
Abstract
In cultured neurons, the authors previously demonstrated that the Na+-K+-Cl− cotransporter is significantly stimulated by elevated extracellular potassium and glutamate, which are important factors in cerebral ischemic damage. These findings led the authors to hypothesize that stimulation of the cotransporter after ischemia might result in Na+, K+, and Cl− influx, and might contribute to neuron damage. In the current study, the authors investigated such a role of the Na+-K+-Cl− cotransporter in focal cerebral ischemia. Cerebral ischemia was induced by 2-hour occlusion of the left middle cerebral artery (MCA) and 24-hour reperfusion in male spontaneously hypertensive rats (SHRs). Immunocytochemical staining and immunoblotting revealed an up-regulation of expression of the cotransporter protein in neurons in cortex at 24 hours of reperfusion. Artificial cerebral spinal fluid (aCSF) or 100 μmol/L bumetanide (a cotransporter inhibitor) in aCSF were continuously microdialyzed through a microdialysis probe into left cortices throughout 2-hour MCA occlusion and 24-hour reperfusion. Compared with the aCSF-treated group, infarction volume was significantly reduced in the bumetanide-treated group (25%, P < 0.05). In addition, brain water content in the bumetanide-treated brains was decreased by 70% (P < 0.05). These results strongly suggest that the Na+-K+-Cl− cotransporter may play an important role in cerebral ischemic neuronal damage.
The electroneutral Na+-K+-Cl− cotransporter transports Na+, K+, and Cl− into cells under physiologic conditions and is characteristically inhibited by loop diuretics such as bumetanide and furosemide (Haas, 1994). To date, two isoforms of the Na+-K+-Cl− cotransporter, NKCC1 and NKCC2, have been identified by cDNA cloning. NKCC1 has a wide range of tissue distribution, whereas the NKCC2 isoform is found only in the kidney (Payne and Forbush, 1994; Delpire et al., 1996; Haas and Forbush, 1998). A high expression level of NKCC1 mRNA in the central nervous system (CNS) has been demonstrated by Northern blotting and in situ hybridization (Payne and Forbush, 1994; Delpire et al., 1996; Plotkin et al., 1997b; Clayton et al., 1998). Immunocytochemical study has shown that the NKCC1 protein is expressed on plasma membranes of neurons throughout the brain, especially in pyramidal cells of the cerebral cortex (Plotkin et al., 1997a, 1997b). Results of studies from the authors' and other laboratories suggest that the cotransporter plays an important role in Cl− accumulation in neurons (Alvarez-Leefmans et al., 1988; Sun and Murali, 1999). The cotransporter-meditated intracellular Cl− accumulation may result in GABA-induced depolarization of dorsal root ganglion neurons and immature cortical neurons (Misgeld et al., 1986; Alvarez-Leefmans et al., 1988; Sun and Murali, 1999, Sung et al., 2000). In addition, Na+-K+-Cl− cotransporter has been suggested to play a role in K+ uptake and cell volume regulation in astrocytes (Walz and Hertz, 1984; Kimelberg, 1987; Mongin et al., 1994; Su et al., 2000). The cotransporter activity in cultured astrocytes is stimulated by elevated extracellular potassium ([K+]o) and inhibition of the cotransporter activity decreases astrocyte swelling and K+ uptake (Walz and Hertz, 1984; Kimelberg, 1987; Mongin et al., 1994; Su et al., 2000).
Little is known regarding how Na+-K+-Cl− cotransporter activity is regulated in the CNS. Glutamate, N-methyl-d-aspartate, and the metabotropic glutamate receptor agonist t-ACPD significantly stimulate cotransporter activity in neurons (Sun and Murali, 1998, 1999). Cotransporter activity in cortical neurons and astrocytes is elevated when intracellular Ca++ increases in the presence of high [K+]o(Schomberg et al., 2001; Su et al., 2000). Because both high [K+]o and elevated extracellular glutamate play important roles in ischemic cell damage, the authors hypothesize that stimulation of the cotransporter in neurons may contribute to overload of intracellular Na+ and Cl− and cell swelling during ischemia. Several studies suggest that the Na+-K+-Cl− cotransporter may be involved in ischemic cerebral cell damage. Twenty-four hours of hypoxia decreases cellular adenosine triphosphate (ATP) content and reduces Na+-K+-ATPase activity, while significantly increasing the Na+-K+-Cl− cotransporter activity in rat brain capillary endothelial cells (Kawai et al., 1996). Significant reduction of brain edema by the Na+-K+-Cl− cotransporter and Cl− channel inhibitor torasemide or its derivative also has been observed in focal cerebral ischemia and traumatic brain injury (Staub et al., 1994; Le Bars et al., 1996). However, no study has yet directly demonstrated a role of the cotransporter in ischemic neuronal damage.
In this study, the authors evaluated whether expression of the cotransporter in brain was altered after ischemia and if inhibition of the cotransporter activity in brain could significantly reduce ischemic brain damage. The results show that expression of the cotransporter protein was up-regulated in neurons after 2-hour ischemia and 24-hour reperfusion. Inhibition of the cotransporter with bumetanide significantly decreased infarction volume and brain edema.
MATERIALS AND METHODS
Animal preparation
Male, spontaneously hypertensive rats (SHRs, Charles River Farms, Wilmington, MA, U.S.A.) weighing 270 to 300 g were anesthetized with 2% halothane for induction and 0.8% to 1.2% halothane plus 60% N2O and 40% O2 for maintenance. Animals were ventilated mechanically with a rodent ventilator (Harvard rodent ventilator, Model 683; Harvard, South Natick, MA, U.S.A.). The left femoral artery was cannulated for continuous arterial blood pressure monitoring and serial blood sampling for determination of pH, Paco2, Pao2, hemoglobin, blood glucose, Na+, and K+ (i-STAT; Sensor Devices, Waukesha, WI, U.S.A.). Temporalis muscle and rectal temperatures were monitored and maintained at 36.6°C to 37.0°C with a heating blanket and a heating lamp.
A total of 56 rats were used in the current study. Four rats died as a result of bleeding after the carotid artery was injured during vessel dissection. Five rats died after 1 to 6 hours of reperfusion because of perforation of the intracranial vessels by the nylon sutures. These 9 rats were excluded from the study. The remaining rats were randomly assigned to the following groups: sham control group (n = 6), [butyl-3H]-bumetanide microdialysis group (n = 6), nontreated ischemic control group (n = 13), artificial cerebral spinal fluid (aCSF)-treated ischemic control group (n = 11), and bumetanide-treated ischemic group (n = 11).
All animal procedures used in the current study were conducted in strict compliance with the NIH Guide for the Care and Use of Laboratory Animals and were approved by the University of Wisconsin Center for Health Sciences Research Animal Care Committee.
Focal ischemia model
Focal cerebral ischemia in rats was induced by occlusion of the left middle cerebral artery (MCA). Briefly, under an operating microscope, the left common carotid artery was exposed through a midline incision. The occipital artery branches of the external carotid artery (ECA) were isolated and coagulated. The superior thyroid artery was dissected and coagulated. The ECA was dissected further distally and coagulated along with the terminal lingual and maxillary artery branches. The internal carotid artery (ICA) was isolated and carefully separated from the adjacent vagus nerve. The extracranial branch of the ICA, the pterygopalatine artery, then was dissected and ligated. A 20-mm-long 3.0 monofilament nylon suture with a blunted tip was introduced into the ECA lumen through a puncture and then gently advanced in the ICA lumen through the ECA to block MCA blood flow for 2 hours. For reperfusion, the suture was withdrawn to restore blood flow. The incision was closed with suture and the animals were reperfused for 24 hours. The sham-operated rats were subjected to the same procedures except for intravascular filament occlusion.
