
Abstract
Cortical spreading depression (CSD) is characterized by reversible neuronal dysfunction in the absence of cell death. Preconditioning by CSD induces tolerance against subsequent lethal ischemia. In this study, we used quantitative reverse transcriptase-polymerase chain reaction and immunocytochemistry to analyze proinflammatory cytokine expression after CSD induced by topical application of potassium chloride (KCl) to the cortical surface of rat brains. Relative to control cortex, we found an increase of tumor necrosis factor-α (mean 62-fold, P < 0.001) and interleukin (IL)-1β (mean 24-fold, P < 0.001) mRNA levels within 4 hours ipsilateral to the site of KCl application. At 16 hours cytokine expression was decreasing toward baseline levels. Ipsilateral cytokine induction was abolished by pretreatment with the noncompetitive N-methyl-d-aspartate antagonist, MK-801. In contrast to focal cortical infarction, cytokine induction in CSD was not accompanied by the expression of inducible nitric oxide synthase mRNA. In immunocytochemical studies, expression of IL-1β protein was localized to ramified microglia in cortical layers I to III of the ipsilateral hemisphere. Our finding that NMDA receptor signaling without subsequent neuronal cell death is sufficient to induce inflammatory cytokine expression in the brain has basic implications for central nervous system immunoregulation. We postulate that cytokine expression in CSD forms part of a physiologic stress response that contributes to the development of ischemic tolerance in this and other preconditioning paradigms.
Keywords
Proinflammatory cytokines such as tumor necrosis factor-α (TNF-α) and interleukin-1β (IL-1β) are strongly induced after focal ischemic and traumatic brain injury (Arvin et al., 1996; Rothwell et al., 1997; Stoll et al., 1998). In several in vitro and in vivo models, cytokines exacerbate hypoxic as well as excitotoxic damage of neurons (Relton and Rothwell, 1992; Hewett et al., 1994; Chao and Hu, 1994; Barone et al., 1997). Neurotoxic cytokine effects appear to be mediated by the downstream activation of the cytokine-inducible nitric oxide synthase (iNOS) pathway (Hewett et al., 1994; Iadecola et al., 1997). However, depending on the time window and dosage of their administration, cytokines can also confer protection against neuronal injury both in vitro (Cheng et al., 1994; Strijbos and Rothwell, 1995) and in vivo (Bruce et al., 1996; Nawashiro et al., 1997).
Cortical spreading depression (CSD) is characterized by a transient suppression of all neuronal activity that repetitively extends from sites of increased extracellular potassium concentration to the entire ipsilateral hemisphere (Martins-Ferreira et al., 2000). The propagation of CSD is inhibited by pretreatment with the noncompetitive N-methyl-d-aspartate (NMDA) receptor antagonist, MK-801, and therefore is likely to be mediated by glutamatergic signaling through the NMDA receptor (Marrannes et al., 1988; Lauritzen and Hansen, 1992; Schroeter et al., 1995). In contrast with paradigms of lethal NMDA neurotoxicity (Choi et al., 1987), healthy cortical neurons can fully compensate for the massive ion shifts caused by CSD (Hossmann, 1996). Therefore, CSD proceeds without cell death (Nedergaard and Hansen, 1988), but induces neuronal tolerance against subsequent lethal ischemic challenge (Kobayashi et al., 1995; Yanamoto et al., 1998).
Although CSD has been shown to profoundly in fluence gene expression in the brain (Miettinen et al., 1997; Kariko et al., 1998), the molecular mechanisms underlying tolerance induction by CSD are not completely understood. To elucidate a potential contribution of proinflammatory cytokines, we used reverse transcriptase-polymerase chain reaction (RT-PCR) to study the expression of TNF-α, IL-1β, and iNOS mRNA after CSD induced by the topical application of potassium chloride (KCl) to the cortical surface in rats. IL-1β expression was also analyzed by immunocytochemistry. As a paradigm of lethal neuronal injury, we comparatively studied infarctions induced by photothrombosis of cortical microvessels (Watson et al., 1985).
