
Abstract
The Wlds mouse mutant demonstrates a remarkable phenotype of delayed axonal and synaptic degeneration after nerve lesion. In this study, the authors tested the hypothesis that expression of Wld protein is neuroprotective in an in vivo mouse model of global cerebral ischemia. This model is associated with selective neuronal degeneration in specific brain regions such as the caudate nucleus and CA2 hippocampal pyramidal cell layer. The extent of neuronal damage was quantified in Wlds compared to wild-type mice after an identical episode of global cerebral ischemia. The results demonstrated a significant and marked reduction in the extent of neuronal damage in Wlds as compared to wild-type C57Bl/6 mice. In the caudate nucleus, Wld expression significantly reduced the percentage of ischemic neuronal damage after global ischemia (Wlds, 27.7 ± 16.8%; wild-type mice, 58.7 ± 32.3%; P = 0.036). Similarly, in the CA2 pyramidal cell layer, there was a significant reduction of neuronal damage in the Wlds mice as compared to wild-type mice after ischemia (Wlds, 17.7 ± 23.0%; wild-type mice, 41.9 ± 28.0%; P < 0.023). Thus, these results clearly demonstrate that the Wld gene confers substantial neuroprotection after cerebral ischemia, and suggest a new role to that previously described for Wlds.
Keywords
The Wlds mutant mouse was discovered in the late 1980s as a spontaneous mutation in a breeding colony of C57Bl/6 mice supplied by Harlan-Olac (Lunn et al., 1989). The gene responsible resulted from the splicing of two genes, Ube4b and NMNAT, within an 85-kb triplication on chromosome 4. The chimeric gene codes for a novel 42-kd protein (Wld) whose function is yet to be defined. Wlds mutant mice show no readily discernable phenotype, yet what distinguishes these mice is a 10-fold delay in Wallerian degeneration after nerve damage (Gillingwater and Ribchester, 2001). The protection from axonal degeneration in these mice was originally identified and subsequently investigated in great detail within the peripheral nervous system. After axotomy, the structure and function of motor axons and presynaptic nerve terminals is preserved in Wlds mice and transgenic mice expressing Wld protein (Mack et al., 2001). However, the protection of synapses is weaker than that of axons (Gillingwater et al., 2002).
Recent studies have shown that the Wlds mutation also provides axon protection in response to neurodegenerative stimuli other than nerve lesion, such as vincristine neuropathy as well as mouse models of motor neuron disease and myelin-related axonopathies (Ferri et al., 2003; Samsam et al., 2003; Wang et al., 2001). Projections of central neurons are also protected after lesions of axonal fiber tracts in Wlds mice, including the perforant path of the hippocampal formation and optic nerve (Perry et al., 1991; Shi and Stanfield, 1996). Less evident is whether Wld is associated with neuronal preservation more generally in the central nervous system (Ludwin and Bisby, 1992).
Global cerebral ischemia in rodents is associated with selective neuronal damage. Moreover, synapses are known to undergo profound changes after ischemia (Hu et al., 1998; Ishimaru et al., 2001; Jourdain et al., 2002). Here, we examined whether expression of Wld is neuroprotective in Wlds mutant mice after transient global cerebral ischemia. The results show that there is a significant reduction in the extent of ischemic damage in vulnerable regions of the brain, namely the caudate nucleus and CA2 region of hippocampus, when the Wld protein is present.
MATERIALS AND METHODS
C57Bl/6J and natural mutant C57Bl/6/Wlds mice of approximately 7 or 8 weeks of age were obtained from Harlan-Olac Laboratories (Bicester, UK). All procedures were carried out under license from the Home Office with the approval of the University Ethical Review Panel and were subject to the Animals (Scientific Procedures) Act of 1986.
For all operative procedures, mice were initially anesthetized with 3% halothane in 70% N2O/30% O2, before halothane levels were reduced to 1.5% and maintained via a facemask, with the animals breathing spontaneously. The body temperature was strictly regulated at 37°C for the duration of the procedure. A midline cervical incision was made and both common carotid arteries were exposed. Taking care not to damage the vagus nerve, both common arteries were isolated using 4/0 silk thread. After a 5-minute stabilization period, both arteries were occluded using microaneurysm clips applied bilaterally for exactly 17 minutes (based on previous studies: Horsburgh et al., 1999a, 2000). Subsequently, both clips were removed and restoration of blood flow confirmed before the incision was sutured closed. After surgery, the mouse was placed in an incubator (28°C) for 2 hours before being returned to the standard animal housing unit. The operator was blinded to the mouse genotype. Equal numbers of C57Bl/6 and Wlds mice were randomly operated on the same day.