Immunocytochemical staining
Three ischemic rats at 24-hour reperfusion and 3 sham-operated rats were deeply anesthetized with an overdose of sodium pentobarbital and transcardially perfused with 4% paraformaldehyde in 0.1 mol/L phosphate-buffered saline (PBS) (pH 7.4). Brains were removed, postfixed in 4% paraformaldehyde overnight at 4°C, and cryoprotected with 30% sucrose in PBS. Brains were cut into coronal sections (40 μm) on a freezing microtome (Leica SM 2000 R; Leica, Nussloch, Germany). Coronal sections (0.2 mm anterior to bregma) were selected and processed for immunocytochemistry. Four consecutive sections from each rat were used and the free-floating method was used for avidin-biotin-peroxidase labeling. Coronal sections were rinsed with PBS and treated with 0.3% H2O2 (v/v) in PBS for 10 minutes at room temperature to quench endogenous peroxidase. After washing with PBS, sections were incubated with a blocking solution (10% normal goat serum, 0.3% Triton X-100 in PBS) for 30 minutes at 37°C. The slices were probed with T4 anti-cotransporter monoclonal antibody (1:100; Departmental Studies Hybridoma Bank, Iowa City, IA, U.S.A.) (Lytle et al., 1995) for 1 hour at 37°C, followed by overnight incubation at 4°C. After rinsing, sections were incubated with biotinylated goat anti-mouse IgG (1:200; Vector, Burlingame CA, U.S.A.) for 1 hour at 37°C and avidin-peroxidase (1:100; Vector, Burlingame, CA, U.S.A.) for 45 minutes at 37°C. Color reaction was developed with 0.05% diaminobenzidime tetrachloride and 0.03% H2O2 in 50 mmol/L Tris-HCl buffer (pH 7.4). For negative immunocytochemical control, a consecutive section was treated with the similar procedures except that the primary antibody was omitted.
Immunofluorescence staining
For double staining, 4 consecutive coronal sections (0.3 mm and 3.8 mm posterior to bregma) from each of 3 ischemic rats at 24-hour reperfusion were used. Sections were incubated with T4 anti-cotransporter monoclonal antibody (1:100) plus either anti-neuron specific enolase polyclonal antibody (1:100; DAKO, Carpinteria, CA, U.S.A.) or anti–glial fibrillary acidic protein (GFAP) polyclonal antibody (1:100; DAKO) for 1 hour at 37°C, followed by overnight incubation at 4°C. After rinsing in PBS, slices were incubated with goat anti-mouse fluorescein isothiocyanate–conjugated IgG (1:100; Sigma) and goat anti-rabbit Texas Red-X conjugated IgG (1:100; Molecular Probes, Eugene, OR, U.S.A.) for 1 hour at 37°C. Fluorescence images were captured by a laser-scanning confocal microscope (Bio-Rad MRC 1024; Hercules, CA, U.S.A.), as described previously (Su et al., 2000). Bio-Rad MRC-1024 Laser Sharp software (version 2.1T) was used to control the microscope and its settings. An identical setting was used to capture the negative control and experimental images.
Gel electrophoresis and immunoblotting
Four ischemic rats at 24-hour reperfusion were deeply anesthetized with an overdose of sodium pentobarbital. Contralateral and ipsilateral cortex, striatum, and hippocampus were dissected from each rat. The tissue was cut into small pieces with fine scissors in an isolation buffer (0.32 mol/L sucrose, 5 mmol/L Tris-HCl, 2 mmol/L ethlenediminetetracetic acid, pH 7.5) containing protease inhibitors, as described previously (Sun et al., 1999). Tissue was lysed by 30 seconds of sonication at 4°C by an ultrasonic processor (Sonics & Materials, Danbury, CT, U.S.A.). Cell debris was removed by centrifugation at 420 g for 5 minutes and the supernatant fraction was saved. Because the tissue volume of striatum and hippocampus is small, protein samples of the supernatant fractions of these tissues were used in immunoblotting. For cortex, crude membrane protein was prepared by further centrifugation of the tissue supernatant at 12000 g for 8 minutes. The crude membrane protein pellet was resuspended in the isolation buffer.
Protein content was determined by bicinchoninic acid method (Smith et al., 1985). Protein samples and prestained molecular mass markers (Bio-Rad) were denatured in sodium dodecyl sulfate reducing buffer (1:2 by volume; Bio-Rad) and heated at 37°C for 15 minutes before gel electrophoresis. The samples then were electrophoretically separated on 6% sodium dodecyl sulfate gels and the resolved proteins were electrophoretically transferred to a polyvinyl difluoride membrane (0.45 μm; Millipore Corporation, Bedford, MA, U.S.A.). Blots were incubated in 7.5% nonfat dry milk in Tris-buffered saline overnight at 4°C, then incubated for 2 hours with a primary antibody. The blots then were rinsed 5 times with Tris-buffered saline and incubated with horseradish peroxidase-conjugated secondary IgG for 1 hour. After 5 washings, bound antibody was visualized using the enhanced chemiluminescence assay (ECL; Amersham). T4 monoclonal antibody against Na+-K+-Cl− cotransporter and anti-βIII tubulin monoclonal antibody (Promega, Madison, WI, U.S.A.) were used for detection of the cotransporter protein and β tubulin, respectively.
Five to 45 μg protein of brain tissue was loaded on 6% sodium dodecyl sulfate gels, and T4 antibody and anti-βIII tubulin antibody were used. After the ECL reaction, the protein bands on the film were scanned using a Hewlett Packard ScanJet (4c/T) scanner (Hewlett Packard, Greeley, CO, U.S.A.). The relative intensity of each protein band was measured using UN-SCAN-It gel software (Silk Scientific, Orem, UT, U.S.A.). A linear curve was obtained with 5 to 45 μg protein for T4 antibody and anti-β tubulin antibody (r = 0.98 and 0.96, respectively). In addition, to establish a linear curve for exposure time of the ECL on the film, the blot with 5 to 45 μg protein was exposed to a film for 10, 20, 30, or 60 seconds. Exposure time was found to be linear for both T4 antibody and anti-β actin antibody between 20 to 30 seconds (r 0.96). Therefore, 15 μg protein was loaded in all immunoblots of the current study and ECL exposure time was 10 seconds. The relative intensity of NKCC1 and β tubulin bands was measured and data were expressed as a ratio of the NKCC1 and β tubulin band intensity.
Brain microdialysis procedures for administration of bumetanide
Animals were anesthetized as described above and placed in a stereotaxic frame. The left parietal area of the skull was exposed and a small burr hole (coordinates: 0.3 mm posterior and 5 mm lateral to bregma) was drilled, through which a microdialysis probe was passed. A 7-mm-long microdialysis probe (membrane length = 2 mm, outside diameter = 0.32 mm, molecular mass cutoff approximately 40,000 Da; Bioanalytical Systems, West Lafayette, IN, U.S.A.) was placed through a cannula into the left cerebral cortex (2.5 mm in depth, 5 mm lateral and 0.3 mm posterior to bregma) (Paxinos and Walson, 1998). Dental acrylic was used to secure the probe to the skull. Placement of the probe was verified by histologic examination subsequent to the experiments.