MATERIALS AND METHODS
Animal experiments
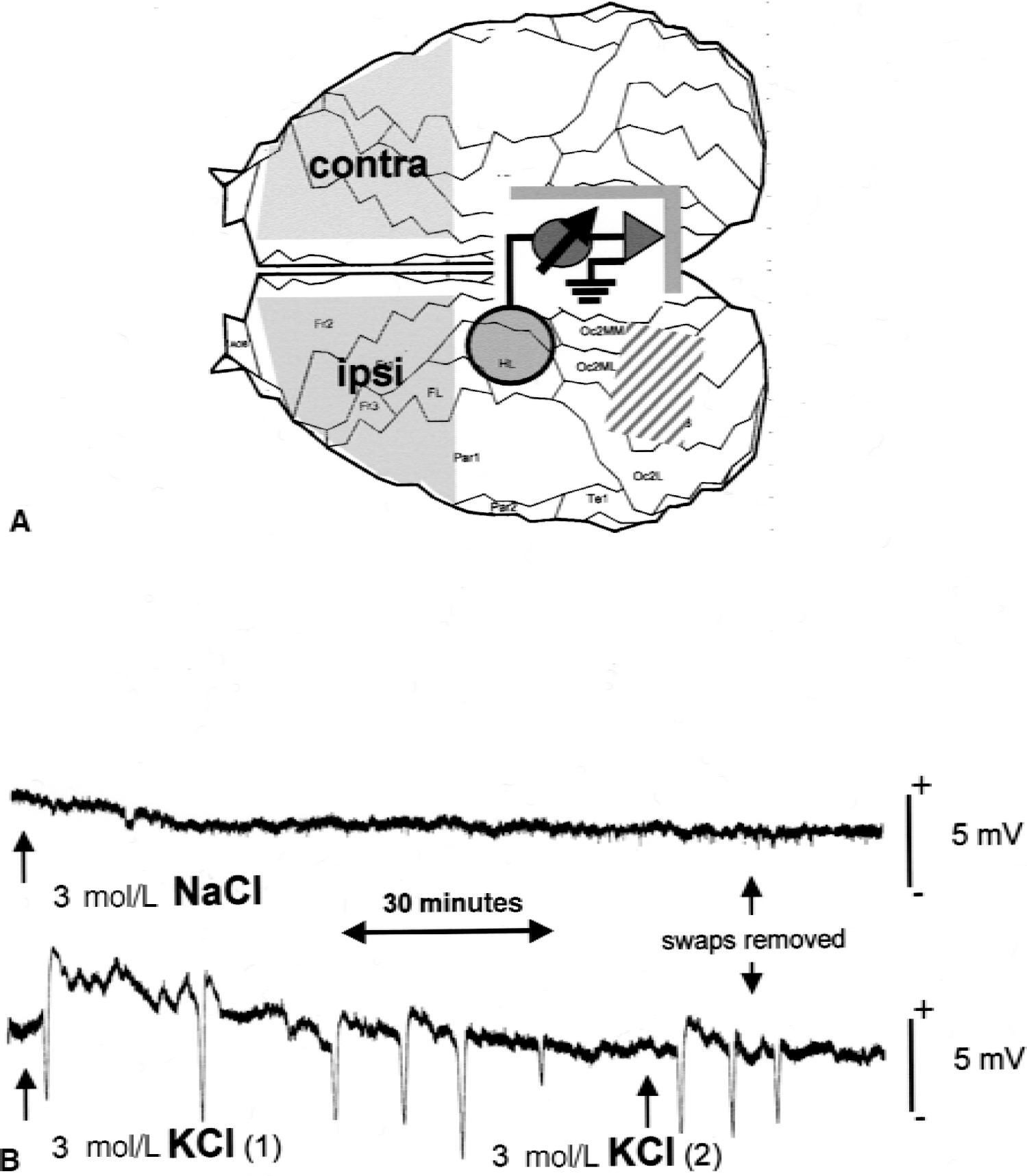
Male Wistar rats (n = 16; 270 to 320 g) were anesthetized with enflurane [2% in an O2/N2 (1:2) mixture] and fixed in a stereotactic frame. Body temperature was measured by a rectal probe and maintained at 36.5°C to 37.5°C with a feedback-controlled heating pad throughout all procedures. After exposure of the skull, two burr holes leaving the dura intact were made over the left hemisphere at the locations indicated in Fig. 1A. A recording electrode filled with artificial cerebrospinal fluid (NaCl 124 mmol/L, KCl 5 mmol/L, CaCl2 2 mmol/L, MgSO4 2 mmol/L, NaH2PO4 1.25 mol/L, glucose 10 mmol/L, NaHCO3 26 mmol/L) was positioned upon the dura. Direct current (DC) potentials were monitored using a home-built amplifier and an analog plotter. Swabs soaked with either 3 mol/L KCl or 3 mol/L NaCl solutions (n = 8 rats in each group) were placed upon the dura over the occipital cortex and in some experiments renewed once during the measurement. After recording of 7 DC deflections in KCl-treated animals, the swabs were removed (usually approximately 100 minutes after their application), and the burr holes flushed with 0.9% saline. In the next 10 minutes, a maximum of 1 or 2 more DC deflections occurred. Typical examples of DC potential recordings in KCl-and NaCl-treated animals are shown in Fig. 1B. For RNA isolation, animals were killed 4 and 16 hours, respectively, after the occurrence of the first DC deflection (n = 4 animals at each time point in both the KCl- and NaCl-treated group).
To clarify the role of NMDA receptor signaling, an additional experiment was performed in which n = 4 rats were injected intravenously through a femoral catheter with the noncompetitive NMDA antagonist, MK-801 (dizolcipine; Research Biochemicals International, Natick, MA, U.S.A.) at a dosage of 2 mg/kg body weight 30 minutes before application of the KCl swab. As control, the remaining rats (n = 4) were pretreated with carrier solution. Cortical tissue was prepared 4 hours after the induction of CSD.
Photothrombosis of cortical microvessels in the right parietal cortex of male Wistar rats was induced as described previously (Jander et al., 1995). For isolation of RNA, animals were killed at 4 and 16 hours after the induction of ischemia, respectively (n = 5 at each time point).