At 72 hours after surgery, mice were perfusion fixed under deep halothane anesthesia. The brain was perfused via the ascending aorta first with heparinized physiologic saline, followed by 4% paraformaldehyde. The brain was kept in the skull overnight, removed the next day, and then immersed in 4% paraformaldehyde for 2 hours before paraffin embedding. Sixmicrometer sections were cut on a microtome and stained with hematoxylin and eosin. The extent of neuronal cell death was quantified in hematoxylin and eosin-stained section. Under double-blind conditions, the numbers of morphologically normal neurons and neurons showing features of ischemic cell change were counted in five different fields in both hemispheres of the caudate nucleus and CA2 pyramidal cell layers of hippocampus using a 10-mm2 grid at ×400 magnification (Graham, 1992; Horsburgh et al., 1999b). The percentage of ischemic neurons in Wlds mice was compared with C57Bl/6 mice using nonparametric statistical tests. This method of quantification has been verified as nonbiased and reproducible in our hands.
The expression or absence of the Wld gene was confirmed using immunocytochemistry of brains and muscle biopsies from each experimental mouse. Perfusion fixed lumbrical muscles were dissected out and fixed for an additional 30 minutes in 4% paraformaldehyde. Muscles were incubated overnight in blocking solution (4% BSA, 0.5% Triton X in 0.1-mol/L PBS) before overnight application (4°C) of the primary antibody raised against the unique 18-amino acid sequence of the chimeric Wld protein (Wld18, 1:500 dilution; Mack et al., 2001). After washing for 5 hours in 0.1-mol/L PBS, the secondary antibody (TRITC-labeled antirabbit) was applied overnight at 4°C. Muscles were then washed again before being whole-mounted on glass slides and viewed using a BioRad (Radiance 2000) confocal microscope. The presence of Wld protein in caudate and CA2 neurons was also determined using immunocytochemistry on paraffin-embedded brain slices from both Wlds and C57Bl/6 experimental mice (see above). Brain