
Abstract
Cerebral ischemia–reperfusion injury is associated with a developing inflammatory response with pathologic contributions from vascular leukocytes and endogenous microglia. Signaling chemokines orchestrate the communication between the different inflammatory cell types and the damaged tissue leading to cellular chemotaxis and lesion occupation. Several therapies aimed at preventing this inflammatory response have demonstrated neuroprotective efficacy in experimental models of stroke, but to date, few investigators have used the chemokines as potential therapeutic targets. In the current study, the authors investigate the neuroprotective action of NR58–3.14.3, a novel broad-spectrum inhibitor of chemokine function (both CXC and CC types), in a rat model of cerebral ischemia–reperfusion injury. Rats were subjected to 90 minutes of focal ischemia by the filament method followed by 72 hours of reperfusion. Both the lesion volume, measured by serial magnetic resonance imaging, and the neurologic function were assessed daily. Intravenous NR58–3.14.3 was administered, 2 mg/kg bolus followed by 0.5 mg/kg · hour constant infusion for the entire 72-hour period. At 72 hours, the cerebral leukocytic infiltrate, tumor necrosis factor-α (TNF-α), and interleukin-8 (IL-8)-like cytokines were analyzed by quantitative immunofluorescence. NR58–3.14.3 significantly reduced the lesion volume by up to 50% at 24, 48, and 72 hours post–middle cerebral artery occlusion, which was associated with a marked functional improvement to 48 hours. In NR58–3.14.3-treated rats, the number of infiltrating granulocytes and macrophages within perilesional regions were reduced, but there were no detectable differences in inflammatory cell numbers within core ischemic areas. The authors reported increased expression of the cytokines, TNF-α, and IL-8–like cytokines within the ischemic lesion, but no differences between the NR58–3.14.3-treated rats and controls were reported. Although chemokines can have pro-or antiinflammatory action, these data suggest the overall effect of chemokine up-regulation and expression in ischemia–reperfusion injury is detrimental to outcome.
Inflammatory processes have been implicated in the pathophysiology of cerebral ischemia–reperfusion injury (IRI) (Barone and Feuerstein, 1999). Experimental IRI studies have shown recruitment and influx of vascular leukocytes, mainly neutrophils, into the lesioned brain in the early postischemic period (Wang et al., 1993). These activated neutrophils promote cerebral ischemic injury by microvascular plugging, thereby reducing cerebral blood flow to tissue “at risk,” and by the production of cytotoxic substances. A strategy aimed at reducing neutrophil accumulation or activation therefore may represent an important therapeutic target to reduce the extent of brain damage after ischemic stroke. However, the regulation of granulocyte activation is complex. Although neutrophils can target and occupy the brain in abundance under the chemotactic influence of a single intracerebrally expressed chemokine, macrophage inflammatory protein 2, they do not become cytotoxic in the absence of the appropriate pathologic signals (Bell et al., 1996).
Several antineutrophil pharmaceuticals have shown efficacy in experimental IRI. Ischemic damage can be attenuated either by neutropenia (Chen et al., 1992) or by impairing neutrophil–endothelial interactions using pharmacological approach (Chopp et al., 1994; Zhang et al., 1994) or genetic deletions of endothelial adhesion proteins (Soriano et al., 1996). Neutrophil inhibitory factor (UK-279,276), a recombinant glycoprotein derived from the dog hookworm, has shown neuroprotective efficacy in IRI by reducing both the extent of ischemic damage and the number of infiltrating neutrophils (Jiang et al., 1995). These studies provide experimental evidence to support the antineutrophil approach for the treatment of IRI.
To date, successful reduction in IRI in animal model studies have not been translated to the clinic. For example, a multicenter, antiintercellular adhesion molecule-1 trial (Enlimomab) failed, with increased morbidity and mortality in the patients receiving the test antibody (Sherman, 1997). However, in the Enlimomab trial, cerebral perfusion indices were not used to dictate patient recruitment. Because experimental antiinflammatory agents like neutrophil inhibitory factor are only neuroprotective in models of IRI and have no efficacy in models of permanent ischemia (Jiang et al., 1995), it is possible that this study failed, at least in part, through poor design. Therefore, it seems prudent to define further the mechanisms involved in inflammatory injury after stroke to identify more specific therapeutic targets.
