
Abstract
A beneficial role of nitric oxide (NO) after cerebral ischemia has been previously attributed to its vascular effects. Recent data indicate a regulatory role for NO in initial leukocyte-endothelial interactions in the cerebral microcirculation under basal and ischemic conditions. In this study, the authors tested the hypothesis that endogenous NO production during and/or after transient focal cerebral ischemia can also be neuroprotective by limiting the process of neutrophil infiltration and its deleterious consequences. Male Sprague-Dawley rats were subjected to 2 hours occlusion of the left middle cerebral artery and the left common carotid artery. The effect of NG-nitro-L-arginine methyl ester (L-NAME) (10 mg/kg, intraperitoneally), an NO synthase inhibitor, was examined at 48 hours after ischemia on both infarct size and myeloperoxidase activity, an index of neutrophil infiltration. L-NAME given 5 minutes after the onset of ischemia increased the cortical infarct volume by 34% and increased cortical myeloperoxidase activity by 60%, whereas administration of L-NAME at 1, 7, and 22 hours of reperfusion had no effect. Such exacerbations of infarction and myeloperoxidase activity produced when L-NAME was given 5 minutes after the onset of ischemia were not observed in rats rendered neutropenic by vinblastine. These results suggest that after transient focal ischemia, early NO production exerts a neuroprotective effect by modulating neutrophil infiltration.
Accumulating evidence suggests that the postischemic influx of neutrophils can contribute to tissue damage in animal models of focal cerebral ischemia-reperfusion (Hartl et al., 1996). Previous investigations have shown progressive neutrophil adherence and infiltration into the brain parenchyma hours to days after the initial insult (Barone et al., 1995; Matsuo et al., 1994; Zhang et al., 1994a). Moreover, strategies that either reduce circulating neutrophils or inhibit neutrophil adhesion to the vascular endothelium significantly reduce the infarct volume produced by transient occlusion of the middle cerebral artery (Chen et al., 1992; Chopp, 1995; Matsuo et al., 1994; Zhang et al., 1994, Zhang et al., 1995).
Nitric oxide (NO), a well-known modulator of tissue blood flow, platelet aggregation, and microvascular permeability (Cooke and Dzau, 1997) may also be an endogenous regulator of neutrophil-endothelial cell interactions (Kubes et al., 1991). In models of focal cerebral ischemia, previous studies have shown that inhibition of NO synthase (NOS) with NG-nitro-L-arginine methyl ester (L-NAME) administered at high doses and/or at early stages after ischemia caused an increase in infarct size, suggesting that NO can play a beneficial role (Kuluz et al., 1993; Margaill et al., 1997; Zhang and Iadecola, 1993). Since these first experiments, it has been established that NO produced by the constitutive endothelial NOS (eNOS) has a protective role. Indeed, mice lacking the gene for eNOS subjected to focal ischemia exhibit a larger infarction than the corresponding wild-type mice (Huang et al., 1995, Huang et al., 1996). Accordingly, vasodilator NO donors such as L-arginine, isosorbide dinitrate, sodium nitroprusside, and 3-morpholinosydnonimine have been shown to attenuate ischemic brain injury (Buisson et al., 1992; Morikawa et al., 1992; Zhang and Iadecola, 1993, Zhang and Iadecola, 1994). However, the precise mechanisms by which endogenous NO can reduce the infarction produced by middle cerebral artery (MCA) occlusion remain to be clarified. Data from extracerebral tissues have shown that L-NAME in models of hepatic (Liu et al., 1998) and myocardial (Hoshida et al., 1995) ischemia-reperfusion promoted both neutrophil accumulation and tissue damage, which suggests that NO is beneficial by modulating neutrophil-mediated injury. In the cerebral microcirculation, it has been shown that inhibition of NOS under baseline conditions (Gidday et al., 1998), after leukocyte activation by LTB4(Lindauer et al., 1996), or after forebrain ischemia in rats (Hudetz et al., 1999) resulted in an increase in leukocyte-endothelium interactions. Moreover, NO donors after cerebral asphyxia in piglets decreased leukocyte adhesion (Gidday et al., 1998). These studies provide insights into the control of leukocyte-endothelium interactions by NO in cerebral tissues under nonischemic or ischemic conditions.
In the present study, we hypothesized that after transient focal cerebral ischemia, endogenous NO production might be beneficial by reducing neutrophil infiltration of the reperfusion period. For this purpose, L-NAME was administered intraperitoneally at 10 mg/kg, a dosage previously demonstrated to be deleterious in our laboratory (Margaill et al., 1997). Because NO may exert its effect not only during ischemia but also during reperfusion, L-NAME was administered at different time points after ischemia. In a second set of experiments, L-NAME was injected in neutropenic rats to evaluate the contribution of neutrophils.
METHODS
All chemicals were purchased from Sigma Chemical Corp. (Saint Quentin, Fallavier, France) unless otherwise specified. All experiments were performed with male Sprague-Dawley rats (300 to 350 g) in accordance with NIH and French Department of Agriculture guidelines (license no. 01352).