slices were dewaxed and incubated in blocking solution (0.5% BSA, 10% goat serum in PBS) before overnight application (4°C) of the Wld18 primary antibody (1:500 dilution; Mack et al., 2001). After several washes, biotinylated antibodies were applied for 1 hour (Vector, biotinylated antirabbit raised in goat), before application of an avidin biotinylated horseradish peroxidase solution (Vector ELITE) for 1 hour and development of color using a DAB kit (Vector). The slices were then counterstained with hematoxylin before dehydration and mounting in DPX.
RESULTS
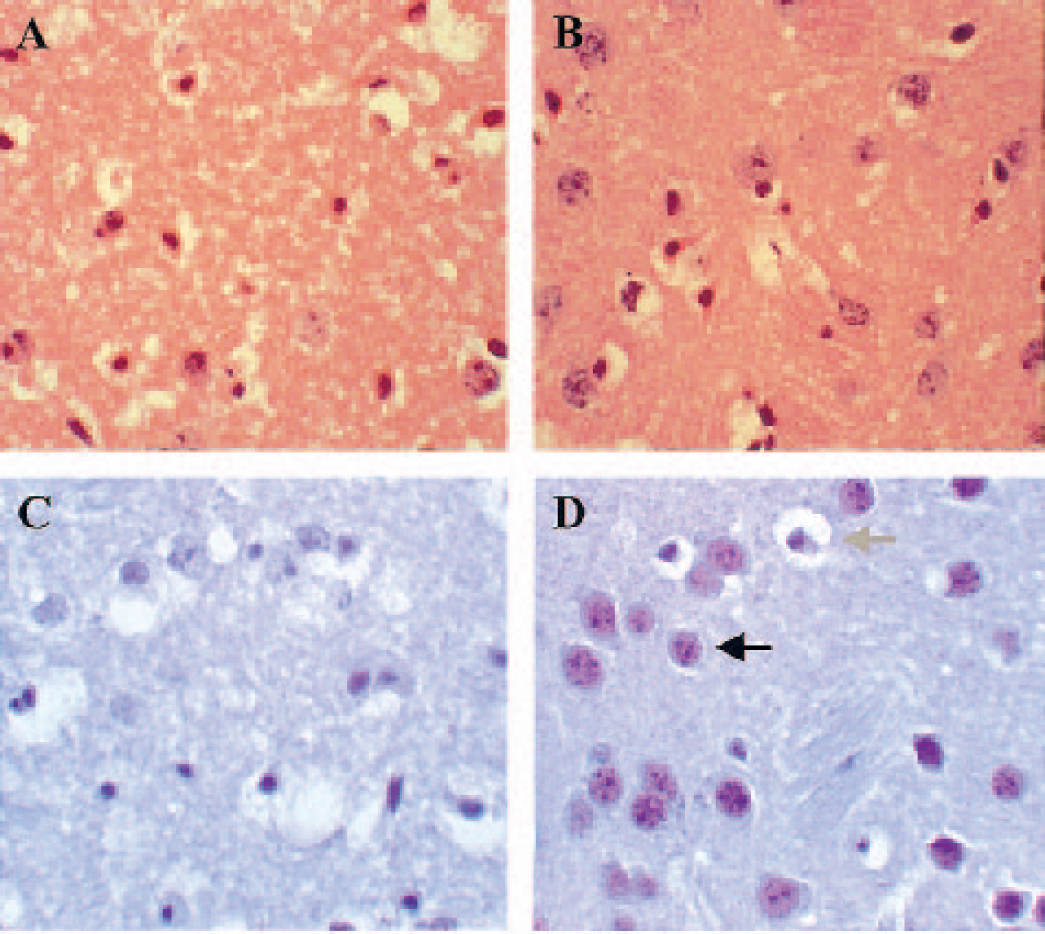
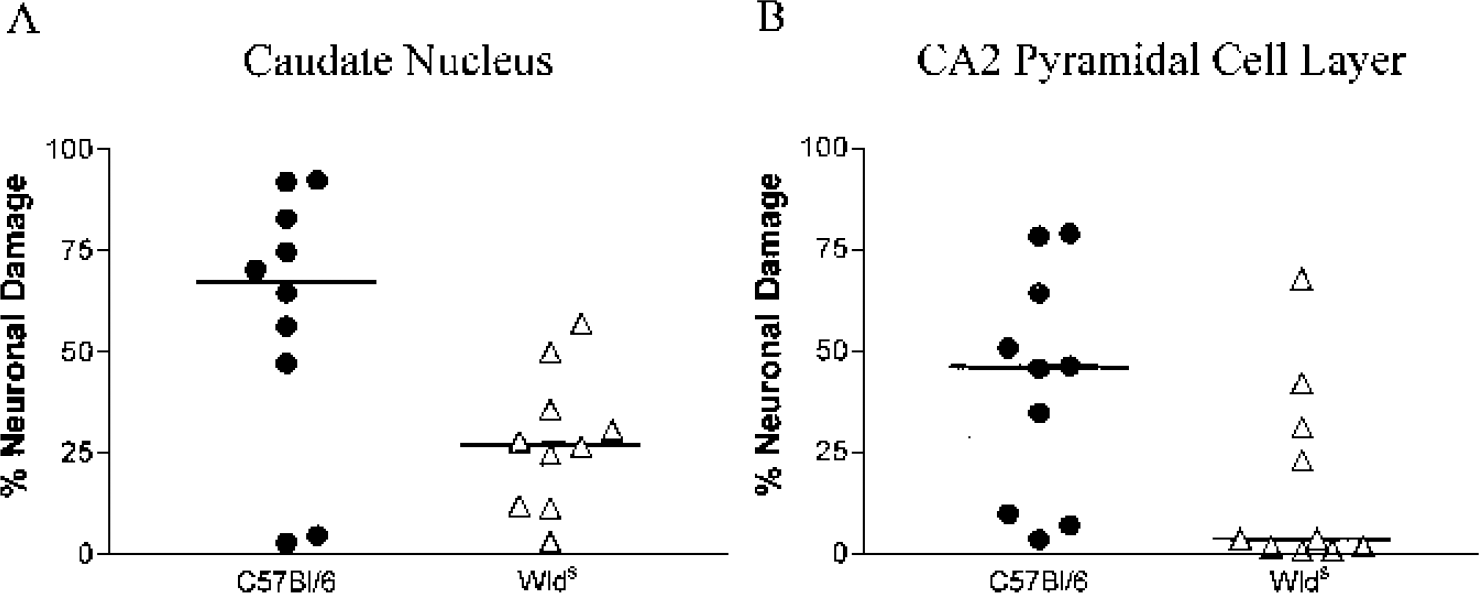
Transient bilateral global ischemia induced by a 17-minute bilateral occlusion of the carotid artery is associated with selective neuronal damage consistently and reproducibly in both the caudate nucleus and the CA2 hippocampal pyramidal cell layer (Horsburgh et al. 1999a; Kelly et al., 2001). It is well documented that there are differences in the rank order of ischemic neuronal vulnerability after global cerebral ischemia in mouse (where the caudate nucleus and CA2 region of the hippocampus are most vulnerable) as compared to rat, gerbil, and humans, where the CA1 region of the hippocampus is most vulnerable. In the present study we quantified, using a nonbiased approach, and compared the numbers of normal and ischemic neurons in the caudate nucleus and the CA2 hippocampal pyramidal cell layer from C57Bl/6 (n = 10) and Wlds (n = 10) mice in hematoxylin and eosin-stained sections. Ischemic neurons were defined in hematoxylin and eosin sections as triangular neurons with densely staining nuclei and intensely eosinophilic cytoplasm. Quantitative assessment of neuronal damage showed a significant and marked reduction in Wlds mice to levels at least half that of controls (Figs. 1A, 1B, 2A). In the caudate nucleus, 27.7 ± 16.8% of neurons were ischemic in Wlds mice, half that in C57Bl/6 mice (58.7 ± 32.3%; P = 0.036). Similarly, quantification of the percentage of neuronal damage in the CA2 