Semiquantitative reverse transcription-polymerase chain reaction
Total RNA was prepared from cortical tissue samples using the TRIzol reagent (GIBCO BRL, Gaithersburg, MD, U.S.A.), according to the manufacturer's instructions. RNA was quantified spectrophotometrically. One microgram RNA isolated from each tissue sample was reverse transcribed using oligo(dT)20 primers and SuperscriptII-Reverse Transcriptase (GIBCO BRL) essentially to the manufacturer's protocol. cDNA equivalent to 20 ng of total RNA was subjected to subsequent PCR analysis as detailed elsewhere (Jander et al., 1998). Rat TNF-α and iNOS primers were purchased from Clontech (Palo Alto, CA, U.S.A.) and Biosource (Camarillo, CA, U.S.A.), respectively. The sequences of the rat IL-1β and glyceraldehyde-phosphate dehydrogenase (GAPDH) primers have been described previously (Gillen et al., 1998). For each primer combination, linear cycling conditions were determined in preliminary experiments as described previously (Jander and Stoll, 1998). Cycle numbers were as follows: TNF-α = 26, IL-1β = 25, iNOS = 25, GAPDH = 18. Controls included RNA subjected to the RT-PCR procedure without addition of reverse transcriptase and PCR performed in the absence of cDNA that always yielded negative results. Representative samples were sequenced on an ABI PRISM 310 genetic analyzer (Perkin Elmer, Oak Brook, IL, U.S.A.) for each PCR product. Polymerase chain reaction products were separated on 1.5% agarose gels containing 10 μg/mL ethidium bromide, photographed using a CSC-camera (Cybertech, Berlin, Germany), and densitometric analysis was performed with TINA 2.1 software (Raytest, Straubenhardt, Germany).
Competitive polymerase chain reaction
Competitive PCR analysis of TNF-α, IL-1β, and GAPDH mRNA was performed using CytoXpress quantitative PCR kits from Biosource, essentially according to the manufacturer's protocol. In initial titration assays, serial dilutions of cDNA corresponding to 50, 5, 0.5 ng starting RNA were coamplified with 1000 copies of an internal calibration standard for 30 cycles and analyzed on 1.5% agarose gels. Dilutions with visibly equivalent intensities of the cDNA and internal calibration standard bands were used for subsequent quantitation in a microplate hybridization assay using colorimetric detection of biotinylated primer incorporated into both cytokine and internal calibration standard amplicons during the PCR reaction.
Statistical analysis
Analysis of variance (Tukey's Multiple Comparison Test) was performed using GraphPad Prism 3.0 software for statistical analysis of competitive PCR data.
Immunocytochemistry
At 4, 8, and 16 hours after induction of CSD, rats were deeply anesthetized and perfused with 4% paraformaldehyde in 0.15 mol/L phosphate buffer (pH 7.4) through the left ventricle. Whole brains were removed from the scull, postfixed in the same fixative overnight at 4°C, and cryoprotected by overnight infiltration with 20% sucrose in phosphate buffer at 4°C. Free-floating 50 μm cryostat sections were washed three times in Tris-buffered saline containing 0.05% Triton X-100 (TBS-T). Endogenous peroxidase was blocked by 30 minutes incubation in 0.3% H2O2 in TBS-T. After three washes in TBS-T, sections were incubated with affinity-purified goat anti-rat IL-1β polyclonal antibody (R&D Systems, Minneapolis, MN, U.S.A.) at 0.5 μg/mL in 2% normal horse serum in TBS-T for 72 hours at 4°C. After three washes in TBS-T, bound antibody was detected using biotinylated horse anti-goat IgG (Vector Laboratories, Burlingame, CA, U.S.A.) and the ABC Elite kit (Vector) with diaminobenzidine as substrate. Control experiments in which the primary antibody was replaced by nonspecific goat IgG yielded negative results. Sections were mounted onto gelatine-coated slides, air-dried, dehydrated with ascending series of ethanol, cleared, and coverslipped with Entellan (Merck, Darmstadt, Germany).
RESULTS
During the recording period of 100 to 120 minutes, topical application of 3 mol/L KCl solution onto the intact dura induced 7 to 9 DC deflections of ~ 5 mV typical for CSD (Fig. 1B). As a control, we applied 3 mol/L NaCl instead of KCl solution onto the cortex. No CSD-like DC deflections were detectable (Fig. 1B) in these animals. To characterize cytokine expression, we prepared cortical tissue samples from frontal cortex ipsi-and contralateral to the site of KCl or NaCl application and isolated total RNA for PCR analysis. At 4 hours after induction of CSD in KCl-treated rats, semiquantitative RT-PCR showed a strong increase of TNF-α and IL-1β mRNA levels that was strictly confined to the ipsilateral cortex (Fig. 2). At 16 hours, cytokine mRNA levels were decreasing toward baseline levels. In NaCl-treated rats, no significant increase of cytokine mRNA levels greater than background levels was found (Fig. 2).
