
Abstract
The authors used mRNA differential display to identify genes whose expression levels are altered in the adult rat hippocampus 24 hours after global ischemia. At this time after challenge, the basic helix-loop-helix transcription factor, SEF-2, and the 26S proteasome complex subunit, p112, were identified as genes whose expression levels are decreased and increased, respectively, in the hippocampus. To determine the spatial and temporal patterns of expression change for each gene, the authors antisense in situ hybridization to paired brain sections of sham-operated and global ischemia-challenged rats at 6, 12, and 24 hours after reperfusion SEF-2 expression was not significantly altered from that of sham-operated controls in any hippocampal subfield at or before 12 hours after challenge. At 24 hours after ischemia, however, SEF-2 expression levels were significantly diminished in the vulnerable CA1 subfield, but not in the less vulnerable CA3 or dentate granule cell subfields. The proteasome p112 subunit gene displayed no change in expression levels at 6 hours after insult; however, an elevated expression was observed at 12 hours after challenge in the dentate granule cell subfield. By 24 hours after challenge, p112 expression was significantly elevated in both the CA1 and dentate granule cell subfields. These results demonstrate that a member of the basic helix-loop-helix family of transcription factors, SEF-2, and the major subunit of the 26S proteasome complex, p112, display altered gene expression in the hippocampus after transient cerebral ischemia.
Brief episodes of transient cerebral ischemia are known to initiate cellular responses that lead to the delayed neuronal death of several neuronal populations in the brain (Pulsinelli et al., 1982; Petito et al., 1987). The CA1 subfield of the hippocampus represents one such population of neurons that is highly vulnerable to global ischemia challenge in both experimental animals and in humans (Kirino, 1982; Pulsinelli et al., 1982; Petito et al., 1987). Accumulating evidence suggests that neuronal damage after the ischemic period likely involves an over-stimulation of Ca2+ ion-permeant glutamate receptors (Benveniste et al., 1988; Desphande et al., 1987; Dienel, 1984; Gorter et al., 1997), leading to acute alterations in neuronal Ca2+ ion homeostasis (reviewed in Choi, 1988, 1994; MacManus and Linnik, 1997). What remains unclear, however, is how Ca2+ ions initiate neurodegenerative pathways within specific vulnerable neurons. Unlike focal ischemia, in which the challenged neurons die rapidly after insult, the sensitive CA1 hippocampal neurons do not immediately die after a transient global ischemia challenge. Rather, these cells initially recover, and only later degenerate (Desphande et al., 1987; Meldrum and Garthwaite, 1990; Pulsinelli et al., 1982). Owing to the delayed nature through which these vulnerable hippocampal neurons die, it has been proposed that alterations in gene expression may either contribute to, or reinforce, the initial expiration signals arising from the global ischemia challenge (Akins et al., 1996; MacManus and Linnik, 1997). This possibility has been supported by reports demonstrating that the postischemic CA1 neurons display hallmarks consistent with apoptosis (Nitatori et al., 1993; Krajewski et al., 1995; MacManus et al., 1993; Okamoto et al., 1993; Heron et al., 1993; Kihara et al., 1994; Honkaniemi et al., 1996; Chen et al., 1996), a cell death cascade in which alterations in the complement of expressed genes are often observed preceding cell death (reviewed in Thompson, 1995; White, 1996).
The link between the Ca2+ ion influx that results after global ischemia and the commitment of the vulnerable hippocampal neurons to cell death has remained elusive. One group of proteins that become activated after ischemia, either directly or indirectly by Ca2+ ions, is protein kinases (Enslen and Soderling, 1994; Enslen et al., 1994, 1995; Hu and Weiloch, 1993, 1994; Finkbeiner and Greenberg, 1997). It is likely that the inappropriate activation of these kinases is involved in the delayed neuronal death of the CA1 neurons, as inhibitors of protein kinases have proven to be neuroprotective in global ischemia (Kindy, 1993). Activation of protein kinases promotes a wide range of effects within a cell that often leads to changes in cellular gene expression (Enslen et al., 1994; Enslen and Soderling, 1994). Because inappropriate Ca2+ ion influx after global ischemia could lead to an alteration in the complement or levels of genes expressed in the vulnerable neurons of the hippocampus, we and others have examined the dynamics of gene expression after global ischemia for several candidate neurotransmitter receptor genes whose altered expression could affect the viability of the sensitive neurons (Pellegrini-Giampietro et al., 1992, 1994; Hsu et al., 1996, 1998; Zhang et al., 1997). The examination of such candidate genes, however, is limited by the availability of defined genes for investigation and may, therefore, be biased toward specific systems.