hippocampal pyramidal cell layer (Fig. 2B) showed a marked reduction in the percentage of ischemic damage in Wlds mice (17.7 ± 23.1%) compared to C57Bl/6 mice (41.9 ± 28.0%, P = 0.023). The levels and variability of ischemic damage in the caudate nucleus and CA2 region C57Bl/6 mice are consistent with that found in other studies within our laboratory using C57Bl/6 mice (unpublished observations).
(
Quantification of the percentage of ischemic neuronal damage in the caudate nucleus (
Lumbrical muscles from all experimental mice were immunocytochemically stained for the presence or absence of Ube4b/Nmnat (Wld) chimeric protein to confirm the identity of the Wlds and control mice on breaking the double-blind coding of the brain sections. Wld expression was present in muscle tissue from all Wlds mice, appearing as punctate staining of myonuclei, and was entirely absent in all C57Bl/6 tissue (cf. Mack et al., 2001; data not shown). Adjacent sections to those used for histologic analysis from both C57Bl/6 and Wlds experimental mice were immunocytochemically stained for the presence of Wld protein. As expected, no Wld protein was detected in any neurons in C57Bl/6 caudate (Fig. 2C) or CA2 pyramidal layer. Nuclear Wld expression was detected in the majority of surviving neurons from caudate (Figure 2D) and the CA2 pyramidal layer (data not shown) in Wlds mice after ischemia. The morphology of Wld-expressing persistent neurons appeared normal, suggesting that brain areas expressing the Wld protein are protected from ischemic degeneration. In contrast, neurons that had the characteristic appearance of ischemic damage did not express Wld protein.
DISCUSSION
This study shows that expression of the Wld chimeric gene is associated with a marked reduction in the extent of neuronal damage after global cerebral ischemia. In contrast to previous studies, which exclusively showed a protective effect of Wld on axons and synapses, this is the first, to our knowledge, to show that Wlds can provide significant protection against ischemia-induced neuronal death.