The authors hypothesize that identification of the signals that lead to leukocyte recruitment and activation would provide useful new targets for therapeutic intervention. Chemokines are a group of low molecular weight chemotactic cytokines that exhibit a variety of proinflammatory activities. It has been postulated that stimulated expression of certain chemokines, such as interleukin-8 (IL-8), by central nervous system cells is crucial for postischemic vascular leukocyte targeting and accumulation and is the major determinant of leukocyte composition in inflammatory infiltrates (Ransohoff and Tani, 1998). Some successful experimental neuroprotection studies using an anti-chemokine approach for the treatment of stroke have been reported. A monoclonal antibody to rat cytokine-induced neutrophil chemoattractant—an IL-8–like neutrophil chemoattractant and homologue of murine macrophage inflammatory protein 2—reduced edema formation 24 hours after reperfusion (60-minute ischemia) and reduced the size of the infarction area 7 days postreperfusion (Yamasaki et al., 1997). Cytokine-induced neutrophil chemoattractant production, mainly by the endothelium, previously had been reported to precede polymorphonuclear leukocyte extravasion into the ischemic lesion (Yamasaki et al., 1995). Similarly, IL-8 production by the reperfused rat brain has been demonstrated and both cerebral edema and infarct were effectively reduced by a neutralizing anti–IL-8 antibody when administered at reperfusion (Matsumoto et al., 1997). These studies highlight the potential of a therapy aimed at targeting IL-8 action in IRI. Such a strategy could be promising in humans for which systemic increases of IL-8 expressing mononuclear cells and plasma IL-8 levels from patients with ischemic stroke have been reported (Kostulas et al., 1998). This work suggests that in cerebral ischemia, as in wound healing, early recruitment of mononuclear cells is responsible for the production of neutrophil chemoattractants, such as cytokine-induced neutrophil chemoattractant and IL-8, which subsequently orchestrate the granulocytic infiltrate responsible for exacerbating tissue damage. Consequently, it is plausible that a broad-spectrum chemokine inhibitor, capable of blocking the recruitment of mononuclear cells and granulocytes, may be more effective than agents that target a single chemokine.
Although there are now a range of antibodies and small molecule receptor antagonists that inhibit leukocyte recruitment by a single chemokine or a small number of closely related chemokines (Horuk and Ng, 2000), few broad-spectrum chemokine inhibitors have been reported. Recently, however, the authors designed and characterized a number of peptides and peptide derivatives that inhibit leukocyte migration induced by a wide range of chemokines in vitro (Reckless and Grainger, 1999). One such derivative—NR58–3.14.3, a retroinverso analogue of a 12-mer peptide from the human monocyte chemoattractant protein-1 sequence—possesses broad pan-chemokine specificity, nanomolar potency, and an acceptable pharmacologic profile (Wilbert et al., 2000).
In the current study, the authors tested the neuroprotective efficacy of NR58–3.14.3 in a rat model of IRI and provided some histologic evidence for its mode of action.
MATERIALS AND METHODS
Sixteen fed, male Sprague–Dawley rats (Charles Rivers; weighing approximately 330 g) were anesthetized with 2% halothane in 70%:30% N2O:O2, and the left middle cerebral artery (MCA) was occluded for 90 minutes using the intraluminal thread approach (Koizumi et al., 1986). Throughout the surgical period, rectal temperature was monitored and maintained at 37°C with a heated blanket and feedback rectal temperature probe and arterial blood gases were measured before and immediately after MCA occlusion (MCAO). During the ischemic period, rats recovered allowing for neurologic deficit assessment (Bederson et al., 1986) before reperfusion. The neurologic deficit score was used to indirectly assess the “quality” of the occlusion; only rats displaying spontaneous right side-circling gait disturbances (score of 3) were used for further study.
After 90 minutes of ischemia, the rats were reanesthetized and the intraluminal thread withdrawn into the external carotid artery stump enabling antegrade perfusion of the left MCA through the patent common carotid artery. For this study, to maximize the potential benefit of the test compound and to reflect possible clinical use as an adjunct to thrombolysis, the treatment was given at the time of reperfusion. The investigator performing the experimental paradigm was blinded to the identity of the test compound, either sterile, endotoxin-free phosphate-buffered saline (PBS) vehicle or NR58–3.14.3 (0.5 mg/mL in PBS) was administered by jugular venepuncture at a dose of 2 mg/kg. This dose had previously shown antiinflammatory efficacy in other models in vivo, including atherosclerosis and intradermal lipopolysaccharide injection (Reckless et al., 2001). A femoral vein cannula was inserted, tunnelled under the skin, and exteriorized at the back of the neck. This was used to administer either PBS vehicle or NR58–3.14.3 (0.5 mg/mL) by constant intravenous infusion (0.5 mg/kg · hour or equivalent volume for control) over the following 72 hours. Rectal temperature was measured at reperfusion and then daily until the end of the experiment.
The ischemic damage was assessed by conventional structural magnetic resonance imaging (MRI), (echo time [TE] = 70 and 30 milliseconds, repetition time [TR] = 3000 milliseconds) on day 1, 2, and 3 post-MCAO.
For all MR procedures, anesthesia was induced and maintained at 1% halothane v/v in oxygen. Rectal temperature was maintained at 37°C. Magnetic resonance imaging was performed at 4.7 T using a SIS-200 imaging spectrometer (Spectrocopy Imaging Systems, Fremont, CA, U.S.A.) and a home-built 75-mm diameter 8-legged birdcage radiofrequency coil. Eighteen contiguous, coronal slices starting at the level of the eyes, running rostral to caudal through the brain, were acquired using a 128 × 128 acquisition matrix covering a field of view of 4 × 4 cm. Each slice was 0.9-mm-thick with an in-plane spatial resolution of approximately 0.3 mm.
After the final MR analysis at 72 hours post-ictus, the rats were decapitated, and brains were removed and snap-frozen in cryo-protecting medium (Cryo-M-Bed; Bright Instruments, Huntingdon, Cambridgeshire, U.K.) at −70°C.
Immunofluorecence staining
Cryosections (10-μm-thick coronal sections) were cut at −10°C for the immunofluorescence detection of granulocytes (neutrophils), CD14+ cells (macrophages), tumor necrosis factor-α (TNF-α), and IL-8.