After surgery, the microdialysis probe was perfused through a dialysis pump (Series 200; Wood Dale, IL, U.S.A.) at a flow rate of 2 μL/min with aCSF (150 mmol/L NaCl, 3 mmol/L KCl, 1.25 mmol/L CaCl2, 0.48 mmol/L NaH2PO4, 21 mmol/L NaHCO3, 0.82 mmol/L MgCl2, 3.4 mmol/L glucose; pH 7.4) and equilibrated for 1 hour. After equilibration, either aCSF or 100 μmol/L bumetanide (Sigma) in aCSF was continuously microdialyzed by microdialysis probe throughout the 2-hour MCA occlusion and 24-hour reperfusion. A bumetanide stock solution (25 mmol/L in 0.1 mol/L NaOH) was prepared and 100 μmol/L bumetanide solution was made in aCSF.
Autoradiography
In 6 nonischemic SHRs, the extent of bumetanide diffusion away from a 2-mm microdialysis membrane was assessed by autoradiography of [butyl-3H]-bumetanide. Twenty μM [butyl-3H]-bumetanide in aCSF was prepared from a 5-mCi/mL stock (specific activity of 15 Ci/mmol; American Radiolabeled Chemicals, St. Louis, MO, U.S.A.). To test the effectiveness of bumetanide delivery by microdialysis, [butyl-3H]-bumetanide was microdialyzed into the left cerebral cortex for 1 hour. The brain then was carefully removed and frozen at −80°C. Frozen brains were cut into serial 20-μm coronal sections, and 1 of every 10 sequential sections was exposed to hyperfilm-3H (Amersham, Piscataway, NJ, U.S.A.) for 2 weeks at 4°C. After the film was developed, brain sections were stained with cresyl violet to localize tissue distribution of [butyl-3H]-bumetanide.
Infarction size measurement
Fourteen male SHR rats were randomly divided into either aCSF-treated or bumetanide-treated groups (n = 7 for each group). After 24-hour reperfusion, rats were anesthetized with 5% halothane plus 60% N2O and 40% O2 and then decapitated. Brains were removed and frozen at −80°C for 5 minutes. Two-millimeter coronal slices were made with a rodent brain matrix (RBM 4000C; Activational Systems, Warren, MI, U.S.A.). Sections were stained for 20 minutes with 2% 2,3,5,-triphenyltetrazolium chloride monohydrate (TTC; Sigma) at 37°C. In a double-blinded manner, infarction volume was calculated with the method reported by Swanson and colleagues (Swanson et al., 1990) to compensate for brain swelling in the ischemic hemisphere. Briefly, the sections were scanned, and the infarction area in each section was calculated by subtracting the noninfarct area of ipsilateral side from the area of contralateral side with an NIH image analysis software. Infarction areas on each section were summed and multiplied by section thickness to give the infarction volume.
Water content measurement
Rats were decapitated under deep anesthesia with 5% halothane plus 60% N2O and 40% O2 at 24 hours of reperfusion in the aCSF-treated (n = 4) or bumetanide-treated group (n = 4). Ipsilateral and contralateral hemispheres were dissected, and wet weight of the tissue was measured. The tissue was dried at 120°C for 24 hours. In a double-blinded manner, the hemispheric water content was calculated as the difference between wet and dry weights and expressed as percentage of wet weight.
Statistical analysis
Values are presented as mean ± SD. Physiologic variables were compared between the aCSF-treated and bumetanide-treated groups during preischemic and postischemic periods by analysis of variance. Infarction volume and water content were compared between the aCSF-treated and bumetanide-treated groups by the nonparametric Mann–Whitney U test. P < 0.05 was considered statistically significant.
RESULTS
Up-regulation of Na+-K+-Cl− cotransporter expression in ischemic brain