The mouse model of transient cerebral ischemia is associated with delayed neuronal death in specific brain regions (notably the caudate nucleus and CA2 pyramidal cell layer). This slowly evolving neuronal damage matures over days after ischemia despite restoration of regional cerebral blood flow, glucose metabolism, and tissue ATP content. We chose to analyze the effects of Wld at 72 hours after ischemia reperfusion, when neuronal damage is mature in wild-type C57Bl/6J mice (Kelly et al., 2001). However, it is unclear whether the presence of the Wld gene has acted simply to delay the time course of neuronal damage or if it has completely blocked the ischemia-induced pathways and subsequent neuronal death. From the limited studies in the CNS, Wlds has been shown to delay the degenerative response of axons after lesions of the perforant path and spinal cord axons (Shi and Stanfield, 1996; Zhang et al., 1996). This is similar to the characteristic effect of Wld in the peripheral nervous system, where Wallerian degeneration is markedly delayed.
The inclusion of a fragment of an E4 ubiquitination factor, Ube4b, in the Wld gene suggests that a mechanism involving altered ubiquitination of proteins may underlie the protective role of Wld (Coleman and Perry, 2002; Gillingwater and Ribchester, 2001; Mack et al., 2001). After transient cerebral ischemia, impairment of protein ubiquitination occurs (Magnusson and Wieloch, 1989). Conjugated ubiquitin accumulates and free ubiquitin is depleted in regions that undergo selective and delayed ischemic neuronal degeneration (Morimoto et al., 1996). Ube4b also regulates multiubiquitination of proteins that may be targeted for degradation by the 26S proteasome. The activity of this 26S proteasome is reduced during ischemia and for 2 days during reperfusion (Asai et al., 2002). It is tempting to speculate that altered ubiquitination contributes to this protective effect of Wld. It is interesting that the Wld gene provides no protection against apoptotic degeneration of the cell body in vitro (Deckwerth and Johnson, 1994). This is consistent with the present findings because there is limited evidence of programmed cell death after global ischemia. However, Wld does confer significant resistance against nonapoptotic degeneration of the axon and synaptic terminals (Finn et al., 2000; Gillingwater et al., 2002; Fig. 3). It has been suggested that Wld rescues axons from a ubiquitin-proteasome pathway rather than a classical apoptosis pathway or expression of Wld in the nucleus induces an inhibitor