The technique of mRNA differential display represents one method by which the population of expressed genes in two or more samples may be compared (Liang and Pardee, 1992). This procedure has been used previously for the identification of the zinc transporter gene (ZnT-1) as an upregulated message in the gerbil hippocampus after ischemia (Tsuda et al., 1997). Differential display produces a fingerprint of genes that are expressed in an individual tissue. A comparison of fingerprint differences between two samples indicates the likelihood that differentially expressed genes exist between the two samples. We have compared the fingerprints of sham-operated animals and 24-hour postischemic animals to identify such genes with potentially altered expression levels at this time after challenge. In this report, we concentrate on two genes identified as having altered expression levels at 24 hours after global ischemia-one predicted to have decreased expression levels, and one predicted to have increased expression levels. These genes, the basic helix-loop-helix (bHLH) transcription factor SEF-2 and the major proteasome complex subunit gene p112, respectively, are the first differential display-targeted genes to be examined in our rat model, and we show here that their expression levels are significantly altered from those of sham-operated control animals in the vulnerable CA1 neurons at 24 hours after ischemic challenge.
MATERIALS AND METHODS
Animal model
Animal experimentation was conducted only after full review and approval of the proposed experiments by local animal care committees in accordance with guidelines established by the Medical Research Council of Canada and the Canadian Council on Animal Care. The four-vessel occlusion ischemia model was performed using a modified version of the method of Pulsinelli and Brierley (1979), as we have described in detail previously (Shinno et al., 1997; Zhang et al., 1997; Hsu et al., 1998). Briefly, male Wistar rats weighing between 200 and 300 g were anesthetized with 1.5% halothane in a 70%:30% N2O and O2 mixture. Rectal temperature was monitored throughout the surgery, and temperature was maintained at 37.5°C using a heat lamp. Bilateral alar foramina of the first cervical vertebrae were exposed, and both vertebral arteries were permanently occluded through electrocauterization. On the second day, anesthesia was induced as on day 1, and bitemporal subdermal EEG needle electrodes were placed in reference to a frontal subdermal electrode. After establishing a baseline EEG level, both common carotid arteries were reexposed and occluded with aneurysm clips for 15 minutes. Animals were included for our studies only if a flat bitemporal EEG was maintained during the duration of the carotid occlusion and a dilated pupil was observed throughout the ischemia in the absence of a lid reflex with light stimulation. Animals that did not meet these criteria were excluded from the study. Sham-operated control animals received the same procedure and anesthesia as test animals but did not experience either electrocauterization of the vertebral arteries or transient carotid occlusion. Each set of animals received the same degree of surgical preparation and recovery paradigms to minimize variations that could result from surgical procedures.
mRNA differential display
Differential mRNA display was carried out as previously described (Liang and Pardee, 1992; Liang et al., 1994) using a kit from Genhunter Corp. (Nashville, TN, U.S.A.). Total cellular RNA was prepared from isolated hippocampi by guanidinium thiocyanate and phenol-chloroform extraction (Sambrook et al., 1989). RNA samples were DNase treated for 15 minutes at 37°C to remove traces of contaminating genomic DNA. Reverse transcription of RNA was performed using poly-T12MN (T12MA, T12MC, T12MG, and T12MT). The resulting cDNA were amplified by polymerase chain reaction (PCR) in the presence of 35S-dATP and Taq DNA polymerase (Pharmacia, Baie d'Urfé, Quebec, Canada), using arbitrary decamers as 5′ primers and the corresponding T12MN as the 3′ primer. Samples were denatured at 94°C for 35 seconds, annealed at 40°C for 120 seconds, and extended at 72°C for 35 seconds, for a total of 40 cycles. Radiolabeled PCR products were electrophoresed on a denaturing 6% polyacrylamide sequencing gel. Select bands that revealed differences in the intensity of the observed PCR products from challenged subjects were recovered from the sequencing gels and reamplified with the appropriate primer sets for a further 40 cycles, in the absence of radioisotope.
Cloning and sequence analysis
Amplified products were electrophoresed through a 1.5% low melting point agarose gel, and the fragments were purified using agarase (New England Biolabs, Mississauga, Ontario, Canada). The primer pairs used for the amplification of SEF-2 were T12MC with AGCCAGCGAA (Genhunter primer AP-1 [Nashville, TN, U.S.A.]), and T12MT with AGCCAGCGAA (Genhunter primer AP-1) for p112. Briefly, the DNA fragment was excised from the agarose gel, and agarase buffer was added. Samples were heated to 70°C for 10 minutes, and after cooling to 40°C, 1.5 U of agarase was added, and the samples were incubated for 1 hour. Samples were then cooled on ice for 10 minutes and centrifuged at 14,000 g for 5 minutes to pellet insoluble material, and the DNA was precipitated with ethanol. Purified products were subcloned into the PCR-2 vector (Invitrogen, Carlsbad, CA, U.S.A.), and positive colonies selected by α-complementation. Insert sequences were obtained using dideoxy sequencing with fluorescein-labeled primers on a Pharmacia A.L.F. automated sequencer. Nucleotide sequences were compared with known sequences by searching GenBank and EMBL databases with BLAST software (Altschul et al., 1990; 1994).