Transient focal cerebral ischemia
Rats were anesthetized with chloral hydrate (400 mg/kg, given intraperitoneally) and allowed to breathe spontaneously. The left MCA was exposed by means of a temporal craniotomy and occluded with a microclip (15 × 0.4 mm; Ohwa Tsuho Co., Tokyo, Japan), and the left common carotid artery (CCA) was concomitantly clamped. The microclip was placed on the MCA at a site proximal to the lenticulostriate artery to induce both cortical and striatal infarction. Two hours after induction of ischemia, rats were reanesthetized, the microclip occluding the MCA was removed, and CCA circulation was restored. Reperfusion in both arteries was checked under a microscope within the first 5 minutes after clip removal. During the surgical procedure and until recovery from anesthesia (2 hours after the onset of reperfusion), body temperature was maintained at 37°C to 38°C by means of a heating blanket. Sham-operated rats underwent the same surgical procedure, but the MCA and the CCA were not occluded. After surgery, rats were returned to their cage. Animals with feeding difficulties were fed with mashed laboratory chow.
Forty-eight hours after ischemia, the animals were killed with an overdose of sodium pentobarbitone and immediately perfused transcardially with 200 mL isotonic saline at 25°C with a pressure of 100 mm Hg to flush all blood components from the vasculature. Brains were removed and sliced into seven 2-mm-thick coronal sections using a brain matrix for the rat. In the fourth section, the infarcted cortical tissue was isolated, frozen, and stored at −40°C for myeloperoxidase determination (see below). A similar segment was isolated from sham-operated rats. The six remaining sections were used to quantify infarct volume as follows.
Measurement of infarct volume
Coronal brain sections were immersed in 2% 2,3,5-triphenyltetrazolium chloride, incubated for 20 minutes at 20°C, and then placed in formalin for one night in the dark. The striatal and cortical areas of infarction were measured on each section using an image analyzer (Imstar, Paris, France). Because the fourth section was used for the myeloperoxidase assay, the cortical and striatal infarct areas corresponding to this section were read on the back of the third section. The distance between coronal sections was used to calculate a linear integration for striatal and cortical infarct volumes. Infarct volumes were corrected for brain edema according to Golanov and Reis (1995).
Evaluation of neutrophil infiltration
Myeloperoxidase activity was used as an index of neutrophil accumulation and determined as previously described by Barone et al. (1991). Frozen tissue samples were quickly weighed and homogenized (Ultra-Turrax, IKA Labortechnik, Germany) in 20 volumes of 5 mmol/L potassium phosphate buffer (4°C, pH 6) followed by centrifugation at 30,000 g (30 minutes at 4°C). The supernatant was discarded and the pellet was washed again as described above. After decanting the supernatant, the pellet was extracted by suspending it in 10 volumes of 0.5% hexadecyltrimethylammonium bromide dissolved in 50 mmol/L potassium phosphate buffer (25°C, pH 6). The samples were immediately frozen in isopentane at −40°C, and three freeze/thaw and sonication (10 seconds) cycles were performed. After the last sonication, samples were incubated at 4°C for 20 minutes and centrifuged at 15,500 g (15 minutes at 4°C). Myeloperoxidase activity in the supernatant was assayed as described by Bradley et al. (1982). Briefly, 0.1 mL supernatant was mixed with 2.9 mL 50 mmol/L potassium phosphate buffer containing 0.167 mg o-dianisidine dihydrochloride and 0.0005% hydrogen peroxide. The change in absorbance at 460 nm was measured at 15-second intervals for 2 minutes with a spectrophotometer (Milton Roy; Rochester, NY, U.S.A.). One unit (U) of myeloperoxidase activity was defined as the amount that degraded 1 μmol hydrogen peroxide per minute at 25°C, and was normalized on the basis of wet weight tissue (U/g wet weight).
Determination of regional cerebral blood flow
Rats were anesthetized with chloral hydrate (400 mg/kg, given intraperitoneally) and placed on a stereotactic frame (David Kopf; Rouchaire, Courtaboeuf, France). The skull was exposed and a hole 2.5 mm in diameter was drilled in the left side at a site 3 mm lateral to the midline and 4.2 mm anterior to the interaural line. These cortical coordinates represented the ischemic penumbra of the infarct. To determine changes in regional cerebral blood flow (rCBF), a laser-Doppler flowmeter (BPM2, Vasamedics; Saint Paul, MN, U.S.A.) with a 2-mm probe (P435. Vasamedics) was used. The probe tip was positioned above the intact dural surface and fixed on a support attached to the skull. Rats were then prepared for MCA and CCA occlusions as described above. Once a stable rCBF was obtained, rats underwent ischemia. rCBF was recorded 5, 15, 30, and 120 minutes after the occlusion and 15 and 30 minutes after the reperfusion. Data are presented as percentage of preischemic rCBF.
Experimental protocols
Experiment 1: Time-dependent effects of L-NAME. This experiment was undertaken to determine whether the deleterious effect of L-NAME was related to the time of injection after ischemia and reperfusion. Rats were assigned to six groups: a sham-operated group (n = 4), an ischemic group treated with the vehicle of L-NAME (distilled water, n = 10), and four groups of ischemic rats treated with L-NAME (10 mg/kg, given intraperitoneally) given 5 minutes after the onset of ischemia (n = 11), or after 1 hour (n = 9), 7 hours (n = 7), or 22 hours (n = 10) of reperfusion. Animals were killed after 46 hours of reperfusion, and myeloperoxidase activity and brain infarction were determined as described above.
