
Abstract
Organophosphate-induced brain damage is an irreversible neuronal injury, likely because there is no pharmacological treatment to prevent or block secondary damage processes. The presence of free glutamate (Glu) in the brain has a substantial role in the propagation and maintenance of organophosphate-induced seizures, thus contributing to the secondary brain damage. This report describes for the first time the ability of blood glutamate scavengers (BGS) oxaloacetic acid in combination with glutamate oxaloacetate transaminase to reduce the neuronal damage in an animal model of paraoxon (PO) intoxication. Our method causes a rapid decrease of blood Glu levels and creates a gradient that leads to the efflux of the excess brain Glu into the blood, thus reducing neurotoxicity. We demonstrated that BGS treatment significantly prevented the peripheral benzodiazepine receptor (PBR) density elevation, after PO exposure. Furthermore, we showed that BGS was able to rescue neurons in the piriform cortex of the treated rats. In conclusion, these results suggest that treatment with BGS has a neuroprotective effect in the PO intoxication. This is the first time that this approach is used in PO intoxication and it may be of high clinical significance for the future treatment of the secondary neurologic damage post organophosphates exposure.
INTRODUCTION
Organophosphate compounds are highly toxic chemicals widely used as pesticides (e.g. malathion and paraoxon (PO)) and as chemical warfare nerve agents (e.g. soman, sarin). Pesticide poisoning is one of the most common poisonings worldwide, estimated at one million cases each year with several hundred thousand deaths. 1 Organophosphates inactivate the enzyme acetylcholine esterase (AChE; EC 3.1.1.7), a serine protease that hydrolyzes the neurotransmitter acetylcholine. Exposure to organophosphates leads to acetylcholine accumulation that causes the overstimulation of cholinergic receptors, which consequently results in the rapid and profound excitotoxicity and dysfunction of cholinergic neurons. This effect is reactivated very slowly, if at all. 2
Exposure to organophosphates damages several brain areas including the entorhinal and piriform cortex, amygdala, and hippocampus CA1/CA3 [ref 3]. Much of the brain damage does not typically occur at the time of the initial lesion, making secondary neuronal damage a major contributor to the neuronal loss. 4 The degree of brain damage depends on the severity of the convulsions and is in direct relation to the increase in peripheral benzodiazepine receptors (PBR) density. 5 Peripheral benzodiazepine receptors are located in the microglia and are used as reliable markers of brain damage. 6 The density of PBRs increases when microglias are activated in response to tissue damage. This increase appears as early as 24 hours post systemic organophosphates exposure and lasts for at least 2 weeks post exposure. 7
Glutamate (Glu) has a substantial role in the propagation and maintenance of organophosphate-induced seizures, thus contributing to the secondary brain damage.
8
Glu was shown to be released in excess in the brain under soman intoxication and had a prominent role during seizures.
9
Furthermore, Glu receptor antagonists in general, and
The standard treatment for organophosphates intoxication consists of pretreatment with pyridostigmine, a reversible inhibitor of AChE and anticholinergic agents such as atropine sulfate.
13
In addition, it was reported that benactyzine (combined with anticholinergic and anti-NMDA properties), or benzodiazepines, could reduce some of the brain damage if administered early enough.14, 15 Yet, such a treatment failed to control the brain damage associated with excitotoxicity of Glu.
9
It also failed to prevent the elevation in PBR density post organophosphate exposure.3, 16
The maintenance of brain extracellular Glu at levels below its excitotoxic threshold is performed not only by Glu transporters located on glia and neurons but also by those present on the anti-luminal side of the brain capillary endothelial cells.19, 20 These transporters remove the excess extracellular brain Glu into the blood stream. 21 In a recent study, we have established the feasibility of accelerating the naturally occurring brain–to-blood Glu efflux by inverting the concentration gradient between the blood versus brain Glu level as a novel neuroprotective treatment. 20 This was achieved by the Glu scavenging properties of the blood-resident enzyme Glu oxaloacetate transaminase (human source—hGOT) which, with oxaloacetate (OxAc), converts Glu into 2-ketoglutarate and aspartate, thereby decreasing the blood concentration of Glu. Blood glutamate scavengers (BGS) provided highly significant brain neuroprotection in rat animal models of closed head injury, ischemic stroke and glioma brain tumors, in which there is a local breach of the blood–brain barrier.22, 23, 24
In the present study, we investigated whether BGS could be used as a neuroprotective treatment in a well-established and reliable animal model of PO intoxication by inhibiting the secondary neuronal damage associated with organophosphates.