Quantitative immunofluorescence analyses (Mosedale et al., 1996) were performed on three slices taken from each of five stereotaxic levels through the MCA territory. The focal and unilateral nature of the pathology in this particular model of stroke enabled the unaffected contralateral hemisphere to be examined for comparison to the lesioned hemisphere in each analysis. Three additional sections neighboring those stained with each antibody were incubated similarly, but without the primary antibody, and served as the negative control for each epitope. Granulocytes (with the morphology of neutrophils) were counted in 4 regions of interest (ROIs; Fig. 1) in both ipsilateral and contralateral hemispheres using a primary monoclonal antibody (mouse IgM anti-rat granulocyte, MCA-967, 1 in 10 dilution, 4°C, overnight; Serotec, Oxford, U.K.) and visualized with a secondary antibody linked to fluorescein isothiocyanate (Jackson, West Grove, PA, U.S.A.).
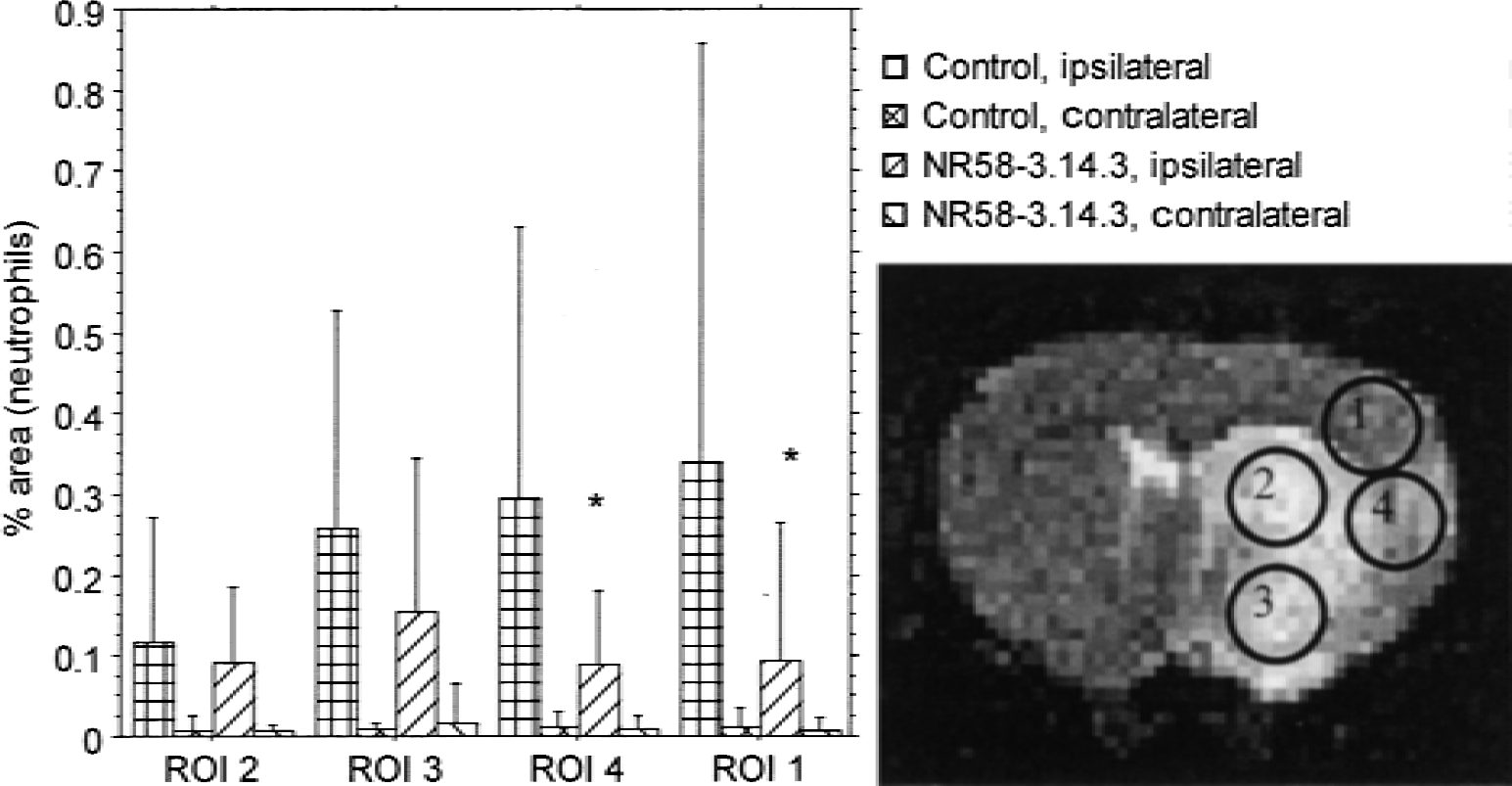
Representative coronal T2-weighted magnetic resonance image taken from the level of the caudate putamen at 24 hours post–middle cerebral artery occlusion. The hyperintense lesion is clearly visible in the caudate and surrounding neocortex. Neutrophils were analyzed from within the four regions of interest (ROIs) indicated on the image in both hemispheres. Significant neutrophil reductions were shown in regions 1 and 4 with NR58–3.14.3 treatment. * P < 0.05, nonparametric Mann-Whitney U test. Data is expressed as mean ± SD, n = 7 per group.