In situ hybridization
Three different time frames of recovery after carotid occlusion were examined: 6, 12, and 24 hours. Each experimental time point consisted of a minimum of three animals for both sham-operated control and experimental groups. At the appropriate time after carotid occlusion or sham operation, animals were anesthetized and killed by transcardiac perfusion with a 4% paraformaldehyde solution according to the method of Simmons et al. (1989). The paired sham-operated animal was perfused on the same day as the ischemic subject for subsequent comparisons. After perfusion, the brain was removed and post-fixed at 4°C overnight in 4% paraformaldehyde plus 15% sucrose as described (Hsu et al., 1996, 1998; Zhang et al., 1997). After post-fixation on the second day, coronal brain sections of 20 μm were obtained by cryostat sectioning and mounted onto subbed polylysine-coated slides. Each slide contained sections from both a sham-operated and an experimental animal that were matched as best as possible to equivalent hippocampal planes. Sections were then processed as described (Simmons et al., 1989; Zhang et al., 1997; Hsu et al., 1998).
Data analysis
After posthybridization, slides were exposed to either XO-MAT-AR or BioMax film (Kodak; Rochester, NY, U.S.A.) for 5 to 15 days. Developed films were analyzed using the quantitative densitometry system (MCID-5.0, St. Catherines, Ontario, Canada). Pixel densities (relative optical densities) were recorded for each region of the hippocampus. The resulting values did not exceed the linear range of the film as determined by carbon 14 standard strips. Statistical evaluations were done by applying a two-tailed Student's t test to paired differences between experimental and control relative optical densities. Alterations in expression were considered to be significant if the observed P value was less than 0.025.
RESULTS
mRNA fingerprinting with differential display