Experiment 2: Effect of L-NAME in neutropenic rats. The effect of L-NAME (10 mg/kg, given intraperitoneally 5 minutes after the onset of ischemia) was examined in neutropenic rats. Neutropenia was induced by vinblastine administration, as described previously by Uhl et al. (1993). For this purpose, animals were anesthetized with chloral hydrate (400 mg/kg given intraperitoneally), and their tail veins were cannulated for injections of vinblastine sulfate (0.5 mg/kg) or its vehicle (saline). This dose of vinblastine was shown to result in a maximum reduction in neutrophil count starting on day 4 after injection and persisting for at least 2 days (Parmentier, unpublished observation). To prevent infection, benzathine benzylpenicillin (Specia; Rhone Poulenc Rorer, Montrouge, France) (300,000 U/kg) and gentamicin (Schering-Plough; Levallois-Perret, France) (10 mg/kg) were given intramuscularly concomitantly with vinblastine administration and again just before the onset of ischemia. Ischemia was performed 4 days after vinblastine injection. Blood samples were taken just before ischemia, and leukocyte counts were performed manually with a hemocytometer and May-Grünwald Giemsa-stained smears.
Rats were then further divided into four groups:
Group 1: vehicle of vinblastine and vehicle of L-NAME (n = 9) Group 2: vehicle of vinblastine and L-NAME (n = 8) Group 3: vinblastine and L-NAME (n = 9) Group 4: vinblastine and vehicle of L-NAME (n = 7).
Animals were killed at 46 hours of reperfusion, and myeloperoxidase activity and brain infarction were determined as described above. Myeloperoxidase activity was concomitantly determined in sham-operated rats (n = 6).
Experiment 3: Effect of L-NAME on rCBF and physiologic variables. rCBF was recorded in rats subjected to ischemia and treated with either L-NAME (10 mg/kg, given intraperitoneally, n = 5) or its vehicle (n = 5) given 5 minutes after the onset of ischemia. In a parallel group of anesthetized rats, the tail artery was cannulated to monitor mean arterial blood pressure, arterial oxygen tension (PaO2), arterial carbon dioxide tension (PaCO2), and pH. Mean arterial blood pressure was recorded before ischemia, 30 minutes after the onset of ischemia, and 30 minutes after the onset of reperfusion. At the same time, arterial blood gases and pH were measured with a blood gas analyzer (Model ABL 330; Radiometer, Copenhagen, Denmark), and rectal temperature was monitored and maintained at 37°C to 38°C by means of a heating blanket. Physiologic variables were assessed in ischemic animals receiving either L-NAME (10 mg/kg, given intraperitoneally, n = 5) or its vehicle (n = 5) at 5 minutes after the onset of ischemia.
Data expression and statistical analysis
Data are expressed as mean ± standard deviation. Comparisons between two groups were evaluated by a two-tailed unpaired Student's t-test. Comparisons between multiple groups were evaluated by analysis of variance, and subsequent group comparisons were carried out by the protected least significant difference Fisher's test. Differences were considered statistically significant at a value of P < 0.05.
RESULTS
Experiment 1: Time-dependent effects of L-NAME
In vehicle-treated rats, the transient occlusion of the left MCA and CCA caused infarcts involving the cerebral cortex and the striatum at 2 days of reperfusion (110 ± 48 mm3 and 44 ± 14 mm3, respectively) (Fig. 1A). When injected 5 minutes after the onset of ischemia, L-NAME increased the cortical infarction by 34% (147 ± 50 mm3 vs. 110 ± 48 mm3 in vehicle-treated rats, P < 0.05) but did not modify the striatal lesion size (49 ± 3 mm3 versus 44 ± 14 mm3 in vehicle-treated rats, P > 0.05). The more delayed treatments with L-NAME at 1, 7, or 22 hours of reperfusion did not significantly change the infarct volumes in the cortex and in the striatum (see Fig. 1A).