MATERIALS AND METHODS
Materials
Paraoxon was obtained from Chem Service (West Chester, PA, USA), Glu dehydrogenase was obtained from Roche Diagnostics (Rotkreuz, Switzerland), glass-fiber filters (GF/B 2.5 cm radius) were obtained from Wathaman (Kent, UK), [3H]PK 11195 was obtained from PerkinElmer (Boston, MA, USA); all other materials were obtained from Sigma-Aldrich (St Louis, MO, USA). His-tagged version of the human glutamate oxaloacetate transaminase (hGOT) cDNA that was cloned from the human hepatoma cell line hepG2 was expressed in
Animals
The experiments were conducted according to the Guidelines for the Use of Experimental Animals of the European Community approved by the Animal Care Committees of the Weizmann Institute of Science. Forty-one male, 9 to 11 weeks old, Sprague–Dawley rats (Harlan Laboratories, Rehovot, Israel), were used in this study. Weight variation of animals at the time of PO exposure was in the range of 300 to 320 g. The experiments began after at least 5 days of acclimation. During acclimation and throughout the study, the animals were housed within a limited access rodent facility, two rats in each cage, and after jugular vein cannulation, animals were housed individually in polypropylene cages (37.5 × 21 × 18 cm). The animals were provided diet
Study Design
Jugular vein cannulation was performed as previously described
25
and the rats were allowed to recover for 3 days. After recovery, PO was injected intramuscularly into the hind limb with a dosing of 450
Clinical Signs
Throughout the PO challenge, infusion, and later on, test animals were maintained within a transparent container and were observed for clinical signs with particular attention paid to the onset, intensity, and duration of characteristic and representative peripheral and central cholinomimetic manifestations. These refer mostly to dyspnea, fasciculation, salivation, and lacrimation, ataxia, paralysis, tonic–clonic convulsions, coma, and death. Recorded clinical signs parameters were graded as absent, mild, moderate, or severe. The latency until evident onset of convulsing seizures, as well as the intensity, scored by using the Racine's scale and respective time of cessation was documented. 26 Clinical signs observation was carried out before PO administration and thereafter at 2.5, 5, 7.5, 10, 15, 30, 60, 120, 180 minutes, 5, 6, and 24 hours post administration and then at least once daily throughout the successive 7-day study period. Animals that did not survive the first 24 hours were excluded from the study.
Histology and Immunohistochemistry
One week post PO challenge, rats were anesthetized and perfused transcardially with chilled phosphate-buffered saline/heparin. Brains were removed and fixed in Bouin solution or in 4% formalin. Samples were processed for paraffin embedding by standard procedures. Six-micron thick coronal brain serial sections were generated with 500
Blood Glutamate Determination Assay
Two-hundred microliters of rat blood was collected 10 minutes before and 40 minutes after PO injection. The blood was added to an ice-cold Eppendorf tube containing 200
Peripheral Benzodiazepine Receptor Assay
Three groups of animals were investigated, the control and treated group of rats (1 week after PO challenge), and in addition, a group of naïve animals. The method of PBR assay was modified from Lavoie
After thawing of brain from −80°C, 50 mL cold Tris buffer (20 nmol/L pH 7.4) was added and the brain was manually homogenized using a dounce homogenizer (Bellco, Chicago, IL, USA). Four 5 mL tubes were filled with the homogenized brain and subjected to two rounds of centrifugation at 40,000
Butyrylcholinesterase Activity Assay
Determination of erythrocyte AChE activity and cholinesterase status are no standard laboratory assays. However, determination of plasma BuChE (EC 3.1.1.8) is routinely used for monitoring organophosphate poisoning. 30 Compared with AChE, BuChE may show different inhibition, reactivation, and aging kinetics. Nevertheless, we are using the BuChE activity assay as a marker for the inactivation of cholinesterase in combination with other clinical signs of organophosphate poisoning.
Briefly, 200
BuChE activity was determined by the method of Ellman
Data Analysis
Results were analyzed by one-way analysis of variance (ANOVA) with Newman–Keuls multiple comparison test. The accepted level of significance for all tests was
RESULTS
In this study, we investigated the effect of the BGS, OxAc, and hGOT on the neuronal damage of rats after PO exposure, a potent AChE inhibitor (see Figure 1). From 31 rats that were exposed to PO and survived up to the end of the study, 15 animals were challenged with saline and 16 were with the OxAc+hGOT infusion. Ten naïve rats were used as a standard for PBR and histology assays. Eleven rats (26.2%), six from the control group (28.6%) and five from the treated group (23.8%), died within first 24 hours and therefore were excluded from the study.