The technique of mRNA differential display was used for the identification of mRNA sequences having an altered frequency of message expression in the hippocampus at 24 hours after transient global ischemia. Examples of our differential display panels are shown in Fig. 1. From these differential display reactions, both the transcription factor SEF-2 and the p112 proteasome complex were identified by 2 different differential display fragments, an observation that prompted their selection for further detailed characterization. The fragment of SEF-2 that was isolated was 212 base pairs in length, and the p112 fragment was 196 base pairs. As also seen in Fig. 1, there are several additional fragments whose differential amplification would suggest that the expression levels of the corresponding gene may also be altered by global ischemia. Efforts to identify and evaluate these fragments are under way. Of the picks isolated from the initial differential display profile, roughly 40% have proven to represent apparent false-positives, as no significant changes in expression from sham-operated animals have been observed through in situ hybridization (data not shown).
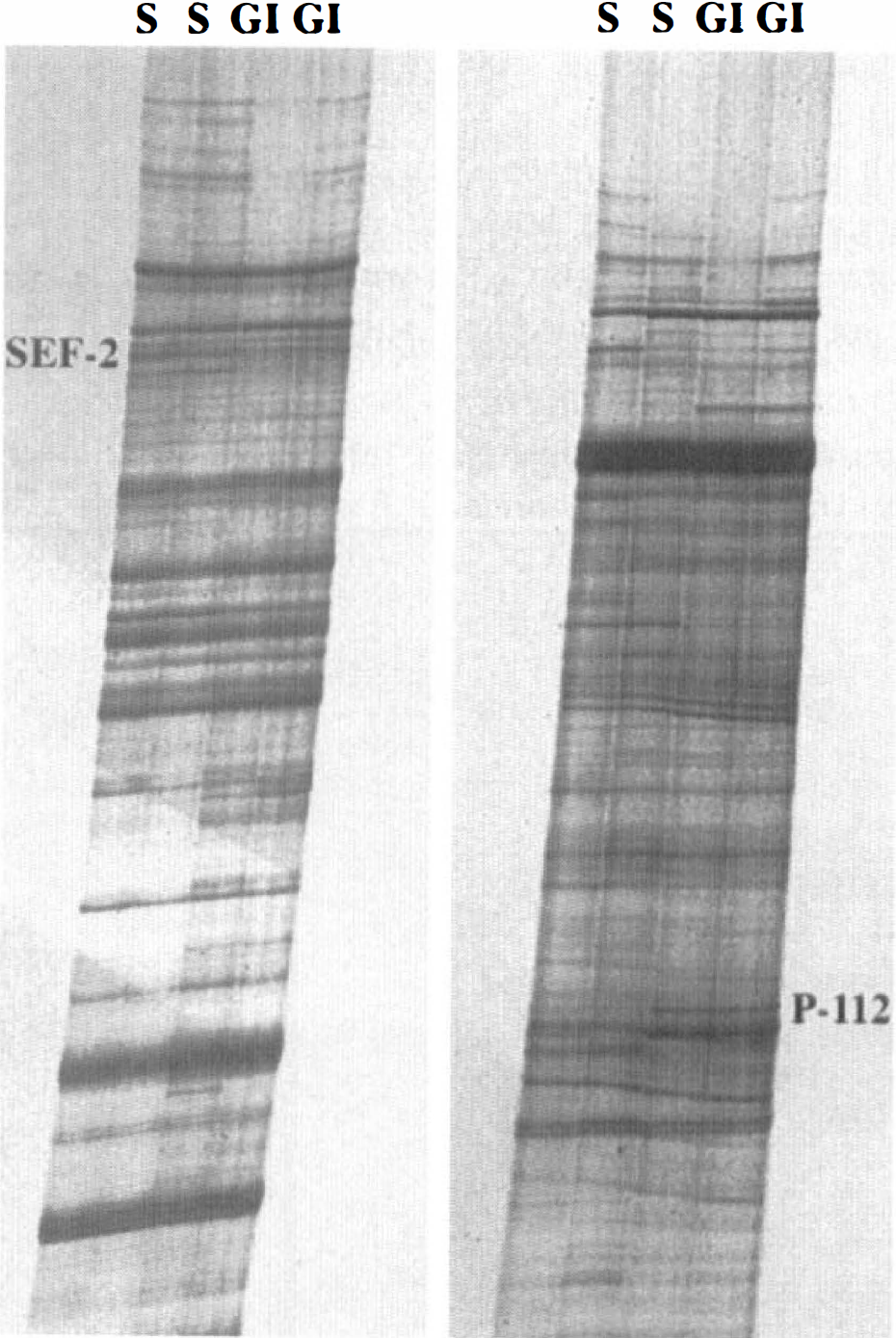
The panels illustrate the amplification results using the technique of differential display on first strand cDNA isolated from control and ischemic subjects. Each panel demonstrates the “fingerprint” pattern of amplification products generated by the polymerase chain reaction (PCR) step of the procedure, using one pair of differential display primers. Two primer pairs (of a total of 20 pairs) are illustrated. Lanes labeled “S” illustrate the hippocampal amplification patterns from two independent sham-operated control animals, while lanes labeled “GI” present the hippocampal amplification patterns from two animals that had first received 15 minutes of global ischemia and were then allowed to recover for 24 hours. SEF-2 was identified from the gel as a gene whose expression level was diminished in the hippocampus at this time after challenge, and p112 was identified as a gene whose expression was increased.
SEF-2 expression after global ischemia
Because total hippocampal mRNA was used as the template for the differential display reactions and would not selectively reveal expression changes in specific hippocampal subfields, we examined the spatial and temporal changes of hippocampal gene expression after global ischemia using antisense in situ hybridization for SEF-2 and p112. Shown in Fig. 2 are antisense in situ hybridization autoradiographs