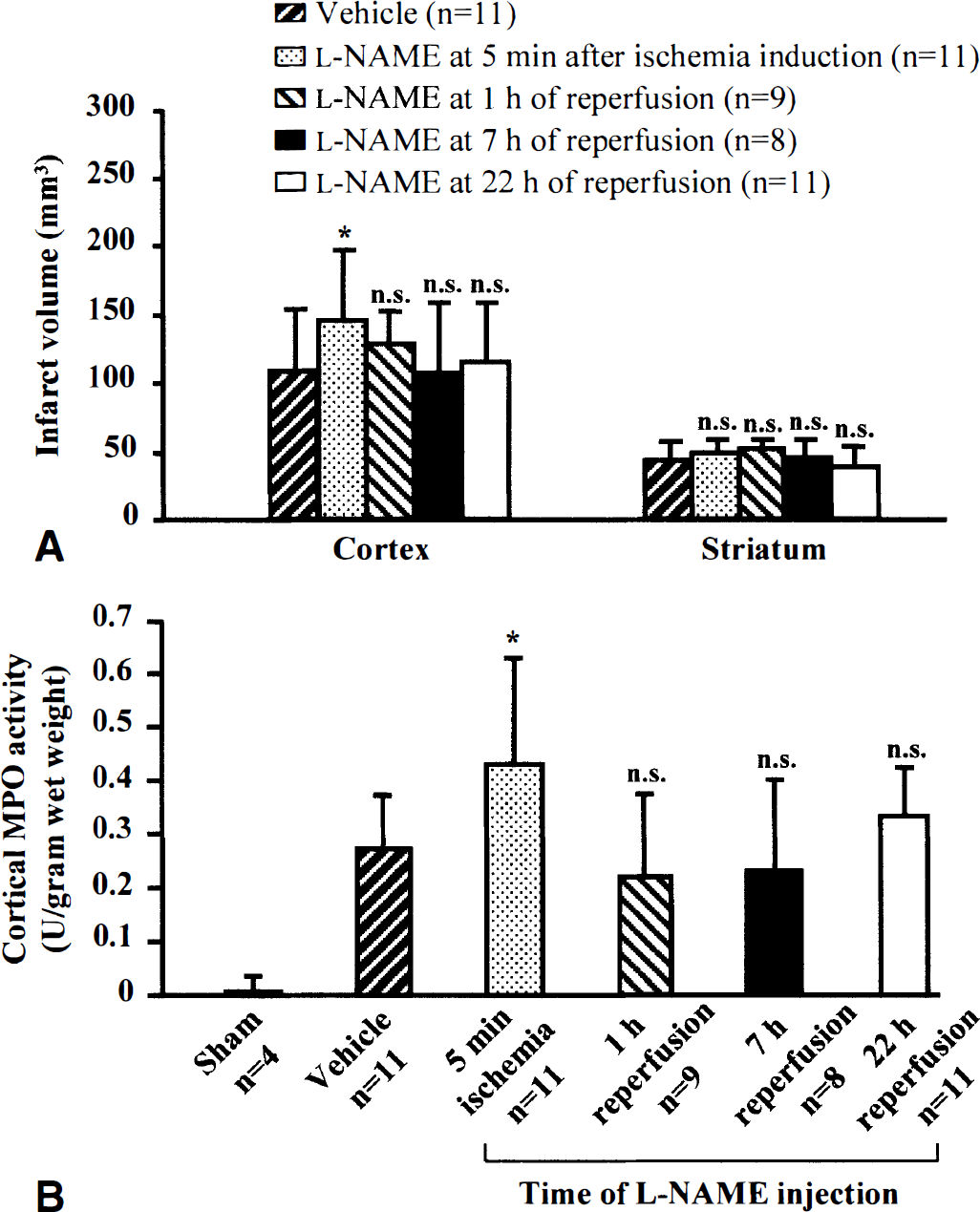
Time-dependent effect of NG-nitro-L-arginine methyl ester (L-NAME) on (
Two days after ischemia, there was a massive increase in cortical myeloperoxidase activity in vehicle-treated rats compared with sham-operated rats (0.27 ± 0.11 vs. 0.01 ± 0.01 U/g wet weight; P < 0.01) (see Fig. 1B). When L-NAME was given 5 minutes after the onset of ischemia, the normal increase in myeloperoxidase activity induced by ischemia was significantly increased by 60% (0.43 ± 0.19 vs. 0.27 ± 0.11 U/g wet weight in vehicle-treated rats; P < 0.05). Delayed treatments after 1, 7, or 22 hours of reperfusion had no effect on myeloperoxidase activity (see Fig. 1B).
Experiment 2: Effect of L-NAME in neutropenic rats
Four days after its injection, vinblastine reduced circulating neutrophils (0.50 ± 0.50 × 109/L vs. 1.80 ± 0.60 × 109/L in saline-treated rats; P < 0.001), but the mononuclear cells (monocytes and lymphocytes) remained unchanged (4.60 ± 1.70 × 109/L vs. 5.48 ± 1.24 × 109/L in saline-treated rats; P > 0.05).
The effect of L-NAME on infarct volumes in vinblastine-treated rats is shown in Fig. 2A. Ischemic rats treated by both the vehicles of L-NAME and vinblastine showed a cortical and striatal infarction at 2 days of reperfusion (109 ± 33 mm3 and 34 ± 4 mm3, respectively). Vinblastine treatment decreased the cortical infarct volume by 42% (63 ± 37 mm3 vs. 109 ± 33 mm3 in vehicle-treated rats; P < 0.05). In contrast, L-NAME given at 5 minutes after the onset of ischemia markedly increased the cortical infarction by 67% (182 ± 73 mm3 vs. 109 ± 33 mm3 in vehicle-treated rats; P < 0.01). However, when L-NAME was injected with the same protocol in vinblastine-treated rats, no increase in the cortical infarction was observed (99 ± 33 mm3 vs. 109 ± 33 mm3 in vehicle-treated rats; P > 0.05). None of the treatments modified the striatal infarct volume.