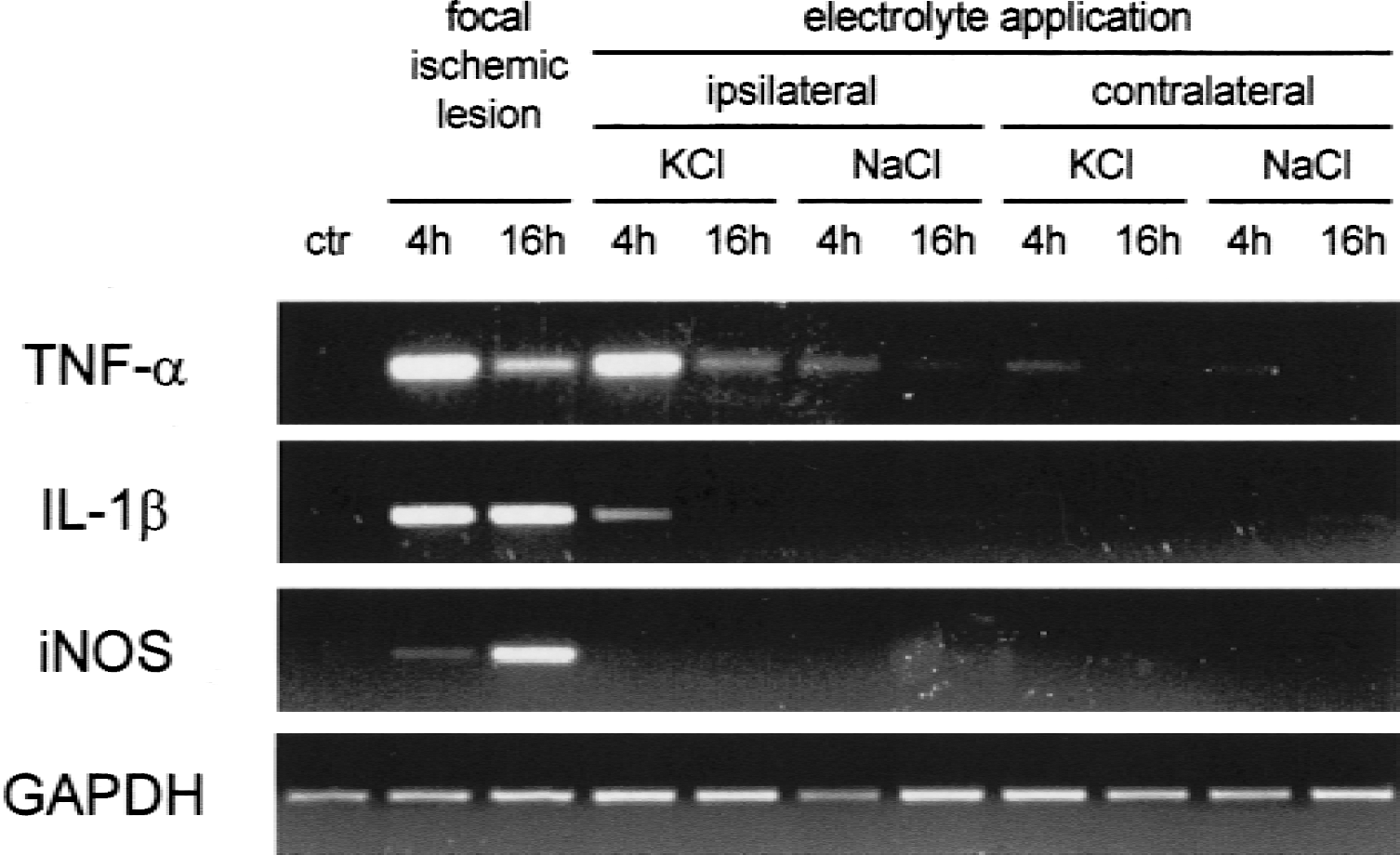
Semiquantitative reverse transcriptase-polymerase chain reaction analysis of tumor necrosis factor-α (TNF-α), IL-1β, and inducible nitric oxide synthase (iNOS) transcripts after topical application of KCl or NaCl to the cortical surface of rats. Glyceraldehyde-phosphate dehydrogenase (GAPDH) mRNA served as a “housekeeping gene” control that is not modulated under the experimental conditions. Tissue samples were removed from frontal cortex ipsilateral or contralateral to the site of electrolyte application at 4 and 16 hours, respectively. For comparison, tissue samples from photochemically induced ischemic lesions 4 and 16 hours after ischemia were analyzed.
In contrast to TNF-α and IL- 1 β mRNA, we found no induction of iNOS mRNA in remote cortex ipsi- or contralateral to the site of KCl or NaCl application (Fig. 2). As positive control for iNOS expression, we used total RNA from focal cortical infarctions induced by photothrombosis of cortical microvessels (Watson et al., 1985; Jander et al., 1995). In line with previous findings in lesions induced by middle cerebral artery occlusion (Iadecola et al., 1995; Galea et al., 1998), we found a clear increase in iNOS mRNA at 4 hours and even more pronounced at 16 hours after ischemia. Likewise, strong increases in TNF-α and IL-1β expression occurred in the focal ischemia model (Fig. 2).
For the more precise quantification of cytokine mRNA levels, we performed competitive PCR analysis. In initial titration experiments (Fig. 3A), increasing dilutions of cDNA were coamplified with a fixed amount (1000 copies) of an internal calibration standard. At 4 hours after KCl application, approximately equal band intensities of TNF-α cDNA and the standard were observed at 5 ng of total starting cDNA, whereas cDNA from NaCl-treated animals only competed at 50 ng starting cDNA. Thus, KCl treatment induced considerable up-regulation of TNF-α mRNA. Similar findings were obtained in the analysis of IL-1β mRNA.