In a separate series of slices, CD14+ cells (predominantly macrophages and some monocytes) were labeled with an anti-rat CD14 monoclonal antibody (3.125 μg/mL Serotec MCA-342, 4°C, overnight) and reported with a secondary antibody linked to rhodamine (tetramethyl-rhodamine isothiocynate (TRITC)), (donkey anti-mouse IgG Fab2 TRITC, Jackson (30 μg/mL, 22°C). Positive cells were counted from five ROIs within the lesioned hemisphere (Fig. 2).
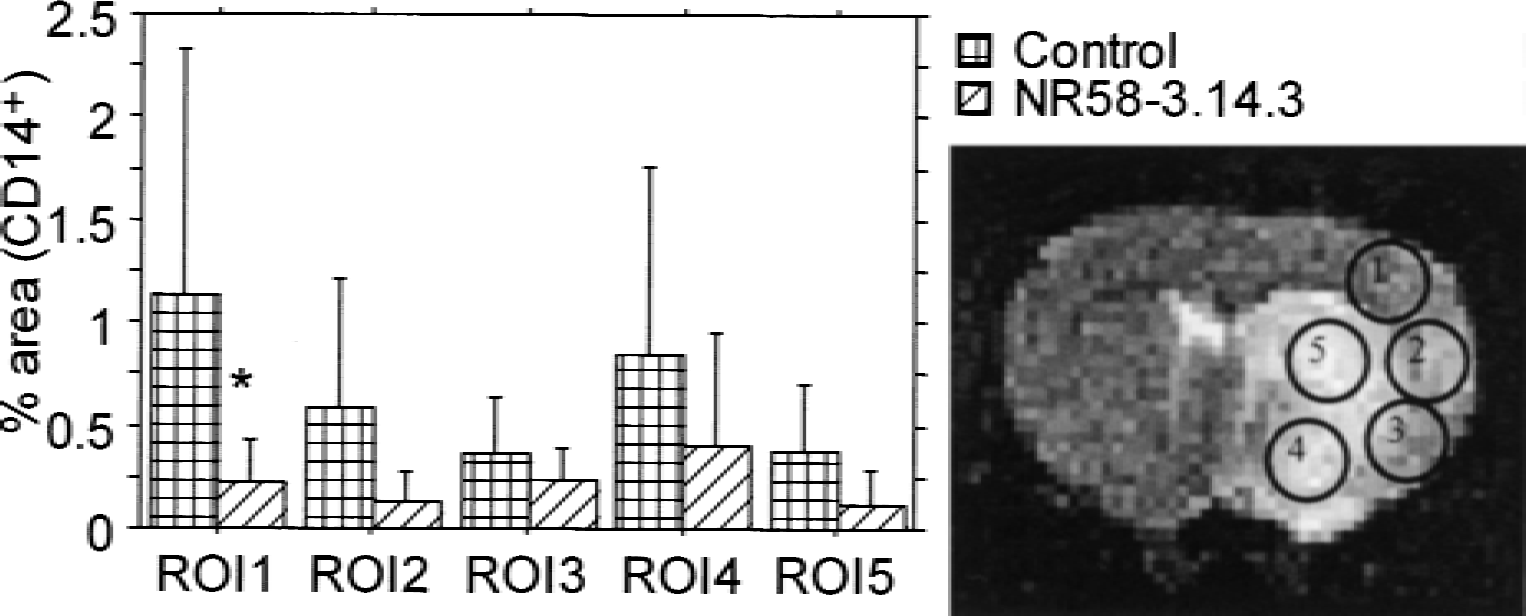
Representative coronal T2-weighted magnetic resonance image taken from the level of the caudate putamen at 24 hours post–middle cerebral artery occlusion. CD14+ cells were analyzed from within the 5 regions of interest (ROIs) indicated within the lesion. Significant CD14+ cells reductions were shown in region 1 only with NR58–3.14.3 treatment. * P < 0.05, nonparametric Mann-Whitney U test.
Again, in a separate series of slices, sections were analyzed for TNF-α and IL-8–like cytokines using rabbit anti-human TNF-α polyclonal antibody (IgG, 5 μg/mL, 4°C, overnight; Santa Cruz Biotechnology, Santa Cruz, CA, U.S.A.; good rat crossreactivity) and goat anti-human IL-8 (R & D Systems, Abingdon, Oxford, U.K.; AB-208-NA, 50 μg/mL, 4°C, overnight, broad specificity to rodent IL-8 related cytokines), respectively. These primary antibodies were reported with a secondary antibodies linked to TRITC (donkey anti-rabbit IgG TRITC, 30 μg/mL, 22°C) and fluorescein isothiocyanate (donkey anti-goat FITC, 30 μg/mL, 22°C). Five ipsilateral ROIs per section were analyzed for TNF-α and IL-8–like cytokines.
For each experiment, Hoechst 33342 (1 μg/mL, Molecular Probes) also was added during the secondary antibody incubation as a nuclear stain.