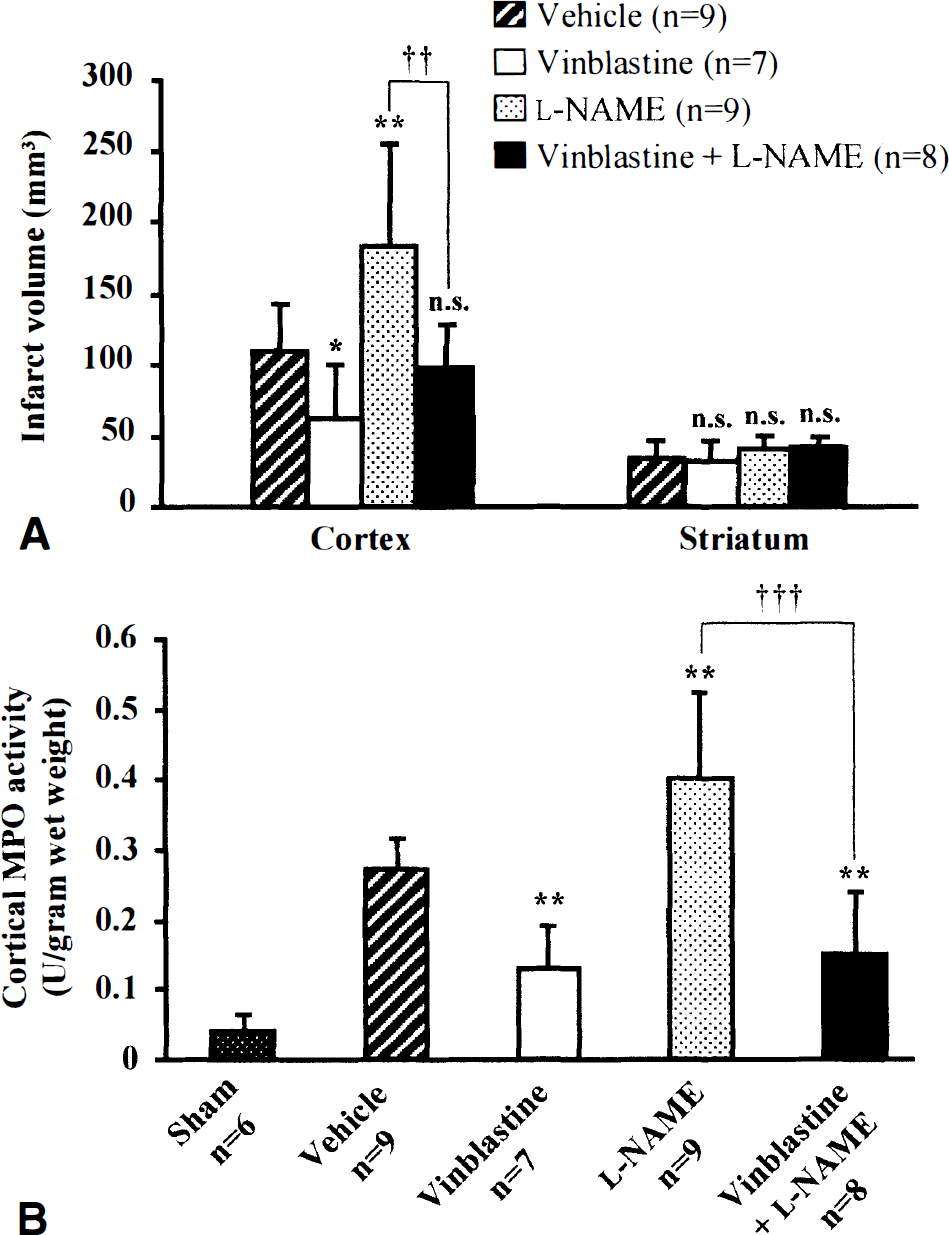
Effect of NG-nitro-L-arginine methyl ester (L-NAME) on (
In the same rats, cortical myeloperoxidase activity was assessed (see Fig. 2B). Vehicle-treated rats exhibited a massive increase in myeloperoxidase activity at 2 days after ischemia compared with sham-operated rats (0.27 ± 0.02 vs. 0.04 ± 0.01 U/g wet weight; P < 0.001). This increase in myeloperoxidase activity induced by ischemia was reduced by 52% in vinblastine-treated rats (0.13 ± 0.06 vs. 0.27 ± 0.02 U/g wet weight in vehicle-treated rats; P < 0.01), but it was exacerbated by 48% in L-NAME-treated rats (0.40 ± 0.12 vs. 0.27 ± 0.02 U/g wet weight in vehicle-treated rats; P < 0.01). However, in vinblastine-treated rats, L-NAME treatment did not affect myeloperoxidase activity (0.15 ± 0.09 vs. 0.13 ± 0.06 U/g wet weight in vinblastine-treated rats; P > 0.05).