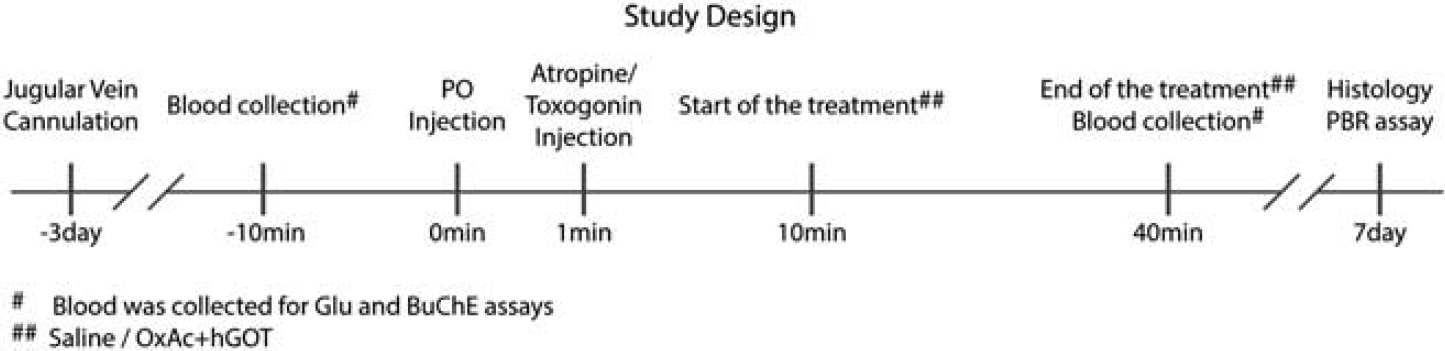
Study design. An illustration of the steps performed in the study.
Effect of Paraoxon Exposure on the Central and Peripheral Cholinergic System
In order to evaluate the action of PO as a potent AChE inhibitor, an equivalent assay verifying the activity of BuChE was performed (see Materials and Methods). We measured the control activity of BuChE (
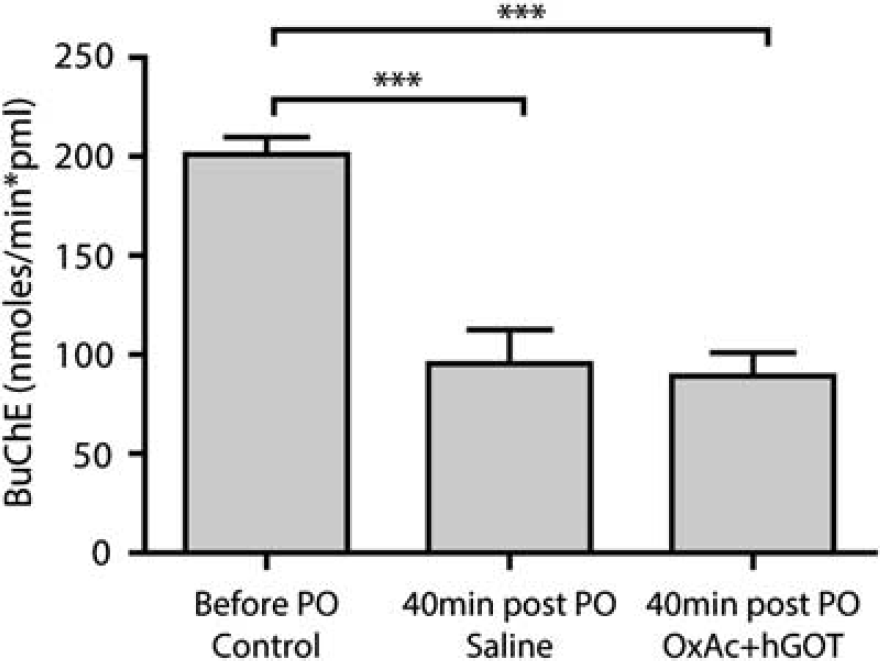
Butyrylcholinesterase activity decreases after paraoxon (PO) challenge. The rate of activity of butyrylcholinesterase (BuChE) is significantly lower 40 minutes after PO injection for both groups as compared with before the injection (control). There is no significant difference between PO and saline and PO and OxAc+hGOT (human glutamate oxaloacetate transaminase) groups indicating no direct effect of the blood glutamate scavenger (BGS) treatment on the PO activity. Control here refers to before the PO challenge (
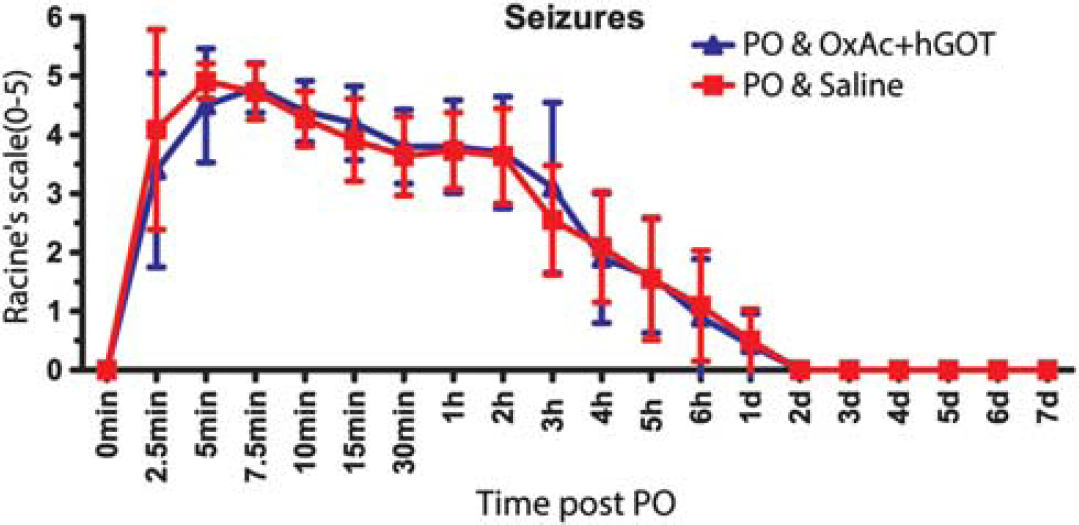
Clinical signs are similar between control and treated groups. Seizures were quantified on a Racine's scale (0 to 5). No significant difference was observed in the control (saline–red squares;