Two-hour MCA occlusion plus 24-hour reperfusion induced a large infarct lesion in SHRs. Infarct tissue was located in the left frontal, parietal, piriform and perirhinal cortices, and striatum (data not shown). The average total lesion volume was 219.7 ± 50.1 mm3 in SHRs (n = 6). No such infarction was observed in the sham-operated SHRs (n = 3, data not shown). In sham-operated SHRs, the T4 antibody immunoreactive signals were localized in cell body and dendrite of pyramidal neurons in cerebral cortex (Fig. 1A and 1B). Expression of the cotransporter in neurons in other cortical layers was low (Fig. 1A). A relatively low level of the cotransporter expression was found in striatum of sham-operated SHRs (Fig. 1E and 1F). After 2 hours of MCA occlusion and 24 hours of reperfusion, an enhanced immunostaining of the cotransporter in neurons scattered throughout ischemic cortex (Fig. 1C and 1D) and striatum (Fig. 1G and 1H). This up-regulation of cotransporter expression was not observed in nonischemic contralateral cortex and striatum (data not shown). Nonspecific signals of the immunocytochemical staining were evaluated in a consecutive section by omitting the T4 primary antibody. Immunoreactive staining of the control sections was negligible, as illustrated in Fig. 1I (cortex) and Fig. 1J (striatum).
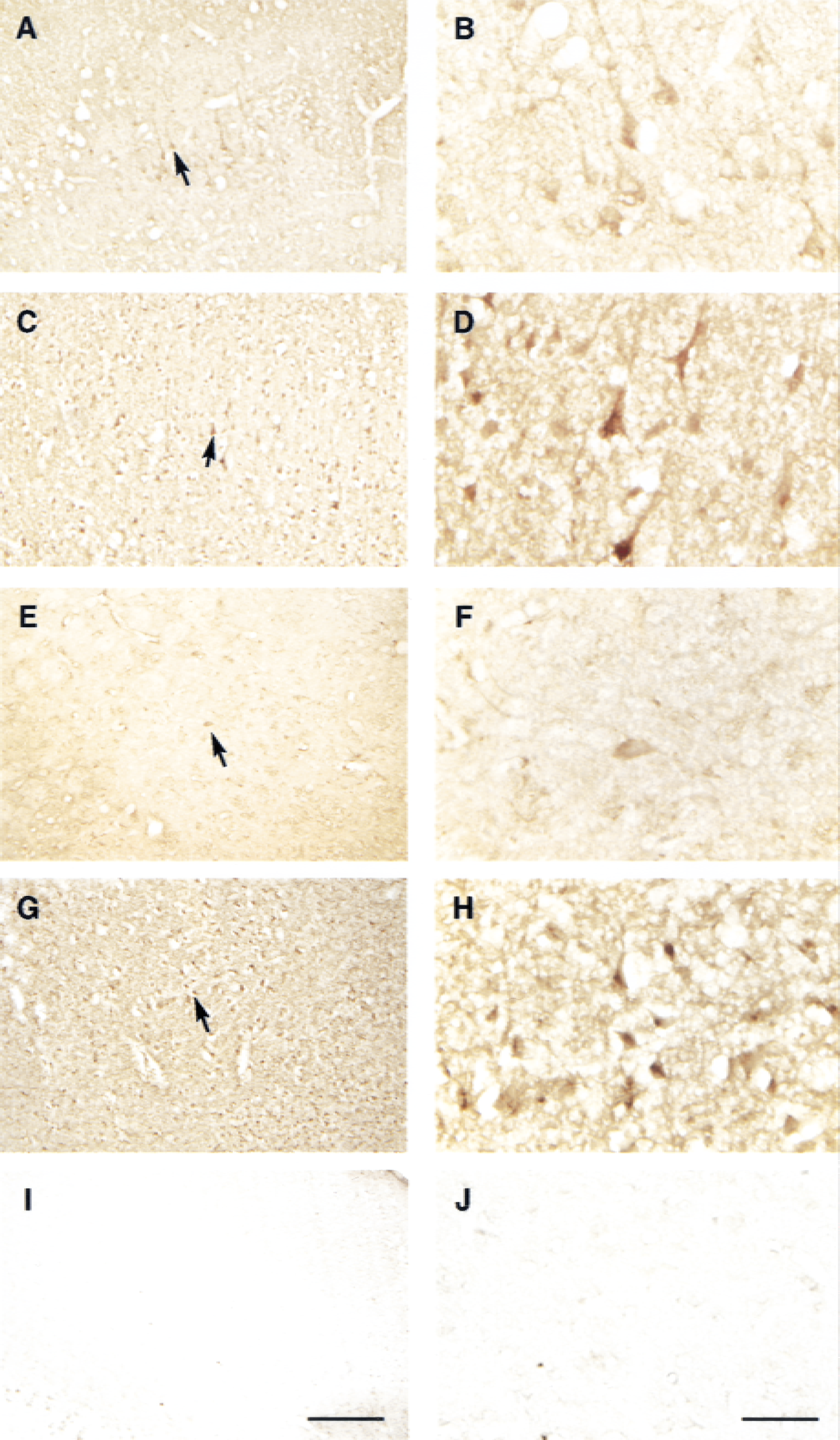
Immunocytochemical staining of Na+-K+-Cl− cotransporter in cortex and striatum. Coronal sections (0.2 mm anterior to bregma) were stained with T4 anti-cotransporter antibody and visualized with avidin-biotin-peroxidase method. Areas in the low-power photographs (arrows,
The morphology of the cells with positive T4 antibody staining in Fig. 1 suggested that the cotransporter is expressed in neurons. To establish this further, fluorescence double labeling was performed. In Fig. 2A, cells in cortical cortex after 2-hour ischemia and 24-hour reperfusion were stained with T4 anti-cotransporter monoclonal antibody. Strong immunoreactive signals of T4 antibody were observed in plasma membranes of cortical neurons with a normal morphology (arrows). Such an intensive staining also was observed in shrunken cells (arrowheads). The same section was double stained with polyclonal antibody against a neuron-specific enolase, a marker protein for neurons (Fig. 2B). Double-staining image revealed that the Na+-K+-Cl− cotransporter and the neuronal enolase were colocalized in neurons (Fig. 2C). To further examine whether expression of the cotransporter could be detected in astrocytes, double labeling of the cotransporter protein and a glial marker GFAP was performed. As shown in Fig. 2D to 2F, an intense staining for the cotransporter was found in the ipsilateral cortical cortex (layer II-III), compared with a low level of GFAP expression. Only faint colocalization signals of these two proteins were found in a few astrocytes (arrowhead, Fig. 2D to 2F). In contrast, strong colocalization signals of the cotransporter and GFAP were observed in many astrocytes in the ipsilateral cortical white matter (Fig. 2G to 2I) and hippocampus (Fig. 2J to 2L). Expression of the cotransporter also was found in perivascular astrocytes (Fig. 2G to 2I). Insets of Fig. 2D to 2F represent a negative control study in which primary antibodies were omitted and the rest of the procedures were the same as in Fig. 2A to 2L. This demonstrates that the images shown in Fig. 2A to 2L are specific immunoreactive signals.
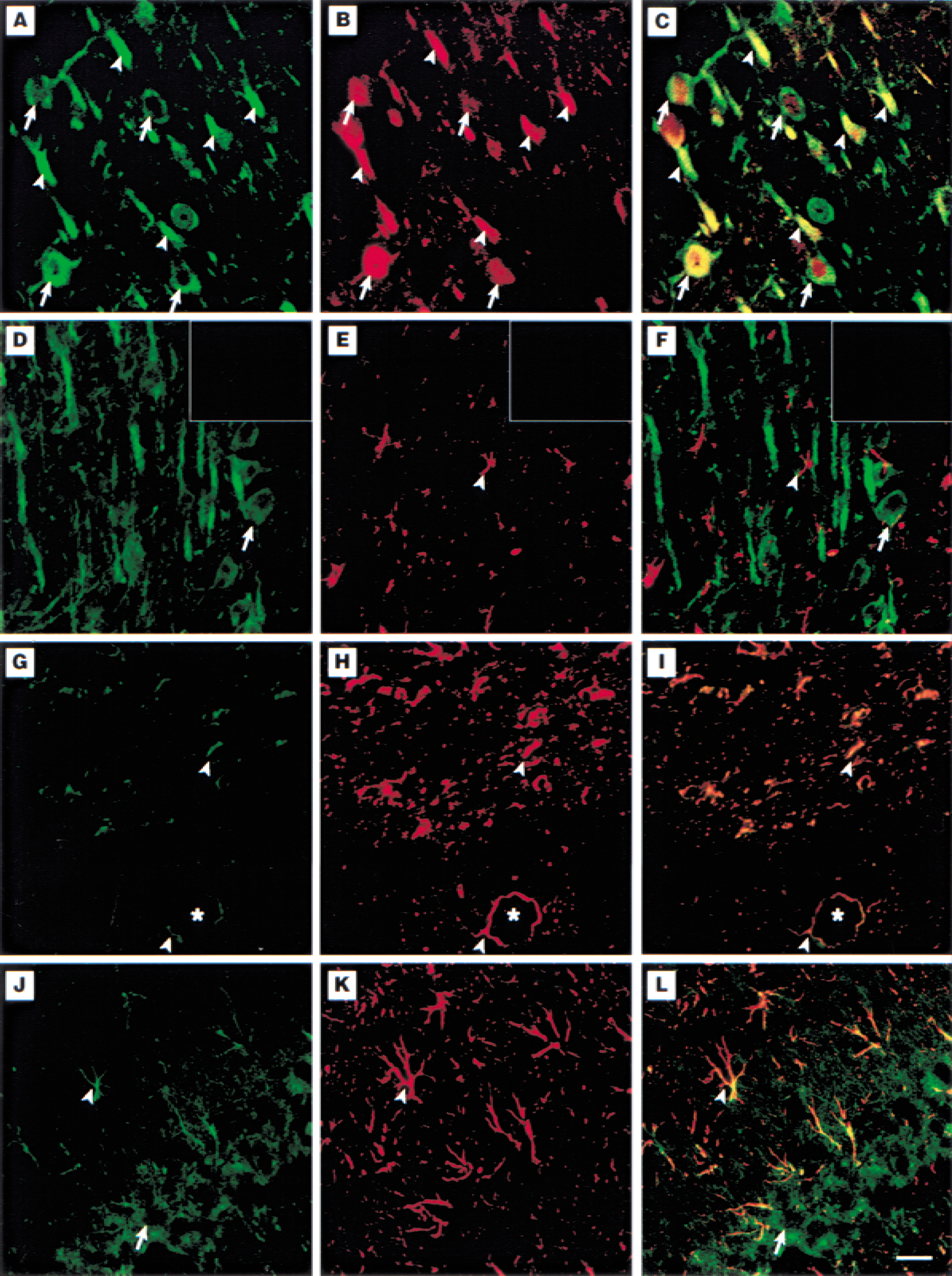
Double labeling of Na+-K+-Cl− cotransporter, neuronal enolase, or glial fibrillary acidic protein (GFAP) in ischemic brain. Coronal sections were from 2-hour ischemia and 24-hour reperfusion rats.