from rat brain sections taken at 6, 12, and 24 hours after global ischemic challenge, using the SEF-2 fragment. Expression levels of SEF-2 were not significantly different from sham-operated control levels in the various subfields of the hippocampus at either 6 or 12 hours after global ischemia, although a decreased trend was observed in the CA1 subfield at 12 hours after insult that failed to reach statistical significance. By 24 hours after challenge, however, a significant decrease in the level of SEF-2 expression was observed in the CA1, but not in other hippocampal subfields. Fig. 3 shows the averaged expression changes for SEF-2 from shamoperated control at these times after global ischemia.
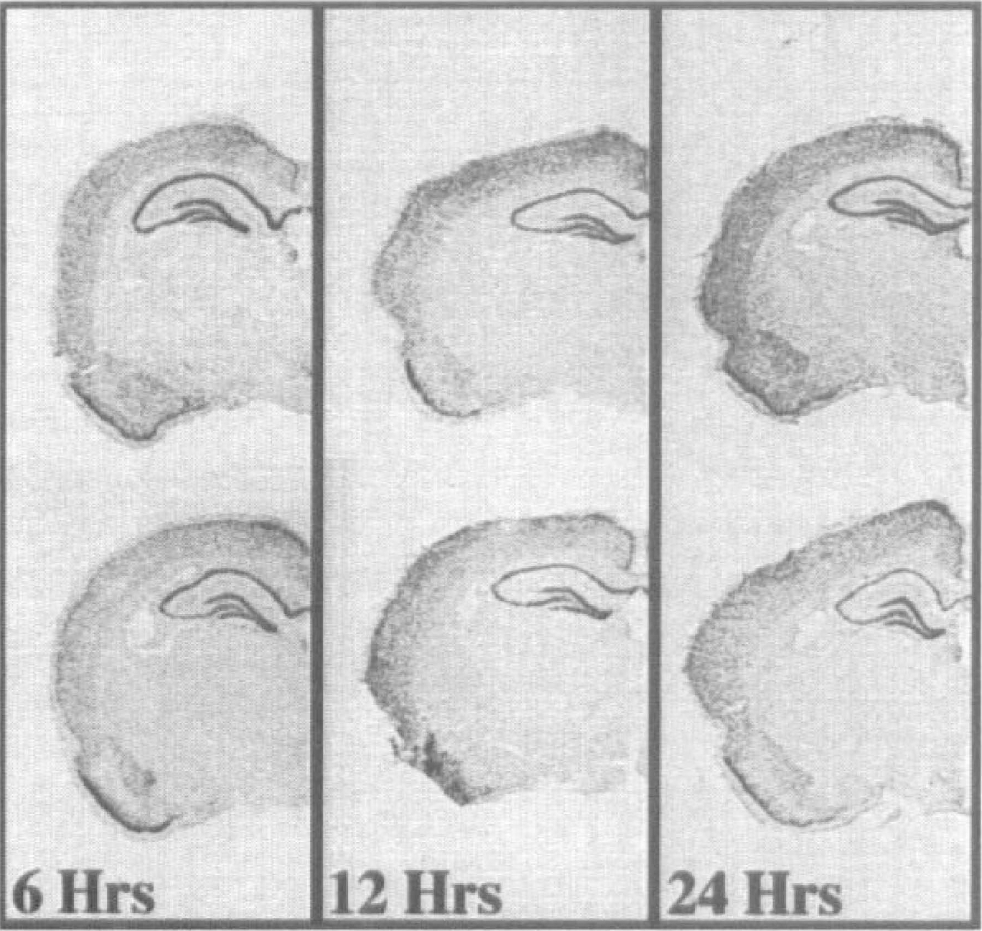
Representative examples of the expression patterns of SEF-2 at 6, 12, and 24 hours after global ischemia. For each panel, the top brain section represents the sham-operated control, and the bottom section originates from a time-matched, ischemia-challenged test animal. Antisense in situ hybridization was conducted as described in the Methods. For each panel, the sham-operated control and test brain sections were cohybridized on the same slide to avoid variability. The time after ischemia is denoted below each pair. A significant decrease in SEF-2 expression is seen at 24 hours after reperfusion in the CA1 subfield in the test animal. Although slightly diminished expression is apparent in the CA3 subfield at 24 hours after ischemia, this decreased expression did not reach statistical significance.
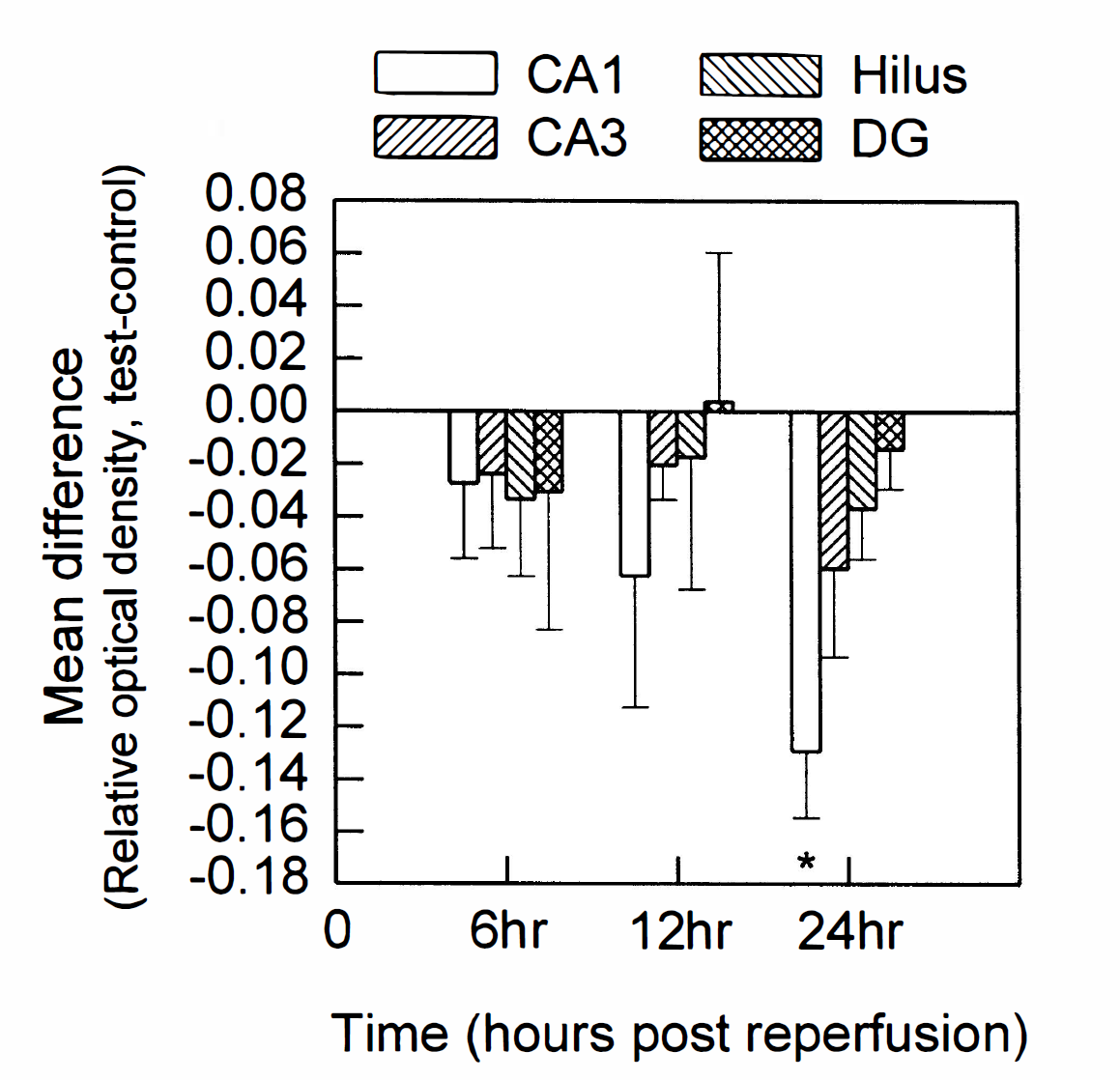
Histogram illustrating the mean and standard deviation for the densitometric analysis of the autoradiographs for anti-sense in situ hybridization with SEF-2. These data consisted of a minimum of 3 animal pairs for each time point, and at least 4 paired slide sections per animal pair. Changes in expression resulting from global ischemia are shown as increases or decreases from the control value. Statistical significance is denoted by an asterisk, at P < 0.025 (Student's paired, two-tailed t test). Hippocampal subfields: CA1, CA3, Hilus (CA4 and hilus), and DG (dentate granule cells). Times after ischemia-reperfusion are shown below the histograms.