In order to evaluate the peripheral blood potency the Glu scavenger hGOT in combination with OxAc to reduce blood Glu level, we monitored the Glu levels before (165±15
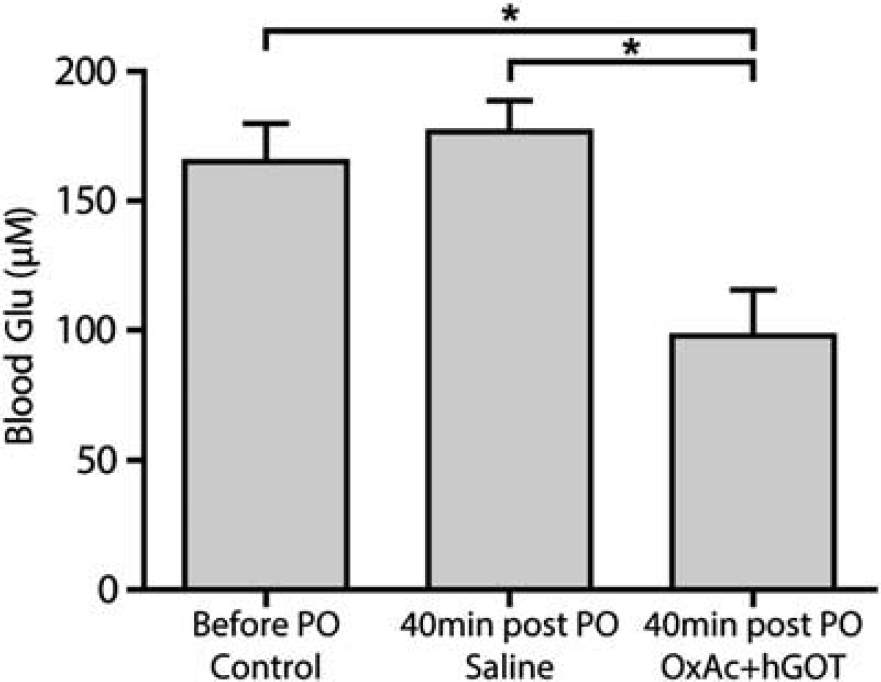
Blood glutamate (Glu) levels decreases with administration of Glu scavengers. The injection of OxAc+hGOT (human glutamate oxaloacetate transaminase) (
Effects of Glutamate Scavengers on the Central Nervous System After Exposure to Paraoxon
Our hypothesis asserts that peripheral effects of Glu scavengers cause beneficial effects by leading to the reduction in the excess brain Glu level responsible for the brain damage associated with organophosphates exposure. Previous investigations have indicated that the measurement of PBR density could be of widespread applicability in the quantification of neural tissue damage in the central nervous system as this astrocytes and microglia receptor is a known indicator of cell stress in many conditions.32, 33 Moreover, given that different areas in the brain are affected upon exposure to organophosphate (see Introduction); we sought to analyze and compare the brains of the naïve versus the two treated groups. Using a radioactive affinity assay, we observed a significantly lower concentration of PBR in the brain of the naïve animals (see Figure 5; 132±13 fmol/mg;