Experiment 3: Effect of L-NAME on rCBF and physiologic variables
In the vehicle-treated rats, occlusion of the arteries induced a rapid reduction in rCBF to 61% ± 3% of preischemic values, which persisted during the 2-hour period of ischemia (Fig. 3). Clip removal produced a progressive increase in rCBF, which was significant after 30 minutes of reperfusion (74% ± 8% vs. 53% ± 6% just before reperfusion, P < 0.05). L-NAME injected 5 minutes after the onset of occlusion did not modify the rCBF time course during either the ischemic period (59% ± 3% of preischemic values) or the first 30 minutes of reperfusion (83% ± 9% at 30 minutes of reperfusion). The mean arterial blood pressure, arterial blood gases, pH, and rectal temperature remained within the normal physiologic range before ischemia, during ischemia, and after reperfusion (Table 1). L-NAME did not modify theses values during ischemia and after reperfusion.
Effect of L-NAME on physiologic variables and rectal temperature
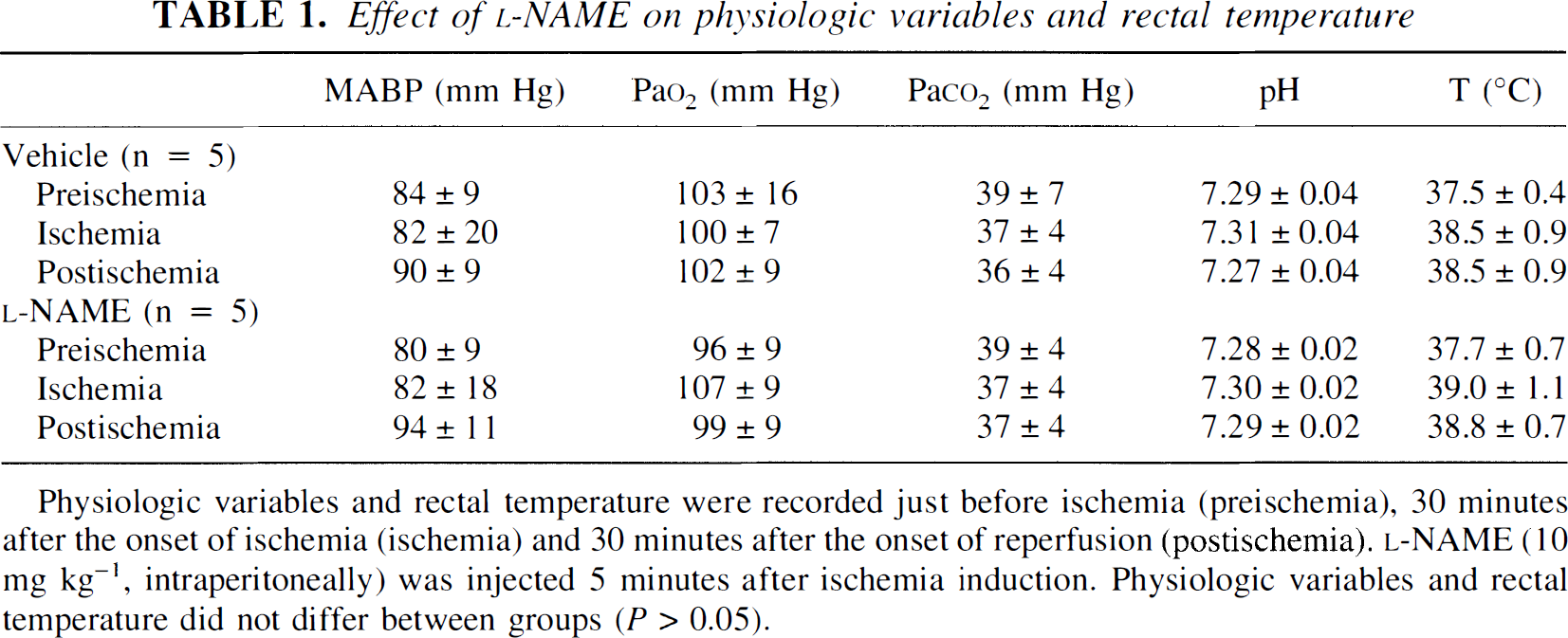
Physiologic variables and rectal temperature were recorded just before ischemia (preischemia), 30 minutes after the onset of ischemia (ischemia) and 30 minutes after the onset of reperfusion (postischemia). L-NAME (10 mg kg−1, intraperitoneally) was injected 5 minutes after ischemia induction. Physiologic variables and rectal temperature did not differ between groups (P > 0.05).