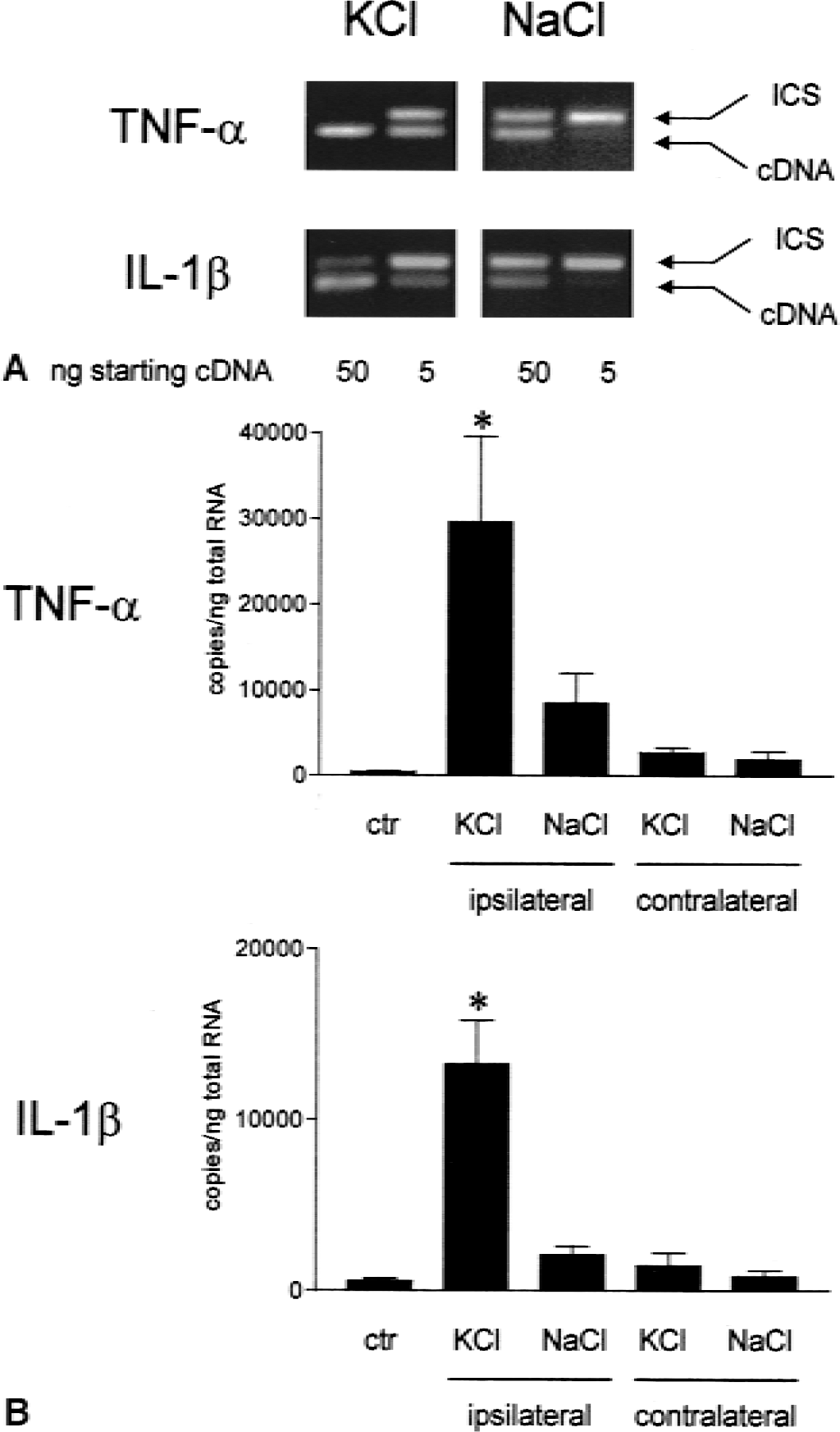
Competitive polymerase chain reaction (PCR) analysis of cytokine mRNA levels at 4 hours after cortical spreading depression induction.
Quantification of the amplicons by microplate detection of biotinylated primer incorporated during the PCR reaction (Fig. 3B) showed a mean 62-fold increase in TNF-α copy numbers in the ipsilateral cortex of KCl-treated rats compared with untreated control cortex (n = 4 animals in each group, P < 0.001). After NaCl treatment, a mean 17-fold increase relative to control cortex was found although this difference did not achieve statistical significance. In the contralateral cortex of both KCl- and NaCl-treated animals, no major increase in TNF-α mRNA was found. IL-1β mRNA levels likewise showed a significant increase in ipsilateral cortex of KCl-treated rats that was however somewhat less pronounced (mean 24-fold, P < 0.001) compared with TNF-α mRNA. Throughout, no differences in GAPDH mRNA levels were observed between any of the treatment groups (not shown).
To compare cytokine expression after CSD with a paradigm of ischemic neuronal cell death, we analyzed photochemically induced cortical infarctions by competitive PCR. In these lesions, mean mRNA induction at 4 hours after ischemia was 93-fold for TNF-α and 80-fold for IL-1β, respectively. Thus, relative to the infarctions, induction in the CSD model reached 67% for TNF-α mRNA and 31% for IL-1β mRNA, respectively.