The authors further studied changes of expression of the cotransporter protein by immunoblotting analysis. As shown in Fig. 3A, an approximately 145.4 ± 1.9 kDa (n = 4) cotransporter protein was detected in cortical cortex, striatum, and hippocampus. The amount of NKCC1 was increased in the ipsilateral cortex after 24 hours of reperfusion compared with one in the contralateral cortex. There were no significant changes in expression of the cotransporter in the ipsilateral striatum and hippocampus. To gain some quantitative information, the authors also examined expression of β tubulin in these regions of the brain. β tubulin (type III) with approximately 47.7 ± 1.9 kDa was detected in brain tissues (n = 4). There was no significant change in β tubulin expression in the ipsilateral cortex and striatum by densitometrical analysis (data not shown, n = 4, P > 0.05). The ratio of the relative intensity of cotransporter to β tubulin bands was increased by 40% in the ipsilateral cortex (Fig. 3B, n = 4, P < 0.05, Mann–Whitney U test). No significant changes of the ratio were found in the ipsilateral striatum (n = 4, P > 0.05, Mann–Whitney U test). The hippocampus is not affected in the focal ischemic model and therefore was used as a negative control. Neither β tubulin nor the cotransporter expression was significantly changed after 24-hour reperfusion in hippocampus (Fig. 3). These results further suggest an up-regulation of the cotransporter protein in ischemic cortical tissue.
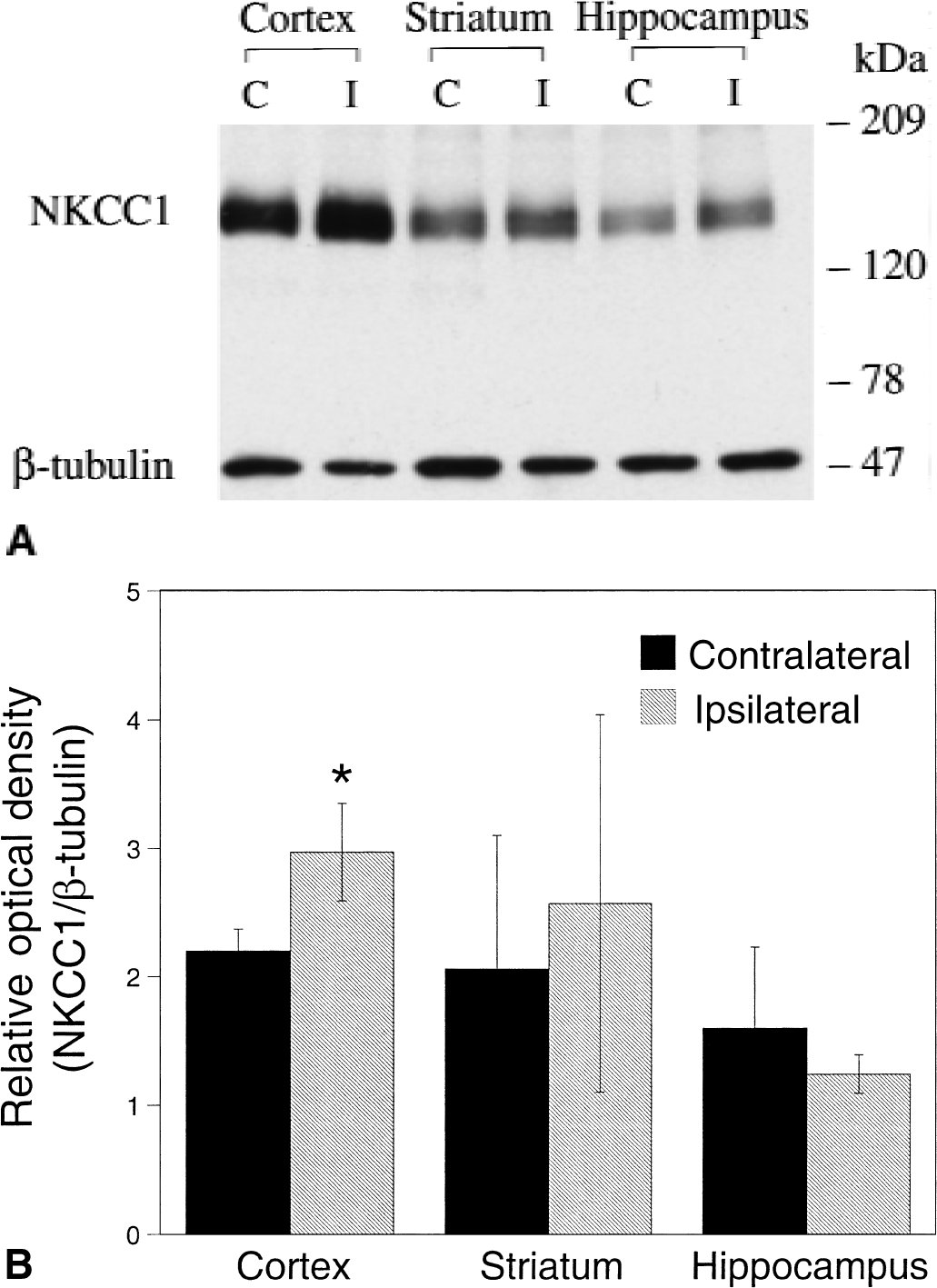
Immunoblotting analysis of Na+-K+-Cl− cotransporter in ischemic brain.
Local administration of [butyl-3H]-bumetanide in cerebral cortex
In Fig. 4A, an autoradiograph of a coronal section illustrates diffusion of 20 μmol/L [butyl-3H]-bumetanide in the left cerebral cortex. Location of the microdialysis probe in the left cerebral cortex is shown in the same coronal section counterstained with cresyl violet (Fig. 4B, upper panel). To identify the approximate location of the labeled tissue, the [butyl-3H]- bumetanide-labeled tissue in Fig. 4A was outlined and superimposed on the cresyl violet-stained section in Fig. 4B. It shows a colocalization of the microdialysis track and labeling of [butyl-3H]- bumetanide in the left cortex. A corresponding coronal section of an ischemic rat was stained with cresyl violet and is shown in Fig. 4B (lower panel). It indicates that infusion of bumetanide through the microdialysis probe was localized in the infarct area after ischemia.