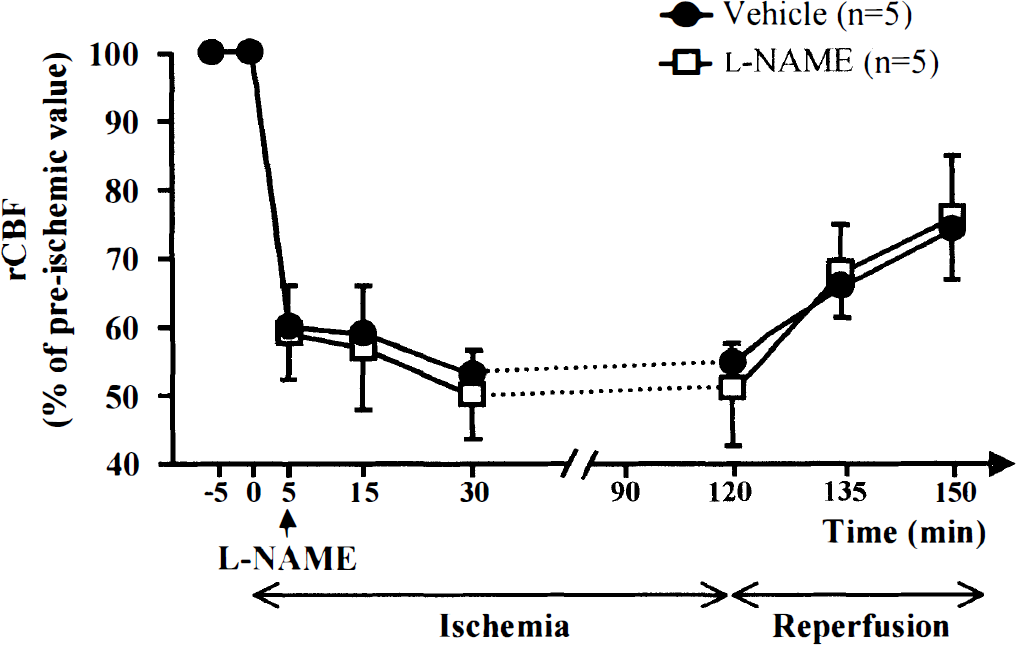
Effect of 2 hours of ischemia and reperfusion on regional cerebral blood flow (rCBF) (mean ± SD) in rats treated with vehicle and NG-NITRO-L-arginine methyl ester (L-NAME). L-NAME (10 mg/kg, given intraperitoneally) or its vehicle was injected at 5 minutes after the onset of ischemia. There was no significant difference between the two groups.
DISCUSSION
Our results demonstrate that administration of the NOS inhibitor L-NAME during the early stage of the 2-hour ischemic period expands brain infarction and induces a significant enlargement of the cerebral neutrophil content at 2 days of reperfusion. Both these effects of L-NAME are totally suppressed in rats rendered neutropenic by vinblastine. Thus, endogenous NO seems to play a beneficial role after transient focal cerebral ischemia by limiting the deleterious effect of neutrophil infiltration into the brain tissues.
A neuroprotective role for NO has been previously demonstrated in models of focal cerebral ischemia by an increase in infarct volumes after early and/or high dosage of NOS inhibitors (Kuluz et al., 1993; Margaill et al., 1997; Zhang and Iadecola, 1993); an expansion of the lesion size in eNOS knockout mice (Huang et al., 1995, Huang et al., 1996); and a reduction in ischemic damage after treatment with L-arginine supplementation or NO donors (Buisson et al., 1992; Morikawa et al., 1992; Zhang and Iadecola, 1993, Zhang and Iadecola, 1994). This beneficial effect has been previously attributed to the vasodilatory properties of NO-more specifically, to an increase in cerebral blood flow during the ischemic period (Morikawa et al., 1994; Zhang and Iadecola, 1993). In our study, it is unlikely that L-NAME enhanced the cerebral infarction by altering the cerebral blood flow or the arterial blood pressure, because these variables did not differ among the two groups of vehicle-treated and L-NAME-treated rats during ischemia and reperfusion. These results are not in accordance with those of previous studies showing that L-NAME induces hypertension and hypoperfusion throughout the brain under basal conditions (Dawson et al., 1993; Kelly et al., 1995) and after global and forebrain ischemia (Humphreys and Koss, 1998; Prado et al., 1993). It is, however, noticeable that these studies used higher doses of L-NAME or intravenous injection, which is a more sensitive way to induce vascular changes than intraperitoneal injection.
There is considerable evidence that cerebral ischemia-reperfusion leads to a progressive increase in neutrophil adherence and infiltration that contributes to the development of ischemic brain damage (Hartl et al., 1996). In our study, the determination of myeloperoxidase activity, an enzyme marker of neutrophils, was used to quantify neutrophil infiltration. This method has been shown to indicate indirectly but precisely significant accumulation of neutrophils into cerebral tissues after ischemia (Barone et al., 1991). In our model, we observed a significant increase in myeloperoxidase activity in the infarcted cortical tissues, indicating significant neutrophil infiltration after 2 days of reperfusion. Moreover, we showed a significant reduction in the cortical infarct size in neutropenic rats. Taken together, these data support an injurious role for neutrophils in our model of transient focal cerebral ischemia.
Kubes et al. (1991) first revealed the role of NO in the regulation of leukocyte-endothelial interactions in noncerebral tissues. They demonstrated that impairment of endothelial NO production in mesenteric venules leads to a drastic increase in leukocyte adherence to the vascular endothelium. From this initial result, other researchers have reported that inhibition of NOS can promote neutrophil-endothelial adherence in peripheral vascular beds under basal and ischemic conditions (Harbrecht et al., 1992; Ma et al., 1993; Persson et al., 1990). Moreover, NOS inhibition in models of peripheral ischemia-reperfusion exacerbates the tissue damage and increases neutrophil accumulation (Kupatt et al., 1997; Liu et al., 1998; Naito et al., 1998; Sato et al., 1997; Tavaf-Montamen et al., 1998). The NO-dependent mechanisms regulating leukocyte-endothelial adherence under basal and ischemic conditions may include the capacity of NO to influence the steady state of the superoxide anion, a well-established proadherent agent (Clancy et al., 1992; Rubanyi et al., 1991; Seth et al., 1994; Suematsu et al., 1994), and the reduction of the expression of adhesion molecules (De Caterina et al., 1995; Gauthier et al., 1994; Khan et al., 1996).