The propagation of CSD can be inhibited by the noncompetitive NMDA receptor antagonist, MK-801 (Marrannes et al., 1988; Lauritzen and Hansen, 1992). To delineate a role of NMDA receptor signaling in CSD-induced cytokine expression, we performed an additional experimental series in which a proportion of the animals received an intravenous injection of MK-801 (2 mg/kg body weight) 30 minutes before induction of CSD (Fig. 4). In MK-801-treated animals, cortical DC deflections after KCl administration were generally absent (not shown). Basic physiologic variables (blood gases, blood glucose, and rectal temperature) were monitored throughout the experiment and revealed no differences between MK-801-pretreated and control rats. Animals were killed at 4 hours after CSD induction and total cortical RNA was analyzed for TNF-α and IL-1 β mRNA expression by competitive PCR. In vehicle-treated animals, a strong induction of both cytokines was found that was almost completely inhibited by MK-801 pretreatment (Fig. 4).
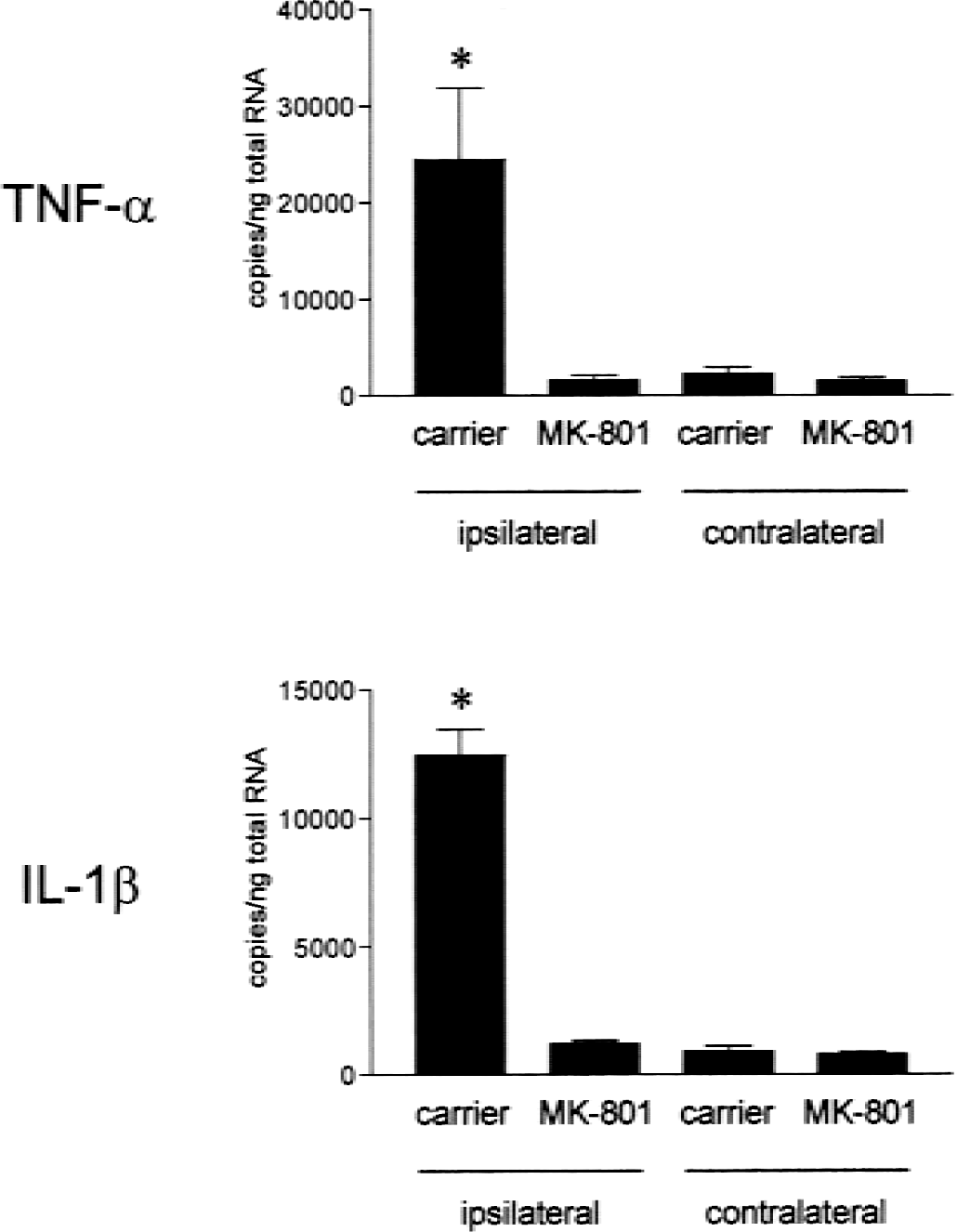
Inhibition of ipsilateral cytokine mRNA induction by pre-treatment with the noncompetitive N-methyl-d-aspartate antagonist, MK-801, assessed by competitive polymerase chain reaction (n = 4 animals in each group). *P < 0.001 compared with MK-801-treated animals.