Image analysis
Magnetic resonance imaging
Image analysis was performed using Dispim software (developed by Dr. Plummer, University College, London, U.K.). Lesion volumes were calculated from T2 maps generated on a pixel-by-pixel basis by fitting the raw image data for each pixel (using TE = 30 and 70 milliseconds) to a monoexponential. Areas of central nervous system lesion were demarcated using an automated contour and threshold process in which a pixel intensity > mean (of the corresponding locus in the contralateral hemisphere) + 2 standard deviations was taken to be pathologic. Pixel intensities in the unaffected hemisphere were used to calculate the “normal” mean for each subject.
Quantitative immunofluorescence
Immunofluorescence images were captured using an Olympus AX 70 microscope (×20 lens, with a mercury light source) and a Hamamatsu C4742–95 digital camera, producing 512 × 512 10-bit images. Automated image capturing programs and analysis were performed, as previously described (Mosedale et al., 1996), using Openlab 2 software (Improvision, Coventry, U.K.).
Statistics
Repeated measures analysis of variance with post hoc Bonferroni tests were used for within and between treatment group comparisons for lesion volume, neurologic deficit, and rectal temperature with time. For the immunofluorescence studies, a nonparametric Mann-Whitney rank test was used for total immunofluorescence and individual ROI immunofluorescence comparisons. P < 0.05 indicated statistical significance.
RESULTS
Two (1 test, 1 control) of the 16 rats had low neurologic deficit scores (<2) at 90 minutes post-MCAO and were removed from further study. Pathologic examination of the brains from these two rats revealed a subarachnoid hemorrhage in one and a bifurcated left MCA (at the level of the Circle of Willis) in the other.
Physiologic variables
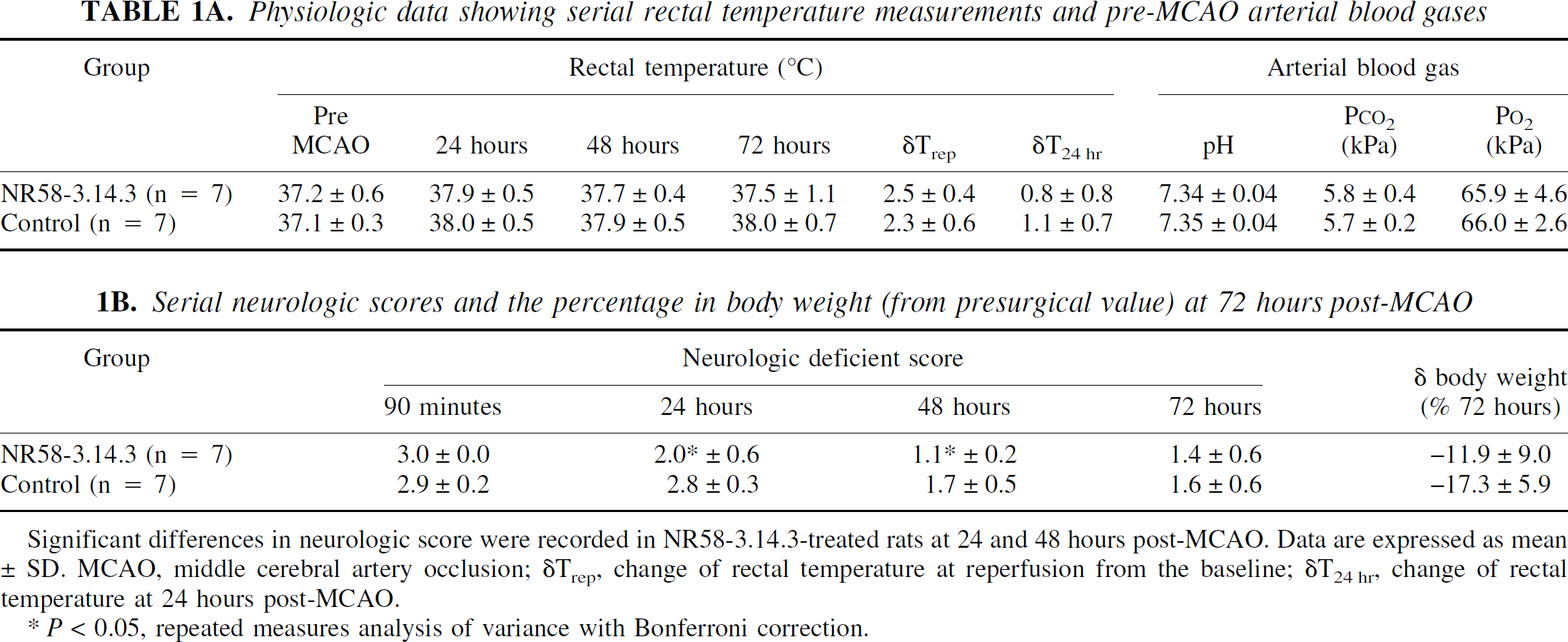
The measured pre-MCAO physiologic variables (rectal temperature and arterial blood gases) were not different between the groups (Table 1A). At reperfusion, there was a marked elevation in rectal temperature in both groups (>2.3°C from baseline, P < 0.001), but there was no significant difference between animals receiving NR58–3.14.3 and those receiving vehicle. At 24 hours post-MCAO, rectal temperature returned toward pre-MCAO values in both groups, but remained slightly elevated through the 72-hour endpoint. This persistent elevation was similar in magnitude between the 2 groups (Control δT72hr = 1.1°C ± 0.6°C versus NR58–3.14.3 δT72hr = 0.4°C ± 0.9°C, mean ± SD, n = 7, P > 0.05).