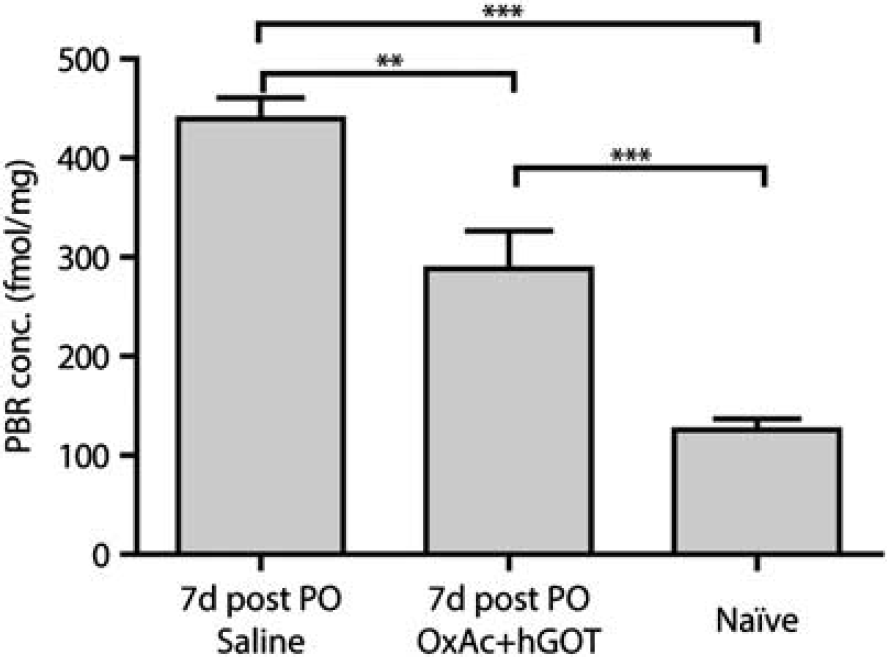
Whole-brain peripheral benzodiazepine receptor (PBR) concentration is lowered upon administration of glutamate (Glu) scavengers. Peripheral benzodiazepine receptor concentration is a good indicator of neuronal stress. Here we see a significant reduction in PBR concentration as a result of the BGS treatment. Although naïve animals (
To further confirm the central protective effects of Glu scavengers, an immunohistological analysis of cell numbers in the piriform cortex, an area highly susceptible to seizure-induced neuropathology was conducted. This area was the only one in which significant morphologic damage in the saline group was found (data not shown). The same results were observed: hence naïve animals had a significantly higher number of traceable cells (see Figure 6C for a representative example; 14.8±0.95 cells/mm2) compared with the brains of OxAc+hGOT-treated animals (see Figure 6A for representative examples; 10.6±1.37 cells/mm2) and the saline groups (see Figure 6B for representative examples; 3.72±0.9 cells/mm2). Nevertheless, in the OxAc+hGOT group, there was a substantial rescue of cell numbers (