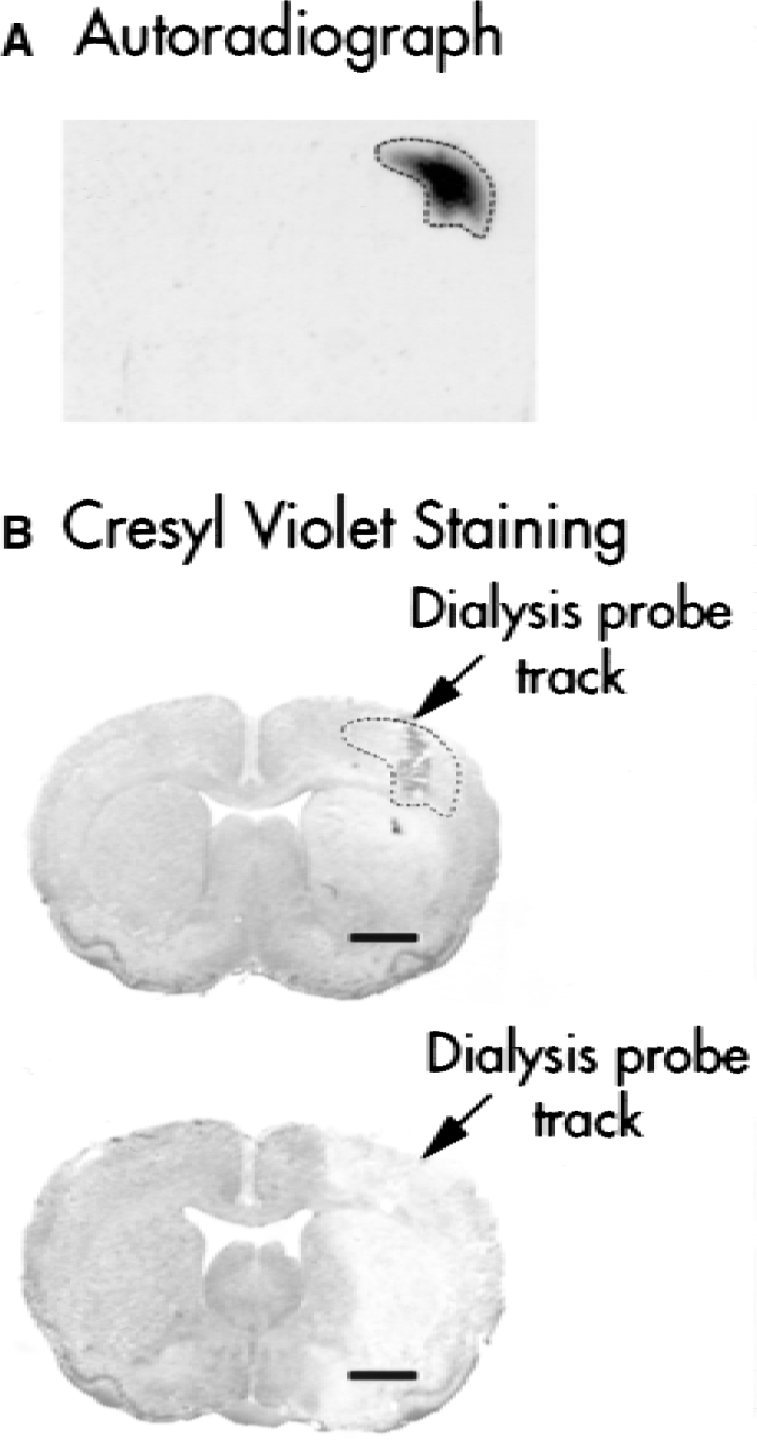
[butyl-3H]-bumetanide autoradiograph.
Physiologic parameters in ischemic spontaneously hypertensive rats
Physiologic variables of animals before and after 2-hour reperfusion after MCA occlusion are shown in Table 1. Mean arterial blood pressure of animals before the ischemic insult was 132.7 ± 7.9 mm Hg. There were no differences in mean arterial blood pressure before and after MCA occlusion in the aCSF-treated animals. The other physiologic parameters were in normal ranges during preischemia and 2-hour reperfusion after MCA occlusion. No statistically significant differences were observed in arterial pH, Paco2, Pao2, hemoglobin, blood glucose, or Na+ and K+ concentrations in the aCSF-treated animals before induction of ischemia and after 2-hour reperfusion. In addition, rectal and temporalis temperatures at 2-hour reperfusion were similar to preischemic levels. Moreover, to monitor possible effects of bumetanide on systemic parameters, the authors compared physiologic variables between the aCSF-treated and bumetanide-treated groups. As shown in Table 1, local administration of bumetanide had no effect on mean arterial blood pressure, plasma pH, hemoglobin, blood glucose, Na+ or K+ concentrations. Similar results were observed at 24-hour reperfusion (data not shown, n = 3).
Physiologic parameters in aCSF-treated and 100 μmol/L bumetanide-treated SHRs
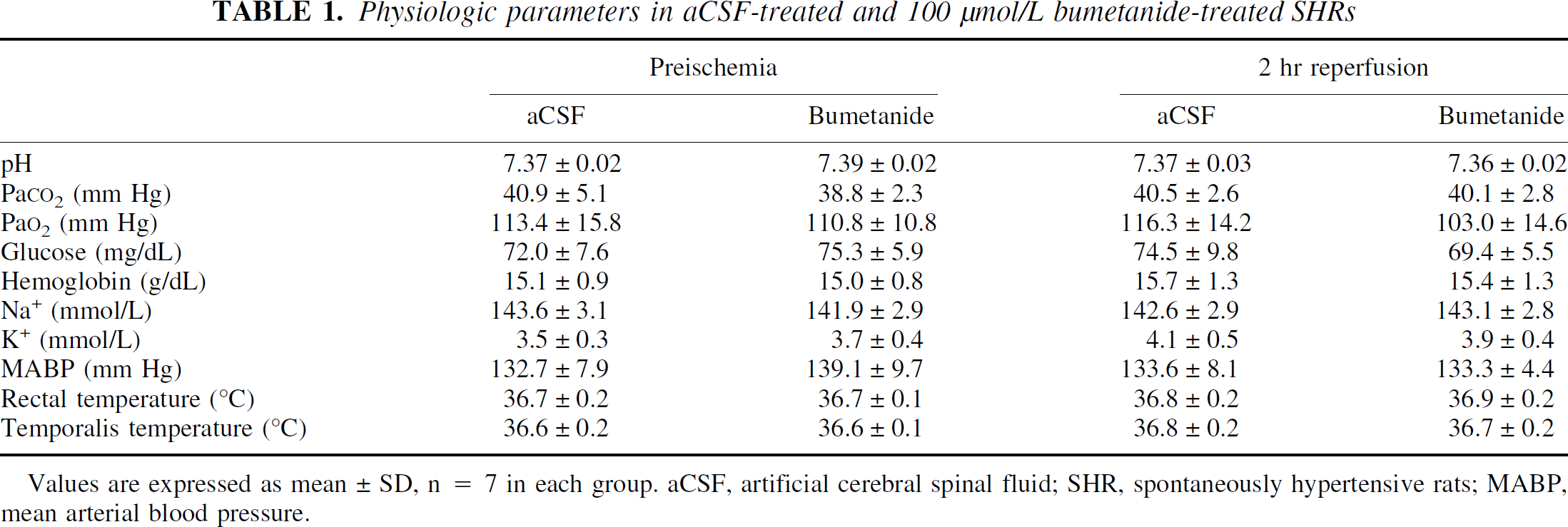
Values are expressed as mean ± SD, n = 7 in each group. aCSF, artificial cerebral spinal fluid; SHR, spontaneously hypertensive rats; MABP, mean arterial blood pressure.
Infarction volume was attenuated in bumetanide-treated brains
In the aCSF-treated animals, the infarct zone is illustrated in Fig. 5. The average total infarction volume (cortical plus subcortical area) in the aCSF-treated group averaged 238.3 ± 38.3 mm3 (n = 7). The average total infarct volume was significantly reduced to 178.3 ± 34.4 mm3 in the bumetanide-treated group (n = 7, P < 0.05). Unlike the aCSF-treated SHRs, 4 of 7 bumetanide-treated rats did not have any infarct at 12 mm posterior to the frontal pole. Regional infarction (cortical) was further analyzed in the aCSF-treated and bumetanide-treated groups. Average cortical infarct volumes were 179.3 ± 35.6 mm3 in the aCSF-treated rats (n = 7) and 134.0 ± 28.1 mm3 in the bumetanide-treated rats (n = 7). These differences were statistically significant (P < 0.05) according to the Mann–Whitney U test.