Physiologic data showing serial rectal temperature measurements and pre-MCAO arterial blood gases
Behavioral measures
Immediately before reperfusion, all rats (except the 2 that were excluded) displayed a neurologic deficit score of 3 (spontaneous right circling gait) on the Bederson scale, and the mean score was not different between the 2 groups (Table 1B). At 24 and 48 hours post-MCAO, the neurologic deficit in the control group remained high, although by 72 hours it was beginning to normalize, which is consistent with previous studies in this model. In contrast, the neurologic deficit was significantly lower at 24 and 48 hours postreperfusion in the animals treated with the chemokine inhibitor, NR58–3.14.3 (P < 0.05 at each time point).
Serial neurologic scores and the percentage in body weight (from presurgical value) at 72 hours post-MCAO
Significant differences in neurologic score were recorded in NR58–3.14.3-treated rats at 24 and 48 hours post-MCAO. Data are expressed as mean ± SD. MCAO, middle cerebral artery occlusion; δTrep, change of rectal temperature at reperfusion from the baseline; δT24 hr, change of rectal temperature at 24 hours post-MCAO.
P < 0.05, repeated measures analysis of variance with Bonferroni correction.
Lesion volumes
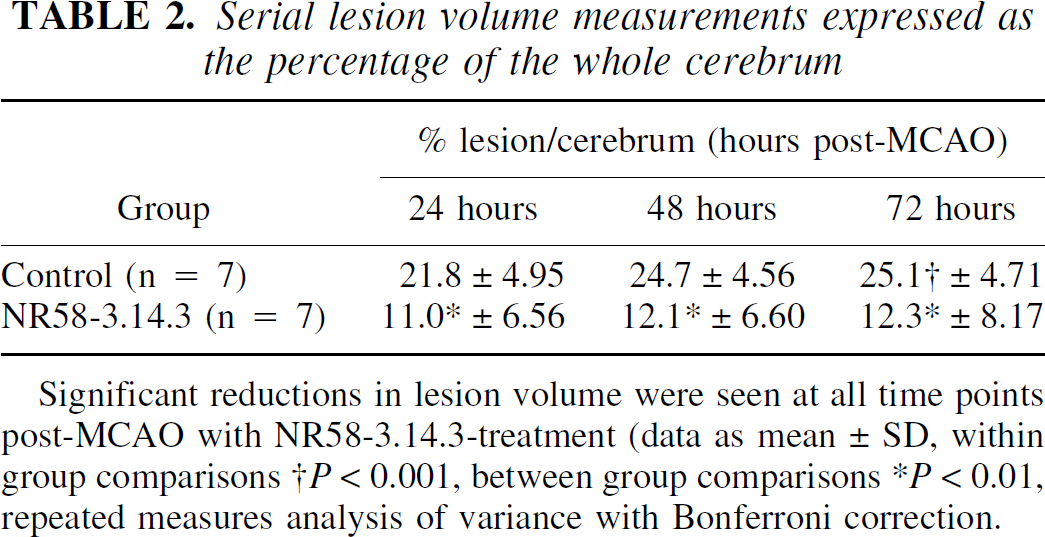
Ischemic lesion volumes on day 1, 2, and 3 post-MCAO are shown in Table 2. In control animals, the lesion continued to increase in size between day 1 and 2 (P < 0.01, repeated measures analysis of variance), but thereafter remaining the same. However, among the rats treated with NR58–3.14.3, the lesion volume was maximal at day 1 and thereafter remained constant until the 72-hour endpoint. Furthermore, the treatment with NR58–3.14.3 markedly reduced the ischemic lesion volume at all 3 time points post-MCAO (P < 0.01).
Serial lesion volume measurements expressed as the percentage of the whole cerebrum

Significant reductions in lesion volume were seen at all time points post-MCAO with NR58–3.14.3-treatment (data as mean ± SD, within group comparisons †P < 0.001, between group comparisons *P < 0.01, repeated measures analysis of variance with Bonferroni correction.
Granulocyte analysis
The number of granulocytes present in the brain (expressed as percentage of tissue area stained) in 4 ROIs within both the ipsilateral and contralateral hemispheres at 72 hours post-MCAO are shown in Fig. 1. In each ipsilateral ROI, granulocytes were seen associated with the vascular endothelium and neural parenchyma but few were seen in the contralateral hemisphere, and these were restricted to the lumen of the blood vessels only. Treatment with NR58–3.14.3 significantly reduced the number of granulocytes within ROIs typically regarded as penumbral in this model (ROIs 1 and 4 within the parietal cortex), where the largest number of granulocytes were found in control brains, but had no effect on the small number of granulocytes that penetrated the “core” zones, that is, ROIs 2 (caudate putamen) and 3 (preoptic area).
CD14+ cell analysis
As with granulocytes, CD14+ cells were seen within the vascular lumen and endothelial lining of pial and other parenchymal vessels within the lesioned hemisphere of control rats, with few, if any, detected in the contralateral hemisphere. Significant staining also was detected in the perivascular space between neural parenchyma and the vascular endothelial cells. Treatment with NR58–3.14.3 reduced CD14+ cells in the lesion penumbra, although this reduction reached statistical significance in only one ROI, the upper parietal cortex (Fig. 2).