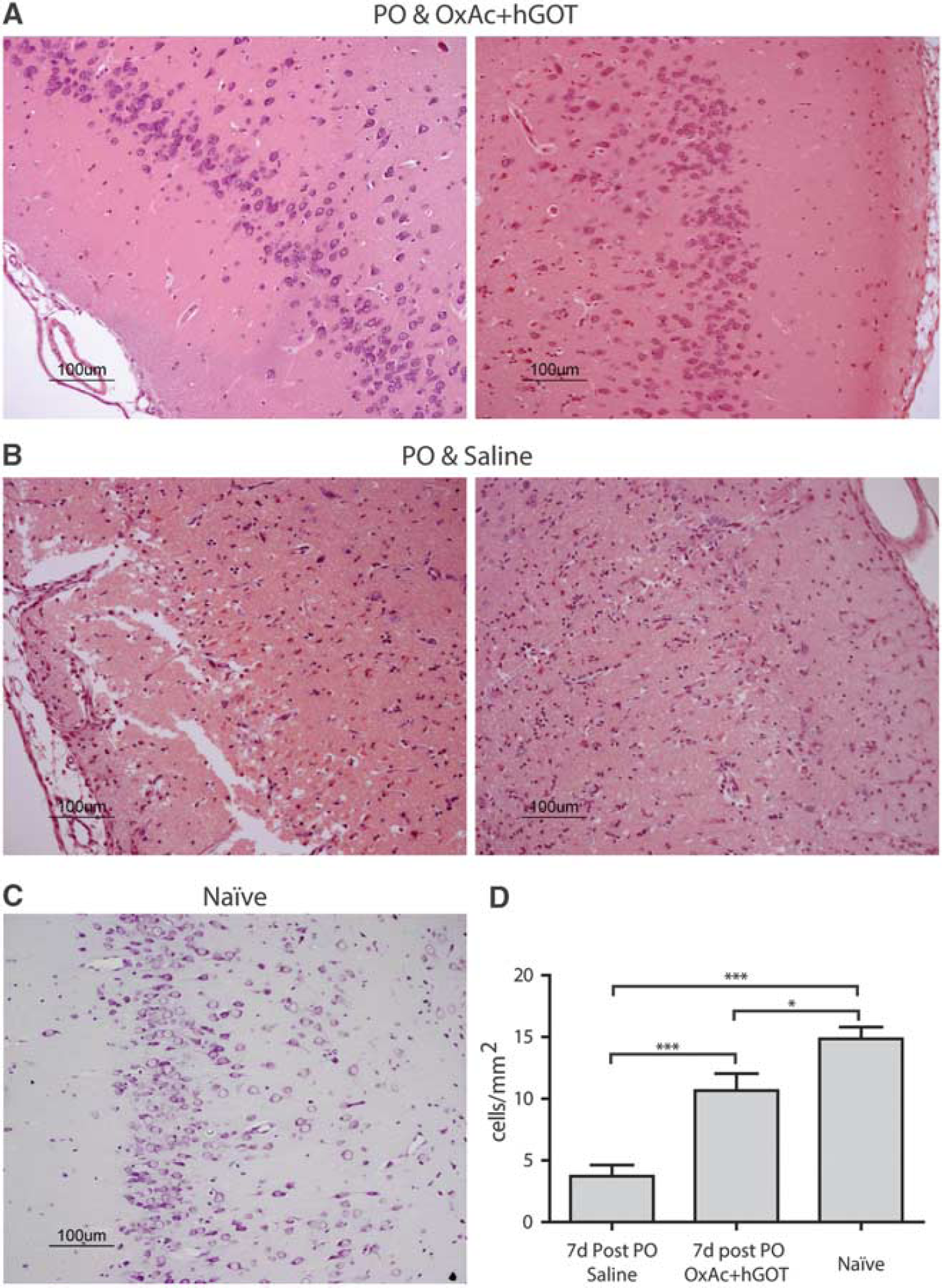
Cell numbers in the piriform cortex are rescued as a result of glutamate (Glu) scavengers. (
DISCUSSION
To date, organophosphate-induced brain damage is an irreversible neuronal injury because no pharmacological treatment is currently available to prevent or block secondary damage processes.34, 35 The major contributors to the secondary neuronal brain damage, manifested in cell death, are thought to be calcium influx and apoptosis as a result of excessive release of extracellular Glu. 36 It has been previously shown that after organophosphates intoxication, there is a release of excess Glu in the brain, which appears to cause the neurologic damage. 3 Understanding secondary neuronal damage processes offer a potential therapeutic window in which progressive neural injury and neuronal cell death may be inhibited, and the extent of disability may be reduced during the first few hours after exposure to organophosphates.37, 38 In this study, we demonstrate for the first time the ability of BGS to reduce the secondary neuronal damage in an animal model of PO intoxication treated with a combination of OxAc and hGOT.