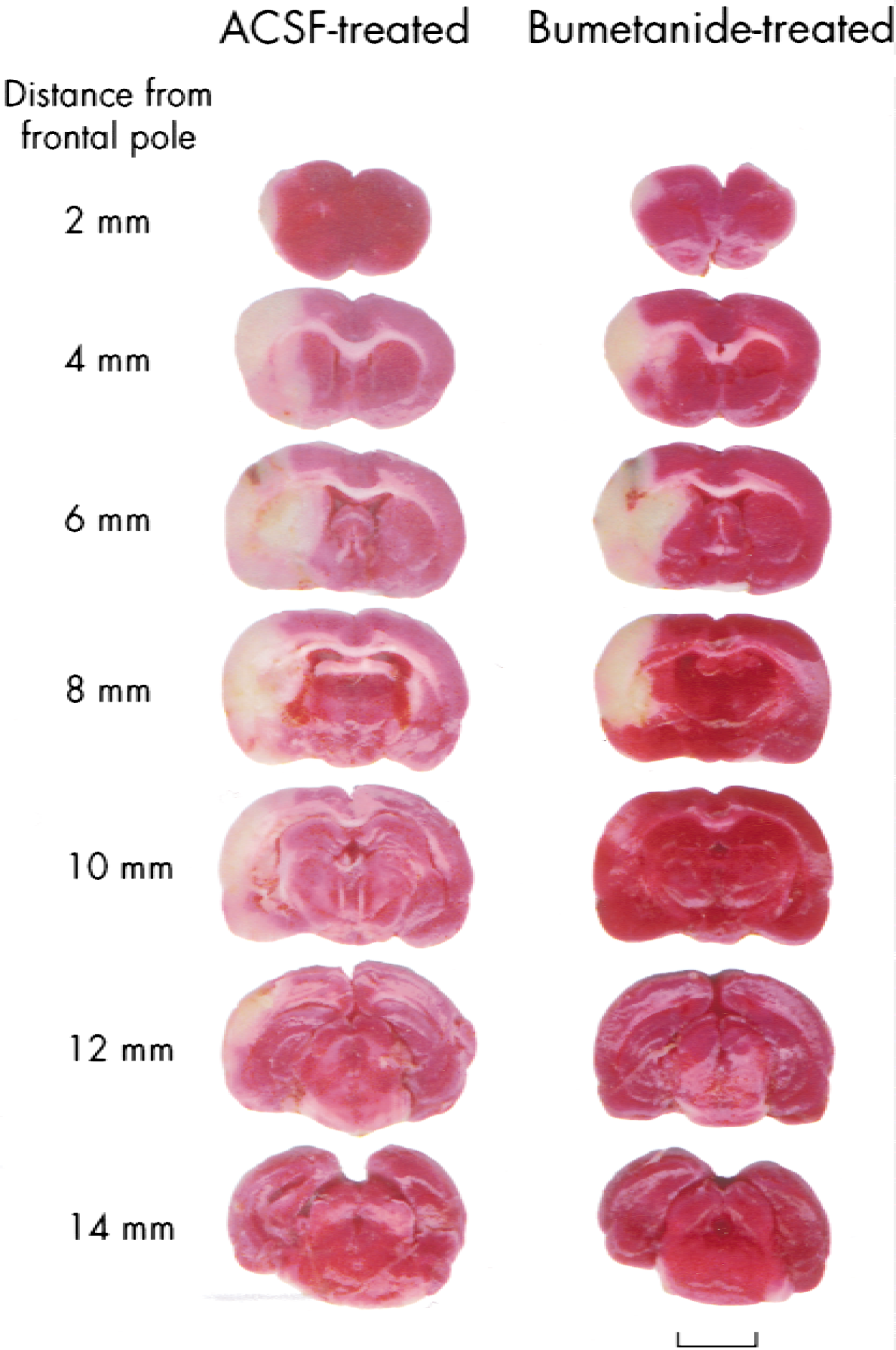
Bumetanide-induced protective effect on infarction volume. Ischemic spontaneously hypertensive rats were treated either with artificial cerebral spinal fluid (aCSF) or 100 μmol/L bumetanide in aCSF through a microdialysis probe throughout 2-hour MCA occlusion and 24-hour reperfusion (Materials and Methods). 2,3,5,-triphenyltetrazolium chloride–stained coronal sections are shown at different levels posterior to the frontal pole. These are representative data of the aCSF-treated group (n = 7) and bumetanide-treated group (n = 7). Scale bar = 5.5 mm.
Effect of bumetanide on brain edema after focal cerebral ischemia
Water content in the aCSF-treated and bumetanide-treated brains after 24-hour reperfusion is shown in Table 2. The water content averaged 78.7% ± 0.2% in the contralateral hemispheres and significantly increased in the ipsilateral side in the aCSF-treated rats (P < 0.05, Mann–Whitney U test). In contrast, in the bumetanide-treated rats, the water content was significantly decreased in the ipsilateral hemisphere (P < 0.05) and not significantly different from the values in the contralateral hemispheres (either of the aCSF-treated or the bumetanide-treated rats).
Effect of bumetanide on water content in brain tissue after focal cerebral ischemia

Values are expressed as mean ± SD, n = 4 spontaneously hypertensive rats in each group. aCSF, artificial cerebral spinal fluid.
P < 0.05 versus contralateral values of the aCSF-treated group.
P < 0.05 versus ipsilateral values of the aCSF-treated group (Mann–Whitney U test).
DISCUSSION
Expression of Na+-K+-Cl− cotransporter in the central nervous system
In situ hybridization reveals mRNA expression of NKCC1 in rat cerebral cortex and hippocampus (Plotkin et al., 1997a). Immunohistochemistry staining with a polyclonal antibody against BSC2 (NKCC1) shows an intense signal in cortical pyramidal cells of Sprague–Dawley adult brain (Plotkin et al., 1997a). In the current study, the authors found that the Na+-K+-Cl− cotransporter protein is expressed in neurons of cortex and striatum in adult SHRs. A double-labeling study shows that the Na+-K+-Cl− cotransporter is colocalized with the neuron-specific enolase. Moreover, an up-regulation of Na+-K+-Cl− cotransporter expression is found in cortical and striatal neurons after ischemia.
Astrocytes have been suggested to play an important role in K+ uptake in the central nervous system (Walz and Hertz, 1984). In cultured mouse and rat astrocytes, Na+-K+-Cl− cotransporter activity is significantly stimulated in elevated extracellular K+, which leads to cell swelling (Walz and Hertz, 1984; Mongin et al., 1994). Therefore, the cotransporter in astrocytes may function in K+ uptake in ischemic brains. The authors' recent study demonstrated that the cotransporter in cultured rat cortical astrocytes is stimulated by high extracellular K+ in a Ca++-dependent manner (Su et al., 2000). In the current study, a few GFAP-positive astrocytes were found in ischemic cortex that is likely because of an ischemia-induced astrocyte cell loss (Liu et al., 1999). Only faint immunoreactive signals for the cotransporter were found in a few cortical astrocytes. The weak staining signal for the cotransporter protein in cortical astrocytes may be because of a low expression of the cotransporter, because much stronger staining intensity for the cotransporter was found in astrocytes of cortical white matter and hippocampas using the same T4 antibody. This implies that astrocytes in different brain regions express different levels of the cotransporter. The current finding suggests that the Na+-K+-Cl− cotransporter in cortical astrocytes could contribute to brain edema during cerebral ischemia and blocking of the cotransporter activity in astrocytes may result in reduction of the ischemic damage.