To extend the studies to the protein level, we performed immunocytochemical labeling studies with various commercially available antibodies against rat TNF-α and IL-1β. While several different TNF-α antibodies yielded variable and overall inconclusive staining patterns, we obtained highly reproducible staining for IL-1β using a rat IL-1β-specific, antigen affinity-purified goat polyclonal antibody on free-floating sections of paraformaldehyde-fixed tissue. IL-1β immunoreactivity was confined to cortical layers I to III of the ipsilateral hemisphere (Fig. 5A to 5C) where it appeared at 4 hours after SD induction, reached its maximum at 8 hours, and decreased toward baseline levels at 16 hours. At higher resolution, IL-1β immunoreactivity appeared to be mainly, if not exclusively, expressed by ramified microglia (Fig. 5B).
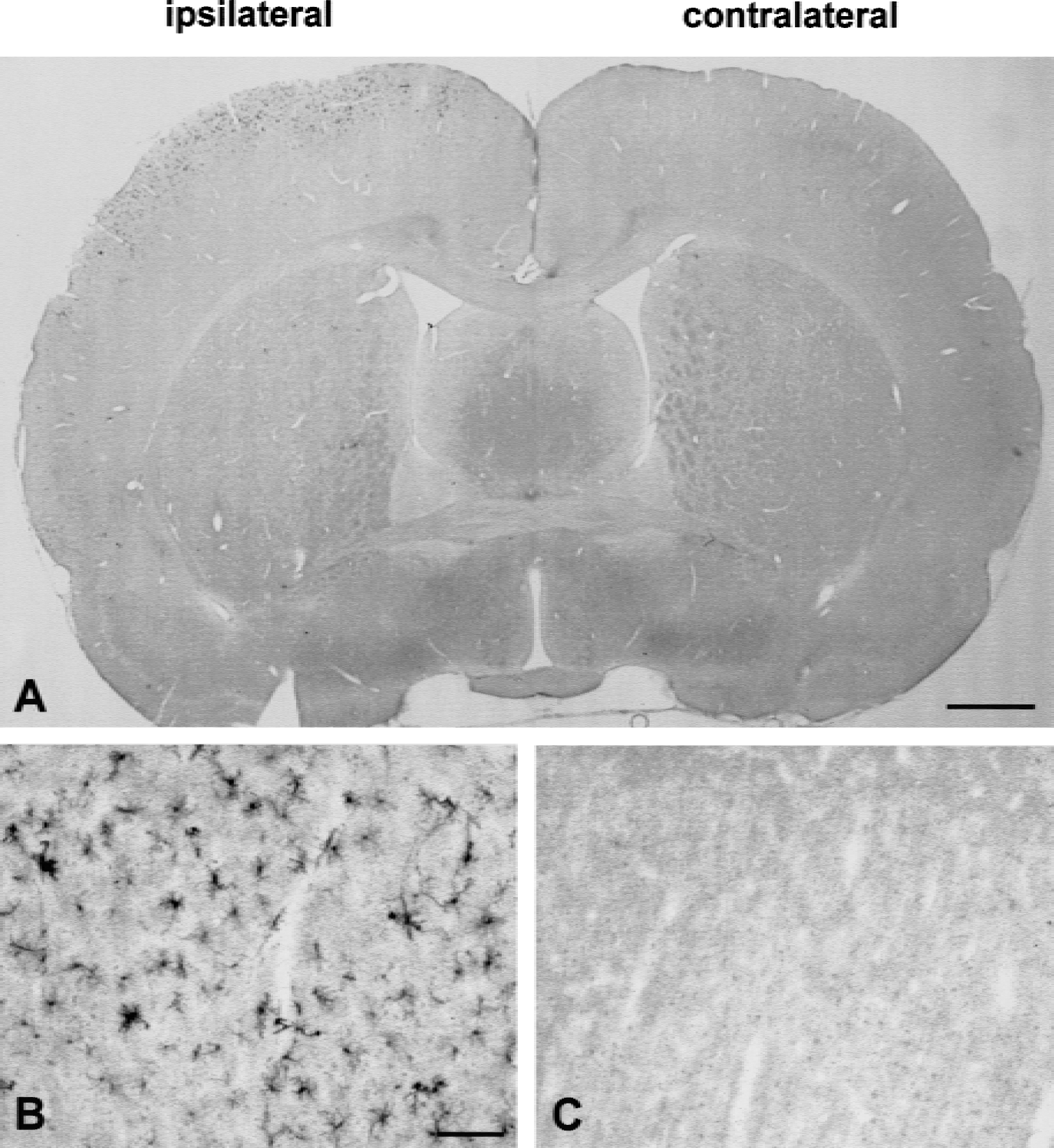
Immunocytochemical localization of IL-1β protein at 8 hours after induction of cortical spreading depression. In line with the mRNA data, expression of IL-1β immunoreactivity is restricted to the ipsilateral hemisphere
DISCUSSION
The current study shows that CSD induced by the topical application of KCl to the cortical surface of the intact rat brain leads to strong induction of the proinflammatory cytokines TNF-α and IL-1β. Although primarily obtained by quantitative RT-PCR analysis of total RNA extracted from defined cortical areas, we were able to corroborate our data at the protein level by the immunocytochemical localization of IL-1β protein. IL-1β immunoreactivity was selectively induced in the ipsilateral hemisphere affected by CSD and was found to be mainly localized to ramified microglia in cortical layers I to III. Because of technical difficulties concerning antibody specificity and poor reproducibility of staining results, we were so far unable to obtain convincing immunocytochemical localization of TNF-α in CSD. Overall, however, the current data indicate that transient neuronal dysfunction without cell death (as it is characteristic for CSD) is sufficient to induce inflammatory cytokine gene expression in the brain. Our findings suggest a novel mechanism of neural-immune interaction that may have basic implications for the regulation and pathophysiologic consequences of inflammatory processes in the central nervous system.