p112 expression after global ischemia
The expression levels of the p112 gene were also found to be altered in the hippocampus after global ischemia; however, the changes were dramatically different from those observed for the SEF-2 gene. Although p112 gene expression was not altered from sham-operated control in any hippocampal subfield at 6 hours after challenge, by 12 hours the expression levels were significantly elevated in the dentate gyrus but remained near control levels in all other hippocampal subfields examined. By 24 hours after insult, p112 expression was significantly induced in both the dentate granule cell and the CA1 subfields. Although a trend toward increased expression was observed in the CA3 and hilus subfields at 24 hours after challenge, this trend did not reach statistical significance (Fig. 4). The averaged expression changes from sham-operated control for the p112 gene are illustrated in Fig. 5.
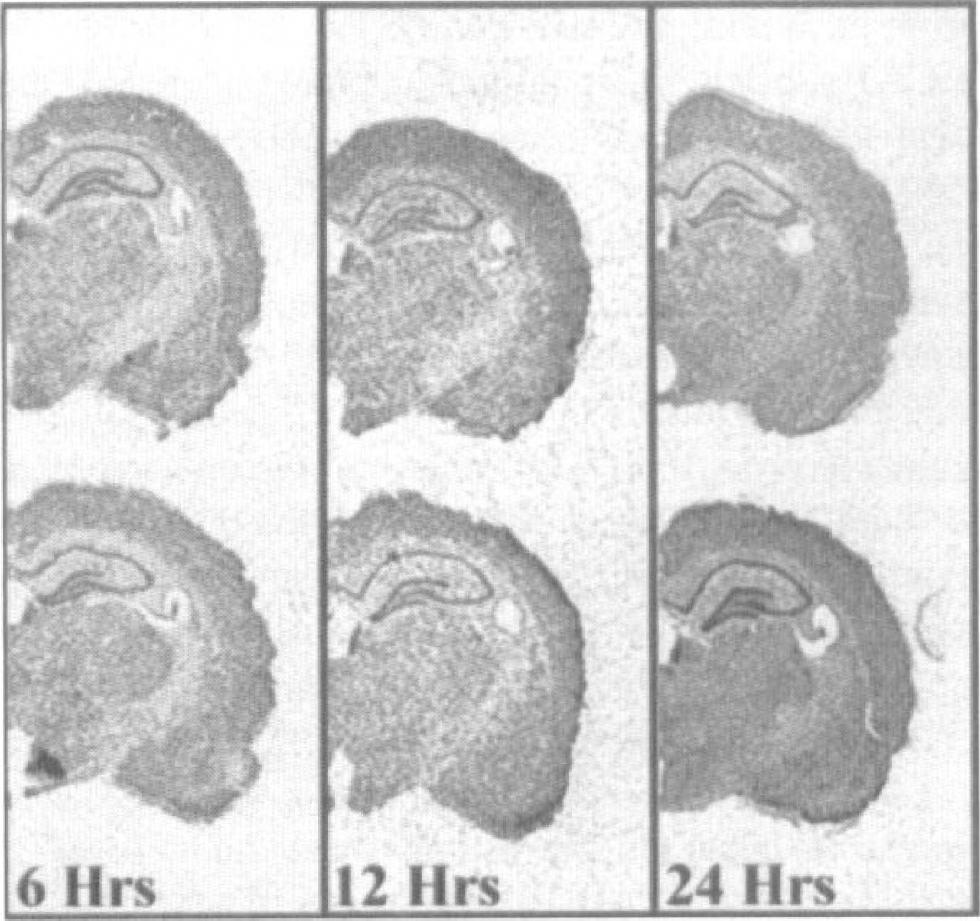
Dynamics of p112 gene expression after four-vessel cerebral ischemia in rat brain. Note the increase in expression in the 512-hour postischemic dentate gyrus, and in the CA1 and dentate gyrus subfields at 24 hours after challenge.