In the cerebral circulation, after the initial observation of Lindauer et al. (1996) that NOS inhibition during leukocyte activation (by LTB4) enhances leukocyte adherence, two other studies have investigated the effects of NOS inhibition on leukocyte adherence during cerebral ischemia-reperfusion. They have reported that superfusion of NO donors (sodium nitroprusside) into the pial vasculature reduces the leukocyte adherence induced by asphyxia in piglets (Gidday et al., 1998) and that L-NAME (20 mg/kg, given intravenously) augments leukocyte adherence induced by forebrain ischemia in rats (Hudetz et al., 1999). Taken together, these studies indicate a role for NO in modulating leukocyte-endothelial adherence in the cerebral circulation under hypoxic and ischemic conditions. However, these studies examined only the effect of NO on leukocyte-endothelial interactions in the pial circulation during the initial hours of postischemic reperfusion (1 or 2 hours). Thus, the time course of the NO-based antiadherent effect after ischemia and reperfusion remains to be established. Moreover, the suppression of NO-based antiadherent effects might lead to an additional increase in tissue damage produced after cerebral ischemia, as has been described after peripheral ischemia (Kupatt et al., 1997; Liu et al., 1998; Naito et al., 1998; Sato et al., 1997; Tavaf-Montamen et al., 1998). This led us to investigate in our neutrophil-dependent model of transient focal ischemia whether L-NAME might exacerbate cerebral infarction by increasing neutrophil infiltration, and whether the time of administration of L-NAME relative to the onset of ischemia was relevant. Neutrophil infiltration and cerebral infarction were examined at 2 days of reperfusion, which corresponds to a much later time than in the studies using peripheral ischemia (generally performed at 2 to 6 hours of reperfusion). This time can be justified by the fact that full development of cerebral infarction is delayed after cerebral ischemia (1 to 2 days), and neutrophil infiltration occurs much later after cerebral ischemia (1 to 2 days) (Barone et al., 1995; Matsuo et al., 1994; Zhang et al., 1994a) than than after peripheral ischemia (6 hours to 1 day) (Smith et al., 1988; Suzuki and Toledo-Pereyra, 1994). The reasons for this delay in neutrophil infiltration after cerebral ischemia remain to be clarified; they might, however, include a late opening of the blood-brain barrier (Belayev et al., 1996). In our study, L-NAME at a dosage that increases infarction enhanced the cerebral neutrophil accumulation at 2 days after ischemia. Both these effects are not observed in neutropenic rats. These results indicate for the first time that NOS inhibition induces a leukocyte-dependent increase in ischemic brain damage after transient focal ischemia.
Our data also demonstrated that the effect of L-NAME on the regulation of cerebral neutrophil accumulation is time-dependent and occurs within the first 3 hours after ischemia. This suggests an early and transient production of NO after focal ischemia, and such a production has indeed been well documented (Iadecola, 1997; Kader et al., 1993). The source responsible for this NO production is likely to be the constitutive isoform(s) of NOS, because we have previously reported that the inducible isoform of NOS appears after 24 hours of reperfusion in our model (Parmentier et al., 1999). Accordingly, a recent study has shown that mice lacking the gene for eNOS or for neuronal NOS exhibit an increase in neutrophil rolling and adherence in the mesentery under baseline conditions (Lefer et al., 1999). Therefore, the transient effect of L-NAME observed in our model might be explained by the fact that endogenous NO produced by the constitutive isoforms of NOS is rapidly impaired after ischemia. Another possible explanation might be that NO acts on neutrophil-endothelial interactions by means of the reduction of the expression of the adhesion molecules, which occurs early and transiently after ischemia.
The role of NO in modulating postischemic neutrophil infiltration demonstrated in the present study may explain, in part, the findings that brain infarction is larger in endothelial NOS knockout mice (Huang et al., 1995, Huang et al., 1996) and after early administration of nonselective NOS inhibitors (Kuluz et al., 1993; Margaill et al., 1997; Zhang and Iadecola, 1993). This can also explain the neuroprotective role of L-arginine supplementation and NO donors (Buisson et al., 1992; Morikawa et al., 1992). In summary, this study provides the first evidence that blocking the acute production of NO aggravates brain damage in a model of transient focal cerebral ischemia by promoting neutrophil accumulation in the ischemic tissues. Thus, during the initial phase after focal ischemia, the antiinflammatory effects of NO exceed its cytotoxic effects, providing a theoretical basis for NO-based therapies in the treatment of focal cerebral ischemia-reperfusion.
Footnotes
Acknowledgment
The authors thank Dr. S. Scott Panter for his comments on this manuscript.