
Abstract
Glial inclusions containing the microtubule-associated protein tau are present in a variety of chronic neurodegenerative conditions. We now report a rapid and time-dependent increase of tau immunoreactivity within oligodendrocytes after focal cerebral ischemia in the rat. The number of tau positive oligodendrocytes in the ipsilateral subcortical white matter increased six- to eightfold by 40 minutes after permanent middle cerebral artery occlusion (MCAO). Tau was detected using antibodies that label both the N- and C-terminal of the protein, suggesting accumulation of full-length protein within these cells. Pretreatment with the spin trap agent α-phenyl-tert-butyl-nitrone (PBN)(100mg/kg) reduced the number of tau-positive oligodendrocytes by 55% in the subcortical white matter of the ischemic hemisphere compared with untreated animals at 40 minutes after MCAO. In contrast, pretreatment with glutamate receptor antagonists MK-801 (0.5 mg/kg) or 2,3-dihydroxy-6-nitro-7-sulpfamoylbenzo(f)quinoxaline (NBQX) (2 × 30 mg/kg), failed to reduce the number of tau-positive oligodendrocytes after 40 minutes of ischemia. The results indicate that oligodendrocytes respond rapidly to an ischemic challenge and that free radical-mediated mechanisms are involved in the cascade leading to increased tau immunoreactivity.
Although traditionally believed to be a neuron-specific protein, the microtubule-associated protein tau has been shown to be present in glial inclusions characteristic of chronic neurodegenerative diseases such as progressive supranuclear palsy, multisystem atrophy and Parkinson's disease (Abe et al., 1992; Ikeda et al., 1993; Iwatsubo et al., 1994; Nishimura et al., 1992; Yamada et al., 1992). Tau within these inclusions is thought to be similar to that present in neurofibrillary tangles in that it is recognized by antibodies directed toward paired helical filament tau (Abe et al., 1992; Iwatsubo et al., 1994; Nishimura et al., 1992; Yamada et al., 1992), and thus such inclusions have been named glial fibrillary tangles (GFT). The formation of neurofibrillary tangles is thought to be central to neuronal degeneration in diseases such as Alzheimer's disease, Parkinson's disease, and progressive supranuclear palsy. Therefore, it is possible to speculate that GFT formation may represent cytoskeletal disturbances within glial cells which subsequently lead to their degeneration and it is interesting to note that oligodendroglial degeneration in multisystem atrophy has been shown to correlate closely with the severity of the disease (Papp and Lantos, 1994). Presently, however, the mechanisms underlying GFT formation remain unknown. Many factors have been hypothesized to play a role in the pathogenesis of chronic neurodegenerative conditions including glutamate excitotoxicity (Choi, 1988) and free radical-mediated injury (Evans, 1993; Nixon and Cataldo, 1994; Halliwell, 1992) although experimental data in direct support of these hypotheses is scarce. By contrast, in conditions of acute brain injury, pathogenic roles for both glutamate excitotoxicity and free radical-induced damage are strongly implicated on the basis of data obtained from experimental models (Cao and Phillis, 1994; Folbergrova et al., 1995; Gill et al., 1992; Park et al., 1988).
Recently, we have reported increased tau immunoreactivity within oligodendrocytes in postmortem brain tissue of patients dying after severe head injury or stroke (Irving et al., 1996b). Increased tau immunoreactivity was detected within 1 hour after injury suggesting that tau accumulates within these cells rapidly after acute injury in the human brain. Similarly in the rat, increased tau immunoreactivity was detected in glia within 2 hours of the induction of focal cerebral ischemia (Dewar and Dawson, 1995). Together these results suggest that tau undergoes rapid alteration within oligodendrocytes in response to acute brain injury although the time course and mechanism of onset is not yet known. With the knowledge that excessive stimulation of glutamate receptors is involved in the processes of ischemic brain damage, we investigated the effect of intracortical perfusion of glutamate on tau immunoreactivity in oligodendrocytes in the rat brain. While intracortical perfusion of monosodium glutamate did induce increased tau immunoreactivity in oligodendrocytes, the perfusion of sodium chloride also stimulated this response (Irving et al., 1996a). We also observed similar effects of glutamate and hypertonic sodium chloride in pure oligodendrocyte cultures (Irving et al., 1995) indicating that increased tau immunoreactivity was not a consequence of neuronal degeneration and that a mechanism other than glutamate receptor stimulation may be involved in mediating this response.
Although the mechanisms through which this occurs is controversial, electrophysiological studies have shown that mature oligodendrocytes respond directly to glutamate exposure in vitro (Ballanyi and Kettenman, 1990; Butt and Tutton, 1992; Patneau et al., 1994; Puchalski et al., 1994). Mature oligodendrocytes do not seem to express N-methyl-D-aspartate (NMDA) receptors (Patneau et al., 1994; Gallo and Russell, 1995). However, glutamate-induced injury has been reported to be mediated through non-NMDA receptors in these cells (Patneau et al., 1994; Puchalski et al., 1994; Yoshioka, 1995). Contrasting studies report that glutamate-induced injury in oligodendrocytes is mediated, not through glutamate receptor activation, but through the actions of free radicals, possibly caused by the action of the glutamate/cystine transporter (Oka et al., 1993) such that increased extracellular glutamate concentrations result in an efflux of cystine leading to glutathione depletion within these cells. Glutathione is an important protective agent against oxidative stress (Bast et al., 1991); therefore its depletion renders the cell vulnerable to oxidative stress. In support of this, oxygen radicals have been shown to cause oligodendroglial cell death directly (Kim and Kim, 1991). The aims of the present study were twofold: first, to determine the time of onset of increased tau immunoreactivity in oligodendrocytes after the induction of focal cerebral ischemia and second, to determine if this response was mediated through glutamate-receptor activation or a free radical-mediated cascade.