Under normal physiologic conditions, Glu, which is released into the synaptic cleft, is quickly removed by excitatory amino-acid transporters located in astrocytes and neurons. However, this widely accepted view of the homeostasis of Glu in brain fluids does not take into consideration the possible contribution of the blood–brain barrier. The maintenance of brain extracellular Glu at levels below its excitotoxic threshold is performed not only by the Glu transporters located on glia and neurons but also by those present on the anti-luminal side of the brain capillary endothelial cells.19, 20 These transporters shuttle the excess extracellular brain Glu to the blood. 21 The method presented in the current study causes a rapid decrease of blood Glu levels. Based on our previous studies, we suggest that the gradient that was created after OxAc/GOT injection leads to the efflux of the excess brain Glu into the blood stream thereby reducing its potential to cause neurologic damage.
Based on previous experiments in our lab and others, we suggest that the combination of a single dose of OxAc and hGOT is optimal for the PO intoxication model. This treatment shows a long lasting effect of several hours in reducing blood Glu levels, 21 and omits the need for repetitive dosing. Additionally, these doses and treatment plan were also proven as beneficial in a rat model of head injury and middle cerebral artery occlusion.22, 24 It is worth mentioning that in contrast to the PO animal model presented in this study, in the above mentioned models, there is a local breach of the blood–brain barrier that could facilitate the efflux of excess Glu.
As BGS treatment takes place in the peripheral blood alongside the detrimental activity of PO, it was important to monitor PO activity in both BGS and saline treated groups to exclude a direct effect of BGS on PO activity. We demonstrate that BuChE activity and the clinical signs of both groups are similar, strongly suggesting that there is no direct effect of OxAc+hGOT treatment on the activity of PO as an AChE inhibitor. Moreover, as early as 30 minutes from OxAc+hGOT injection, the level of the blood Glu level was significantly lowered in the treated group, compared with the control group, which was injected with saline. Thus and based on previous studies showing increased brain Glu levels after organophosphate exposure, 9 we suggest that a gradient is formed between the blood and brain Glu levels, a necessary condition for the central nervous system Glu efflux.
Having demonstrated the blood Glu-lowering activity of the OxAc+hGOT treatment, we investigated the ability of BGS to reduce the excitotoxic effect of excess brain Glu levels after PO intoxication. 39 Testing for PBR density, which is a well-established marker of the brain neuronal damage, we show that our treatment significantly reduced the elevated whole-brain PBR density in the treated animals, as compared with the control group. To further analyze if the elevated level of PBR in treated animals manifested at a whole-brain level, an immunohistochemical analysis was performed. The results from the comparison of the treated and the naïve animals clearly demonstrated that even though the number of neurons in the naïve rats was higher than both groups, the OxAc+hGOT treatment succeeded in preventing the death of neurons in the piriform cortex, an area highly susceptible to seizure-induced neuronal damage. Interestingly, unlike previous studies3, 5, 7 where different brain regions were damaged, in our study only the neurons in the piriform cortex were affected. We do not know what the source of this selectivity is, but can hypothesize that the difference in the organophosphates used, the different model system and experimental setup might explain some of the variability.
In contrast to our expectation and other studies treating organophosphate poising, 18 BGS treatment did not reduce the seizure intensity or the survival of the treated animals. We hypothesize that as our treatment is relatively moderate, a stronger dosing regimen or pretreatment could have reduced mortality or seizure intensity of the treated group and not only the secondary brain damage.
Based on data obtained from several
The clear advantage of our BGS technology is that there seem to be no direct intervention with any of the brain Glu receptors or Glu transport systems. The administration of OxAc+hGOT only temporarily affects the blood Glu levels and this creates a gradient between the levels of Glu in the blood and in the brain, causing a rapid efflux of excess Glu from the brain to the blood without affecting any other brain functions. 22 As plasma Glu levels normally fluctuate by ∼50% during the circadian cycle, 40 we speculate that in the transition of the BGS technology into clinical trials, no severe side effects are expected.
CONCLUSION
We have demonstrated that the
Footnotes
The authors declare no conflict of interest.
ACKNOWLEDGMENTS
We would like to thank Tamara Berkutzki for her professional support in the histology analyses and Professor David Mirelman for his excellent advice on style and content of the manuscript.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.