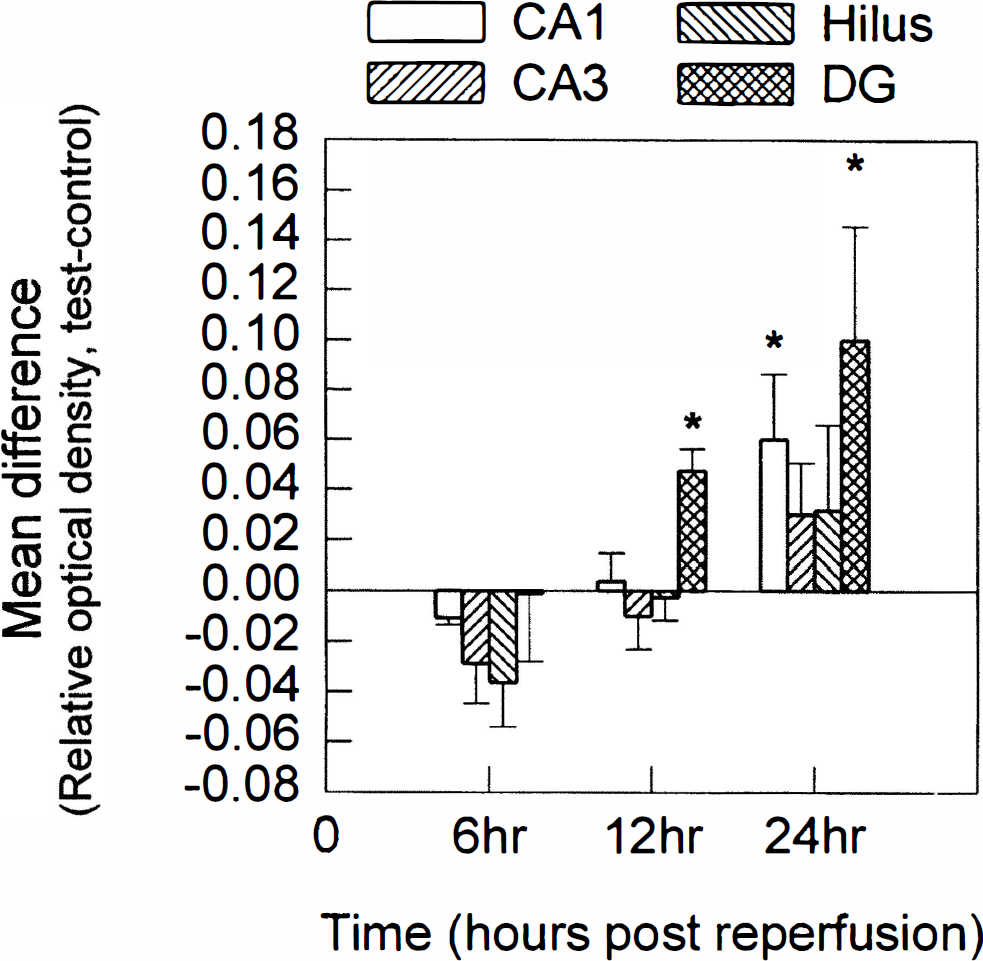
Histogram illustrating the mean and standard deviation for the densitometric analysis of the autoradiograph for antisense in situ hybridization with the p112 gene. As in Fig. 4, analysis was conducted on at least four paired sections originating from 3 to 4 animals for each time point. Statistical significance is denoted by an asterisk, at P < 0.025 (Student's paired two-tailed t test). Hippocampal subfields: CA1, CA3, Hilus (CA4 and hilus), and DG (dentate granule cells). Times after ischemia-reperfusion are shown below the histograms.
DISCUSSION
Transient global ischemia represents a challenge to the brain that initiates degenerative pathways in some neuronal populations, culminating with the death of vulnerable neurons. In our model, this neuronal death is morphologically observed under the light microscope at approximately 3 days, but not at 24 hours, after the challenge (Shinno et al., 1997; Zhang et al., 1997). Owing to the delayed nature of the neuronal expiration and reports suggesting programmed cell death systems may be involved in the course of neuronal expiration after global ischemia (Nitatori et al., 1993; Krajewski et al., 1995; MacManus et al., 1993; Okamoto et al., 1993; Heron et al., 1993; Kihara et al., 1994; Honkaniemi et al., 1996; Chen et al., 1996), we speculated that an alteration could occur in either the complement or the levels of genes being expressed in the vulnerable cells, and that these changes could contribute to the degenerative pathways. In this study, we examined the spatial and temporal hippocampal expression of two genes-SEF-2 and p112-whose expression levels were suggested, by mRNA differential display analysis, to be altered in the hippocampus at 24 hours after insult. We chose to perform differential display at 24 hours after insult to improve the likelihood of finding genes involved in the pathologic processes of CA1 degeneration, as the morphology of the CA1 neurons is maintained at this time after insult, and to minimize the potential contribution of nonneuronal cells, such as microglia, whose presence in the 24-hour postischemic hippocampus is minimal (Gottlieb and Matute, 1997). Thus, the changes in gene expression that we observe do not likely result from either increased or decreased cell prevalence in the hippocampus at this time after challenge.