IL-8–like cytokines and TNF-α analysis
The levels of IL-8–like cytokines and TNF-α (reported as percentage of area stained) are shown in Fig. 3. Consistent with previous reports, the level of IL-8–like cytokines were markedly increased in the ipsilateral hemisphere compared with the contralateral side (background level of fluorescence only. TNF-α also was increased, although to a lesser extent compared with IL-8–like cytokines. Treatment with NR58–3.14.3 did not affect levels of IL-8–like cytokines or TNF-α averaged over all 4 ROIs, nor in any of the ROIs individually (P = 0.43, Mann-Whitney U Test) or TNF-α (P = 0.19, Mann-Whitney U Test). However, the effects of NR58–3.14.3 on the levels of proinflammatory cytokines at earlier time points post-MCAO cannot be excluded.
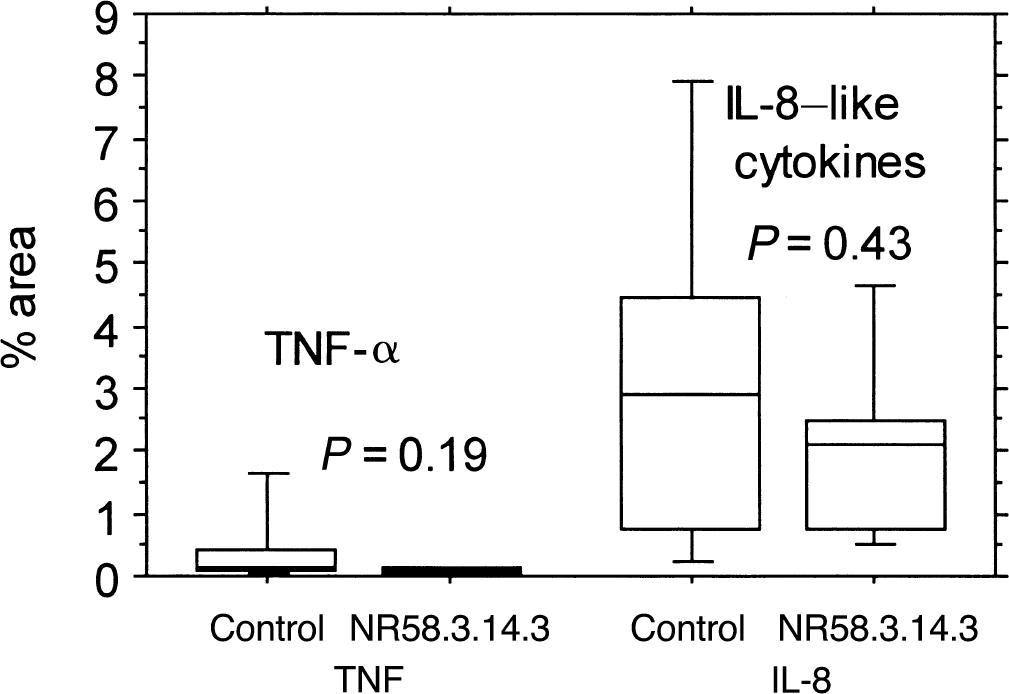
Box and whisker plots showing the percentage area stain for total tumor necrosis factor-α (TNF-α) and interleukin-8–like cytokines from within the same 5 ipsilateral regions as Fig. 2. Immunofluorescence staining from within the contralateral hemisphere was of background level only. Data is shown as median with 25th and 75th percentiles. No differences between the two treatment groups were found.
DISCUSSION
In the current study, the authors report neuroprotection of the adult rat brain after treatment of experimental IRI with a broad-spectrum chemokine inhibitor. In control rats after a 90-minute MCAO, the lesion volume was maximal at 48 hours post-ictus with most of the MRI-observable damage (∼88%) developing within the first 24 hours. This expansion of the lesion to 48 hours was typically observed in the parietal cortex and follows a similar pattern to other transient MCAO models in rats. In NR58–3.14.3-treated rats, ischemic lesion volumes were maximal on day 1 post-MCAO, but were reduced by as much as 50% when compared with controls. With continuous administration, this observed neuroprotection by NR58–3.14.3 persisted through to the 72-hour endpoint. The effect of NR58–3.14.3 on the ischemic lesion volume measured by MRI was associated with a marked functional improvement on days 1 and 2 post-MCAO.
All rats in the current study showed a marked hyperthermia at the time of reperfusion that persisted, albeit to a much lesser degree, until the 72-hour endpoint. However, treatment with NR58–3.14.3 did not affect the extent of this hyperthermia at any time point. Hyperthermia in the intraluminal thread model manifests from the unavoidable occlusion of the hypothalamic and anterior choroidal arteries with the intraluminal thread (He et al., 1999; Li et al., 1999). This thread-induced hyperthermia is a disadvantage of this particular model, only the continuous monitoring of brain and body temperature and maintenance of normothermia during the acute phase will discount this physiologic variable from contributing to the observed pathology (Colbourne et al., 1996).