The initial rise of both TNF-α and IL-1β mRNA at 4 hours after KCl administration was comparable to focal ischemia lesions induced by photothrombosis of cortical microvessels but was more rapidly decreasing at 16 hours. Likewise, immunocytochemical findings in photochemically-induced ischemia (Jander, Schroeter, and Stoll, unpublished observations) and the middle cerebral artery occlusion model (Davies et al., 1999) indicate that the expression of IL-1 β protein is more sustained in focal ischemic lesions than in CSD. As the main difference between the two paradigms, we found strong coinduction of iNOS mRNA in focal ischemia but not after CSD. Both the lack of iNOS expression after CSD and its strong induction in focal ischemia are in accord with previous immunocytochemical data (Iadecola et al., 1995; Caggiano and Kraig, 1998; Galea et al., 1998). Therefore, it appears that CSD as a model of transient neuronal dysfunction without cell death induces proinflammatory cytokines without concomitant iNOS expression, whereas the coinduction of cytokines and iNOS is characteristic of the pannecrosis evolving because of focal brain ischemia. Although protective effects of iNOS in tissue injury responses have been reported (Guo et al., 1999; Sinz et al., 1999), most findings in cerebral ischemia suggest an overall harmful role of iNOS in subacute stages after injury (Dirnagl et al., 1999). In vitro, the exacerbation of hypoxic and excitotoxic neuronal cell injury by inflammatory cytokines depends on the downstream action of iNOS (Hewett et al., 1994). Accordingly, studies in knockout mice showed a critical role of iNOS in delayed infarct growth after focal brain ischemia (Iadecola et al., 1997). The differential coinduction of iNOS may therefore critically determine the pathophysiologic consequences of inflammatory gene induction in CSD and focal ischemia, respectively.
The induction of cytokine mRNA after CSD was almost completely inhibited by pretreatment of the animals with the noncompetitive NMDA antagonist, MK-801. In line with this observation, numerous studies have implicated glutamatergic signaling through NMDA receptors with subsequent intracellular entry of Ca2+ ions in the initiation and propagation of CSD (Marrannes et al., 1988; Lauritzen and Hansen, 1992). Thus, CSD shares basic cellular mechanisms with excitotoxic cell death induced by high concentrations of glutamate (Siesjo and Bengtsson, 1989; Hossmann, 1996), although the massive ion shifts occurring in CSD are compensated without leading to any irreversible neuronal damage (Nedergaard and Hansen, 1988). However, CSD activates a cellular stress response with induction of immediate early genes and heat shock proteins (Kobayashi et al., 1995; Kariko et al., 1998) and leads to the development of ischemic tolerance. The mechanisms of tolerance induction by CSD may involve neurotrophins such as brain-derived neurotrophic factor that show widespread up-regulation in the ipsilateral cortex after focal KCl application (Kokaia et al., 1993; Kawahara et al., 1997; Matsushima et al., 1998; Kariko et al., 1998). In CSD, it is an open question if the inhibition of cytokine synthesis by MK-801 reflects prevention of CSD propagation or a direct effect on cellular activation processes underlying inflammatory gene induction. Notably, similar to the in vivo situation in CSD, tolerance induction by sublethal doses of NMDA also has been shown in vitro (Marini et al., 1998; Grabb and Choi, 1999).
In certain in vitro models, both TNF-α and IL-1β can protect neurons against subsequent hypoxic or excitotoxic injury (Cheng et al., 1994; Strijbos and Rothwell, 1995). In ischemic injury of both brain (Bruce et al., 1996) and heart (Kurrelmeyer et al., 2000), increasing evidence suggests protective roles of endogenous TNF-α in vivo. Protective effects of TNF-α involve activation of NF-κB with subsequent induction of manganese superoxide dismutase (Mattson et al., 1997; Van Antwerp et al., 1998). Furthermore, neurotrophins are highly inducible by inflammatory cytokines (Lindholm et al., 1987; Hattori et al., 1993; Strijbos and Rothwell, 1995; Heese et al., 1998). In hippocampal slice preparations in vitro, long-term potentiation as a basic mechanism of neuronal plasticity was accompanied by substantial induction of IL-1β that depended on NMDA receptor signaling and in turn enhanced the maintenance of long-term potentiation (Schneider et al., 1998). Taken together, it is therefore conceivable that under certain circumstances proinflammatory cytokines participate in a physiologic stress response that contributes to the development of tolerance and postlesional plasticity. Interestingly, tolerance induction by brief nonlethal brain ischemia (that is, ischemic preconditioning) recently has been shown to be likewise accompanied by significant up-regulation of TNF-α and IL-1β mRNA (Wang et al., 2000a, b). Thus, preconditioning stimuli as different as CSD and mild transient ischemia apparently activate similar molecular effector pathways that may critically depend on the production of proinflammatory cytokines.
Footnotes
Acknowledgments
The authors thank B. Blomenkamp, D. Steinhoff, and A. Tries for excellent technical assistance.