Effectiveness of bumetanide diffusion and specificity of bumetanide effect
Bumetanide is a member of sulfamoylbenzoic acid loop diuretic family and exerts its diuretic action by blockade of the Na+-K+-Cl− cotransporter in the thick ascending limb of the loop of Henle (Haas, 1984). The goal of this study was to transiently inhibit the Na+-K+-Cl− cotransporter activity in brain and minimize bumetanide-mediated systemic effects. In addition, the systemic half-life of bumetanide is short (5 to 7 minutes in humans administered intravenously) (McEvoy et al., 1994). Thus, it would require repeated drug administration during a 24-hour experimental period. Although bumetanide has been reported to cross the blood–brain barrier (Ennis et al., 1996), it would be difficult to determine the drug concentration in brain tissue after systemic administration. Therefore, a continuous microdialysis of 100 μmol/L bumetanide into brain was used to locally inhibit the Na+-K+-Cl− cotransporter. Microdialysis of bumetanide has no effect on the systemic physiologic parameters. This implies that intracerebral microdialysis of bumetanide has no significant effect on peripheral organ function.
In rats microdialyzed with 1 to 20 μmol/L [butyl-3H]-bumetanide, there was a dose-dependent increase in the volume of [butyl-3H]-bumetanide–labeled tissues (data not shown). Microdialyzing 20 μmol/L [butyl-3H]-bumetanide for 1 hour produces 20 mm3 of [butyl-3H]-bumetanide–labeled tissue. Given an infarction volume (220 mm3) after 2-hour MCA occlusion and 24-hour reperfusion in SHRs, administration of 20 μmol/L bumetanide for 1 hour would only affect a small percentage of infarct tissue. Because the concentration gradient of bumetanide across the microdialysis membrane is the primary driving force for bumetanide diffusion, the authors increased the bumetanide concentration to 100 μmol/L and extended the total microdialysis duration to 26 hours to increase the distance of bumetanide diffusion. The authors also found that there is protection in the striatum where no drug is anticipated to be delivered to this area. Possible explanations for this effect include that the diffusion of the drug could be increased after ischemia or a reduction of infarct, or edema, or both (see below) in cortex by bumetanide could indirectly decrease cell damage in striatum. This speculation needs to be examined experimentally.
In this study, the exact bumetanide concentration in brain is not known. The recovery rate of a given microdialysis probe is affected by factors such as perfusion flow rate, dialysis membrane area, temperature, and diffusion coefficient of perfused molecules (Benveniste and Huttemeier, 1990). The recovery rate of amino acids using an identical microdialysis probe at a perfusion rate of 2 μL/min has been reported to be 6% to 9%in vitro (Sanchez-Carbente and Massieu, 1999). It can be assumed that the tissue bumetanide concentration was substantially less than 100 μmol/L in the current study. Bumetanide is a potent inhibitor of Na+-K+-Cl− cotransporter, but it also blocks other cotransporters, such as K+-Cl− and Na+-Cl−, at greater concentrations (Russell, 2000). Na+-Cl− cotransporter expression has only been detected in peripheral epithelia (Russell, 2000). In addition, the Ki value of bumetanide is 55 μmol/L to 100 μmol/L for K+-Cl− cotransporters (KCC1, KCC2, and KCC3;Gillen et al., 1996; Payne, 1997; Race et al., 1999), compared with a Ki of 0.1 μmol/L for the Na+-K+-Cl− cotransporter (Russell, 2000). As described above, the authors believe that the tissue bumetanide concentration is below the Ki for K+-Cl− cotransporters, and the protective effect of bumetanide observed in this study is likely through its action on the Na+-K+-Cl− cotransporter.
The role of Na+-K+-Cl− cotransporter in ischemia-mediated damage
Loop diuretics, such as furosemide, were considered useful agents for the treatment of brain edema and intracranial hypertension, particularly in combination with mannitol (Roberts et al., 1987). Hemispheric brain swelling after cold injury was significantly reduced (approximately 20%) with torasemide (10 mg/kg), a more lipophilic loop diuretic than furosemide (Staub et al., 1994). The efficacy of torasemide against brain swelling in brain traumatic injury was thought to be primarily because of inhibition of vasogenic brain edema (Berger et al., 1994; Staub et al. 1994), because the increase in tissue water content from trauma was significantly reduced (Staub et al. 1994). In addition, a torasemide derivative, N-[4-(cycloheptylamino)pyrid-3-yl] sulphonyl-N'-cycloheptyl urea (1 mg/kg), was found to temporarily reduce cytotoxic edema (Le Bars et al., 1996). Similarly, in the current study, the increase in water content after ischemia was reduced by 70% in the bumetanide-treated group. This suggests that the Na+-K+-Cl− cotransporter may contribute to brain edema. A reduction of brain edema by inhibition of the cotransporter activity may decrease the elevated intracranial pressure caused by cerebral edema and significantly improve penumbral blood flow. The latter could subsequently reduce infarct volume.
Molecular mechanisms underlying the cotransporter in ischemic neuron damage are not yet clear. In addition to brain edema formation, the Na+-K+-Cl− cotransporter may contribute to neuronal damage through other mechanisms, such as overload of intracellular Na+ and Cl−. Na+ and Cl− influxes are involved in glutamate-mediated neurotoxicity (Rothman, 1985, Choi and Rothman, 1990). Protection of injured neurons can be achieved by preventing Na+ entry, or Cl− entry, or both (Rothman, 1985; Rosenberg and Lucas, 1996). This view is supported by the current study in which a 25% decrease in infarct volume occurs when the cotransporter activity is inhibited. Moreover, the authors observed an up-regulation of the cotransporter expression in neurons of ischemic cortex at 24-hour reperfusion.
The lethal Cl− entry may contribute to excitotoxic cell death (Rothman, 1985; Choi and Rothman, 1990). An ischemia-induced increase in [Cl−]i of CA1 pyramidal cells and interneurons was observed in hippocampal slices after ischemia and reoxygenation (Inglefield et al., 1998). The Na+-K+-Cl− cotransporter plays an important role in intracellular Cl− accumulation in rat cortical neurons under physiologic conditions (Sun and Murali, 1999). A recent study by Sung et al. (2000) shows that intracellular Cl− concentration is reduced by 50% in dorsal root ganglion neurons from NKCC1 knockout mice. Interestingly, the authors recently found that the Na+-K+-Cl− cotransporter in cortical neurons was significantly stimulated by activation of ionotropic N-methyl-d-aspartate and group I metabotropic glutamate receptors (Sun and Murali, 1999; Schomberg et al., 2001). Under ischemic conditions, stimulation of the cotransporter, or up-regulation of cotransporter expression, or both, could contribute to the lethal Cl− entry. Therefore, blocking of the cotransporter activity by bumetanide could inhibit the cotransporter-mediated Cl− influx in neurons, consequently reducing ischemic neuronal damage.
In summary, Na+-K+-Cl− cotransporter was expressed in cortical and striatal neurons of SHRs. The level of the cotransporter expression was up-regulated in ischemic brains. Inhibition of the cotransporter activity by bumetanide significantly attenuated ischemic infarct volume and brain edema. The results strongly support the prospect that the Na+-K+-Cl− cotransporter may play an important role in cerebral ischemic damage.
Footnotes
Acknowledgments:
The authors thank Dr. Mark Haas for providing the method and instruction on purification of [3H]-bumetanide. The authors also thank Dr. Peter Lipton and Douglas Kintner for helpful comments and suggestions, and Janet Sailor for proofreading the manuscript.