As a first step toward understanding the mechanism by which NR58–3.14.3 mediated neuroprotection, the authors used immunohistochemical techniques in the current model to measure granulocytes and all macrophages (including microglia) within the ischemic brain. The authors observed a reduction in the number of granulocytes and macrophages in the penumbral ROIs only, that is, those cortical regions in close proximity to potential sites of middle cerebral artery and anterior cerebral artery anastomoses (Coyle and Jokelainen, 1982). There were no treatment-induced differences in the number of leukocytes within ROIs classically regarded as “core,” that is, within the striatum and basal ganglia. This observation lends support to the hypothesis that inflammatory cells contribute to the recruitment of penumbral regions to the final infarction, a process that may be suppressed in the NR58–3.14.3-treated rats. The relative contributions of hematogenous macrophages and granulocytes and activated microglia to this penumbral recruitment is likely to depend upon the degree of lesion reperfusion and blood–brain barrier disruption (Mabuchi et al., 2000). However, experiments to investigate the origins of the CD14+ cells the authors observed are plagued with technical difficulties because the distinction between hematogenous and activated brain macrophage phenotypes based on currently available immunologic markers is unreliable and the results should be treated with caution.
The unequivocal involvement of vascular leukocytes to IRI pathology during the acute reperfusion stage has recently been directly visualized with real-time fluorescence microscopy techniques. After only 15 minutes of reperfusion after a 2-hour MCAO, a persistent leukocyte adhesion and rolling in pial venuoles was observed (Ritter et al., 2000). The timing of this acute leukocyte and endothelial cell interaction (postreperfusion) depends upon the duration of the MCAO and occurs later after a milder ischemic insult (Ishikawa et al., 1999). Activated leukocytes may impair the cerebral blood flow by disturbance of the microcirculation through aggregation and vascular plugging.
Consequently, a reduction of microcirculatory polymorphonuclear leukocyte plugs and the amelioration of the no reflow phenomenon (del Zoppo et al., 1991), would be one possible mechanism through which NR58–3.14.3 could have such subacute effects. Indeed, this seems most likely, because NR58–3.14.3 requires opening of the blood–brain barrier to enter the brain parenchyma, a phenomenon demonstrated in similar IRI models (Gao Huang et al., 1999). This potential mechanism of action of NR58–3.14.3 should be investigated in future studies using contrast-enhanced and perfusion-weighted MR techniques.
Selective central nervous system expression of chemokines and other cytokines as a direct consequence of ischemic injury appear to orchestrate the activation and chemotaxis of distinct inflammatory cell types to the injury site (Glabinski and Ransohoff, 1999). Preventing the transcription of these signaling cytokines by the pharmacologic inhibition of the transcription factor NF-κB (Buchan et al., 2000, Philips et al., 2000) or with the use of p50 (a NF-κB subunit) knockout animals (Schneider et al., 1999) affords neuroprotection after experimental stroke and illustrates the general importance of cytokines to the overall inflammatory reaction. In vitro NR58–3.14.3 blocks the migration of human mononuclear and human myelo-monocytic leukemia cells induced by CC (such as monocyte chemoattractant protein-1) and CXC (such as IL-8) chemokines with ED50s in the 2 to 30 nmol/L range (Reckless et al., 2001). NR58–3.14.3 is an inhibitor of chemokine function and has little or no effect on the migration caused by other cytokines, such as transforming growth factor-β, or chemoattractants, such as C5a or fMLP. Thus, the reduced number of inflammatory cells in the penumbra of the ischemic lesion after treatment with NR58–3.14.3, despite unaltered levels of IL-8 like cytokines, is consistent with a similar chemokine–inhibitory mechanism for NR58–3.14.3 during IRI in vivo as is in transwell migration assays in vitro. These data lend support to the hypothesis that IL-8 is a key modulator for leukocyte recruitment to the lesion in IRI. However, the authors can only speculate as to whether suppression of CD14+ cells from the blood is the major chemokine-dependent mechanism that is blocked by NR58–3.14.3, because it is not known to what extent NR58–3.14.3 may have affected the resident microglia cells that also stain CD14+.
Individual chemokines and other cytokines of the inflammatory cascade can have both pro-and antiinflammatory activities on various cell types and form a signaling network that determines the overall level and cellular composition of the inflammatory response. This complexity makes the dissection of the role of individual cytokines in cerebral ischemia very difficult. The application of in vivo transgene technologies with specific gene “knockouts” of single cytokines can allow a more directed experimental approach. However, with the use of a broad-spectrum chemokine inhibitor, the authors have been able to conclude that although individual chemokines may have pro-or antiinflammatory effects in IRI, the cumulative impact of the chemokine network is to promote inflammation. But there also remains the possibility that NR58–3.14.3 is neuroprotective through, as yet, undefined pharmacologic effects quite distinct from chemokine inhibition and that it only delays the lesion maturation and does not prevent it. Nevertheless, within the limitations of the current study, NR58–3.14.3 treatment leads to the attenuation of the postischemic inflammatory response, reduces lesion volume, and improves neurologic function, thereby demonstrating the potential of this broadband pharmacologic approach.
Footnotes
Acknowledgments:
The authors thank Dr. Paul Kinchesh, Dept. of Chemistry, Queen Mary and Westfield College, University of London and the University of London Intercollegiate Research Service (ULIRS) for the provision of MRI facilities, and NeoRx Corporation, Seattle, Washington, U.S.A. for the NR58–3.14.3.