The identification, and subsequent confirmation, of induced expression levels of the proteasome complex subunit, p112, in the hippocampus after global ischemia is an observation consistent with the hypothesis that the rates of proteolytic degradation in the hippocampus are enhanced after global ischemia. The 26S proteasome, of which p112 is a subunit, is involved in the degradation of short-lived proteins, proteins with aberrant structures, and polyubiquinated proteins, and it may play a role in governing cell cycle progression (Ciechanover, 1994; Hochstrasser, 1995). The p112 gene displayed elevated expression levels in dentate granule cells at 12 hours after insult, and in both the dentate granule cells and the CA1 subfield at 24 hours after insult. Should such increases in expression lead to similar increases in translated protein, then at least one constituent of a major system involved in the processing of intracellularly degraded proteins and RNA molecules would be elevated. Because global ischemia induces certain proteases (Yokota et al., 1995; Roberts-Lewis et al., 1994; Camargo-De-Morais et al., 1996; Kinoshita et al., 1997) and cytoskeletal and nuclear protein degradation occurs in the hippocampus after global ischemia (Seubert et al., 1989; Lee et al., 1991; Roberts-Lewis et al., 1994; Harada et al., 1997), the concomitant induction of a proteolytic processing system would seem reasonable. Whether the increase in the p112 subunit represents a signal promoting enhanced proteolysis or a cellular response to compensate for increases in fragments to be processed remains to be determined. Our observation that p112 induction occurs in the less vulnerable dentate granule cells as well as in the CA1 subfield suggests that this induction is a general hippocampal response, rather than a response restricted to, or directly linked to, the expiration process of the CA1 neurons. It also remains to be determined whether other subunits of proteasome complexes are similarly induced in the hippocampus after global ischemia, as our characterization of p112 represents the first molecular investigation of this protein class after global ischemia.
Human SEF-2 was originally identified as immunoglobulin transcription factor 2 (ITF-2 or E2–2) (Henthorn et al., 1990) and is highly conserved in rat (SEF-2), mouse (ME-2), and dog (TFE). SEF-2 is a member of a large family of structurally related transcription factors, the bHLH transcription factors, which are broadly divided into three subgroups: (1) the E proteins, which display a broad expression pattern and exhibit the ability to form homodimers and heterodimers with other bHLH transcription factors; (2) specific factors involved in neurogenesis (NeuroD, neurogenin, MASH, and MATH), myogenesis (MyoD, Myf-5, myogenin, and MRF4/Myf6), and hematopoiesis (Scl/Tal-1, Tal-2, and Lyl-1); and (3) the Id factors, which form heterodimers with other bHLH factors and act to inhibit their DNA binding (Benezra et al., 1990). SEF-2 (E2–2) is a member of the E-protein class of bHLH transcription factors and is widely expressed in peripheral tissues such as heart, placenta, skeletal muscle, and lung (Pscherer et al., 1996). SEF-2 expression in the brain is more restricted, however, being highly enriched in brain regions that accommodate high levels of neuronal plasticity, and also being prevalent during development in neuronal populations that are terminally differentiating (Soosaar et al., 1994). Investigations of whether the expression levels of other members of the bHLH family are affected by global ischemia are in progress.
This report provides the first illustration that the hippocampal expression of a member of the bHLH family of transcription factors is affected by global ischemia. This finding is of particular interest, as delayed neuronal death is associated with the cell population where the decreased expression of SEF-2 was most apparent. The interaction of SEF-2 with transcriptional regulators is highly tissue specific. Of particular interest is the interaction of SEF-2 with the positive regulation of somatostatin receptor II (SST2) gene expression (Pscherer et al., 1996). Somatostatin receptor II mediates inhibitory growth signalling through association with the tyrosine phosphatase, SHP-1 (Lopez et al., 1997; Vaysse and Susini, 1997). Loss of SEF-2 function could therefore lead to a corresponding decrease in SST2. Interestingly, both immunohistochemistry and antisense in situ hybridization have demonstrated that SST2 gene expression in the CA1 subfield of the hippocampus is decreased after global ischemia (Schindler et al., 1997; Thoss et al., 1995). In the absence of sufficient growth inhibitory signals, perhaps provided or supported by a decreased prevalence of SEF-2, these differentiated CA1 cells may receive conflicting signals, attempt to reenter the cell cycle, and thus initiate programmed cell death systems as previously proposed (Finkbeiner and Greenberg; 1996). We therefore find it of particular interest that the decreased expression of SEF-2 after global ischemia is particularly pronounced in the vulnerable neurons of the hippocampus at times that precede morphologically identified cell compromise.
Footnotes
Acknowledgments
The authors thank Lucy Teves and Richard Logan for technical assistance, and Werner Muller for DNA sequencing.