunity to identify novel genes important for brain regeneration or plasticity. These would be amenable to the differential gene expression analyses, but would be profiled at later poststroke time points or under treatment conditions shown to facilitate such brain regeneration and recovery.
Gene therapy
In recent years, the emerging technology of gene therapy has provided some insights into the use of in vivo gene transfer. Therapeutic neovascularization for ischemic diseases has been particularly encouraging. In animal models, angiogenesis and improved outcome in ischemic tissues has been produced by intramuscular injections of plasmid or adenoviral vectors encoding vascular endothelial growth factor (VEGF) (Byun et al., 2001; Gowdak et al., 2000). Similarly, in clinical trials, VEGF gene transfer augments the population of circulating endothelial progenitor cells and transiently increases plasma levels of VEGF (Kalka et al., 2000). Furthermore, myocardial transfer of naked plasmid DNA phVEGF (165) has been found to augment perfusion of ischemic myocardium and reduce the size of defects documented at rest by single-photon emission computed tomography imaging (Vale et al., 2000). Ex vivo gene transfer, using the modification of cultured cells and subsequent implantation into a host organism, is a proven strategy for recovery from central nervous system injury. Recovery from long-term rodent hemiparkinsonism by implantation of tyrosine transfected myoblasts after 6-OHDA lesioning was found to improve behavioral deficit for up to 13 months (Cao et al., 2000).
In vivo gene transfer, the delivery of a gene directly to recipient somatic cells, has also been explored for neuroprotection and recovery from central nervous system injury. The delivery of the protooncogene bcl-2 has been examined in gerbil models using adeno-associated virus vectors. Transduction both pre-and postforebrain ischemia was found to prevent DNA fragmentation in hippocampal CA1 neurones, commonly associated with cell death induced by ischemia (Shimizaki et al., 2000). The adenoviral transfection of the endogenous cytokine antagonist, IL-1ra, has also demonstrated neuroprotection in transient focal cerebral ischemia and reperfusion in the mouse (Yang et al., 1997, 1999). In addition, the neuroprotective potential of HSP70 on brain injury, including that produced by brain ischemia, has been demonstrated. Transgenic animals and gene transfer technology used to overexpress HSP70 clearly protects the brain (Yenari et al., 1998, 1999).
THE ELUSIVENESS OF NOVEL GENE TARGETS AND THE FUTURE
Initially, the movement to discover differential gene expression in brain destruction or protection was believed to be driven by the novel or unknown genes that would provide pathways to completely novel and proprietary therapeutics. Although this may be true, the complexity of understanding and applying resources to gene fragments in the hopes of ultimately reaching this therapeutic nirvana has not yet occurred. We have identified several potentially novel genes that have been differentially expressed in ischemic or tolerant brain tissue but have not made further progress with these to date. Basically, the odds for success are much better for the pursuit of known genes as therapeutic targets, as resourcing can be based on a solid biologic rationale. Unknown genes often may not provide a significant rationale to move forward without the full-length gene in the current resource restricted research environment. Many other factors can cause investing resources into work on unknown genes costly, risky, and difficult to pursue. Some of these include the absence of any understanding of an identifiable function for the unknown protein, and if it is, in fact, associated with a novel gene, and lack of any information that the novel gene/protein has any involvement in the pathophysiology of stroke. In spite of all this, it is clear that among these potential unknowns clearly are the potential novel therapeutic targets of the future. As such, novel and unknown genes might be a potential site of collaboration between the academic and industrial laboratories, thus providing potential novel gene products that can be evaluated for tissue distribution, function, and relevance in tissue injury and protection.
Clearly the methodology is currently available to identify differential gene expression in stroke in addition to other conditions of pathophysiology and disease. However, this methodology needs to be used with the caveats discussed above. If one does operate cautiously as we have suggested, there is the potential for many significant opportunities for biologic discovery, not only related to stroke but in many other conditions such as end organ failure in various cardiovascular diseases, in areas such as oncology, and perhaps extending further to other complex problems such as substance abuse, tolerance, addiction, and drug dependency.
Footnotes
Acknowledgments:
The authors thank Sue Tirri for assisting in the preparation of this review, and Dr. Andy Medhurst for his helpful discussions on differential expression technology.