of degeneration (Coleman and Perry, 2002). Alternatively, however, the inclusion of NMNAT in the chimeric Wld gene may point to a different mechanism of neuroprotection, because NMNAT is capable of inhibiting poly(ADP-ribose) polymerase 1 (PARP1), a DNA repair enzyme that is known to contribute to cell death during ischemia/reperfusion when extensively activated by DNA damage (Schweiger et al., 2001; Ying et al., 2002).
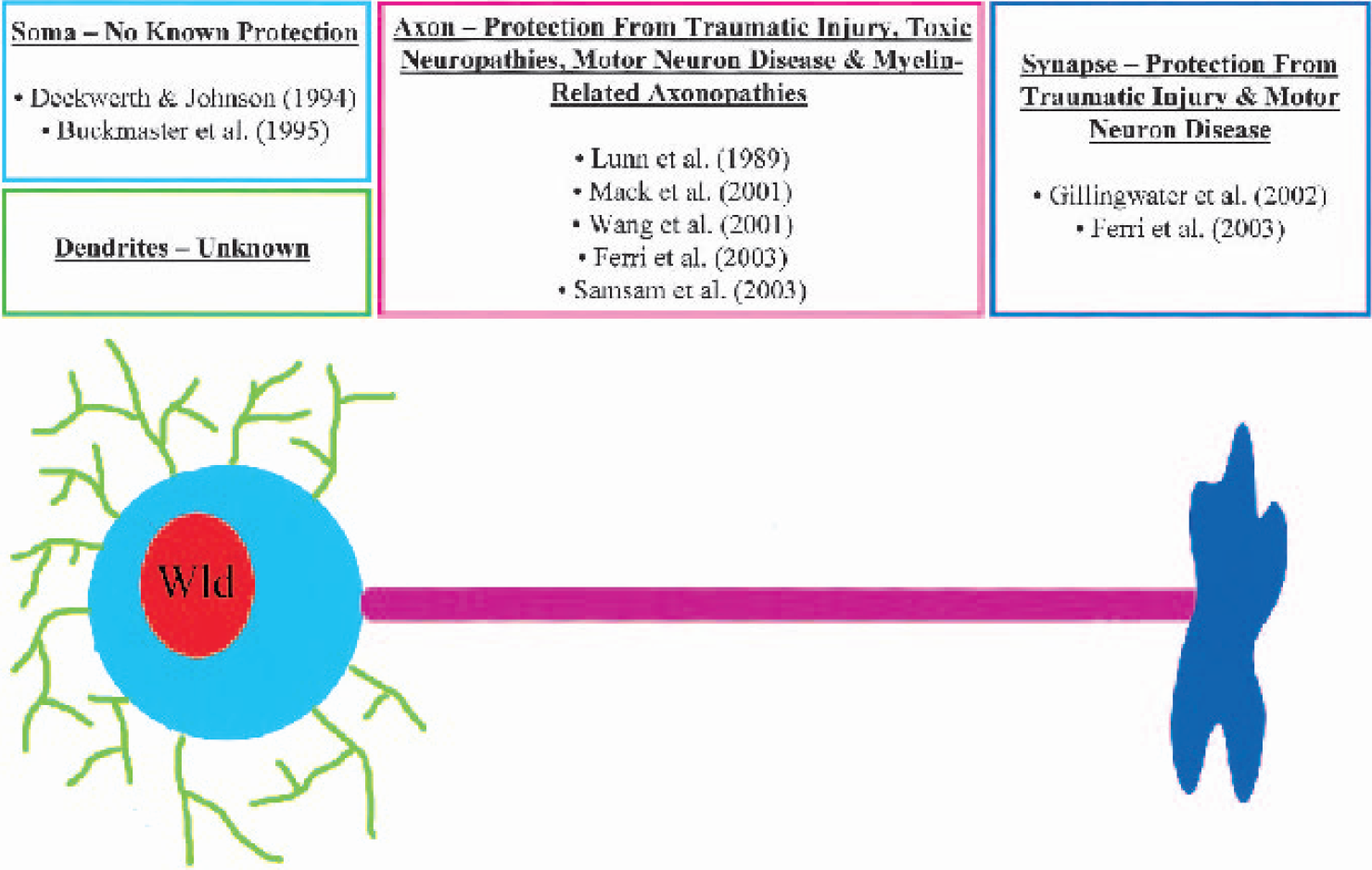
Schematic diagram summarizing previous evidence of neuroprotection conferred by the Wld gene (Gillingwater and Ribchester, 2001).
Another potential mechanism by which Wld is neuroprotective may be via an effect on synaptic neurotransmitter release or levels. Increased release of glutamate from presynaptic nerve terminals and a dramatic increase in extracellular concentrations of glutamate results in major postsynaptic neuronal cell death after ischemia (Choi and Rothman, 1990; Rossi et al., 2000). Consequently, it may be envisaged that preservation of structure and function of presynaptic terminals by the Wld gene, as has been demonstrated at the neuromuscular junction (Gillingwater et al., 2002), after global ischemia reduces the amount of excess neurotransmitter release, resulting in reduced levels of excitotoxic neurodegeneration. Alternatively, the demonstration that Wld can alter cerebral metabolism and, more specifically, significantly reduce levels of cytosolic glutamate supports an effect of Wld on neurotransmitter levels (Tsao et al., 1999). However, peripheral nerve axotomy leads, within a few days, to accelerated spontaneous transmitter release from presynaptic motor nerve terminals of mice or rats expressing the Wld gene (Ribchester et al., 1995; Ribchester RR, Thomson D, Coleman MP, 2003). It will therefore be of great interest to establish whether increases in the extracellular levels of neurotransmitters are delayed or reduced in Wld-expressing mice after ischemia.
The present data add a novel role as a neuroprotectant to the well-characterized WLD phenotype of slow Wallerian degeneration. Understanding how this mutant gene confers protection now has wider ranging implications, including axon pathologies (in stroke), as well as several other forms of neurodegenerative disease (Coleman and Perry, 2002; Gillingwater and Ribchester, 2003).
Footnotes
Acknowledgements
The authors thank Dr. Michael Coleman (Cologne) for the generous gift of Wld18 primary antibody, Jane Knox for helping with brain sectioning, and Derek Thomson for assistance with animal care.