MATERIALS AND METHODS
Focal cerebral ischemia
Twenty-three adult male Fischer 344 rats, each weighing between 247 and 300 g were used in this study. The animals were initially anesthetized with a mixture of 5% halothane, 70% nitrous oxide, and 30% oxygen. A tracheostomy was performed to allow artificial ventilation and anesthesia was subsequently maintained between 1 and 1.25% halothane. Cannulation of a femoral artery and vein allowed the monitoring of blood pressure and arterial blood gases (Table 1), samples for analysis being taken immediately after vessel cannulation (pre-occlusion) and immediately after induction of ischemia (post-occlusion). Animals were maintained normocapnic, normoxic, and normothermic throughout the experimental period (Table 1). The left middle cerebral artery (MCA) was exposed using a subtemporal approach based on the method originally described by Tamura et al., (1981). The exposed MCA was occluded by bipolar electrocoagulation from the area where it crossed the inferior cerebral vein to proximal to the origin of the lenticulostriate branch. All side branches including the lenticulostriate were coagulated and the artery transected. The muscle and skin were then sutured and the animals maintained under anesthesia for appropriate intervals. For the time-course study, animals were killed at 20 minutes (n = 3); 40 minutes (n = 4) or 80 minutes (n = 3) after middle cerebral artery occlusion (MCAO).
Physiological variables
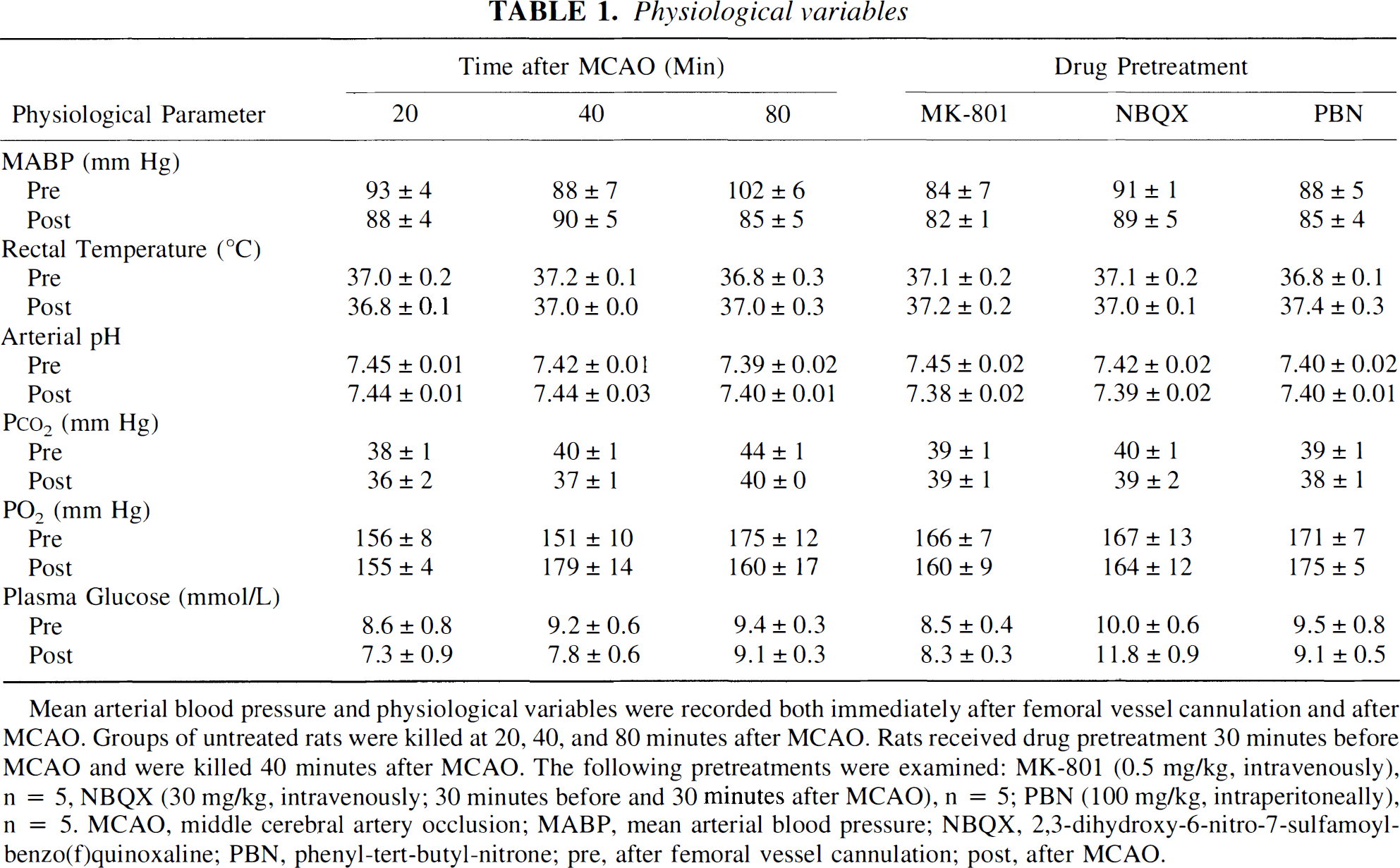
Mean arterial blood pressure and physiological variables were recorded both immediately after femoral vessel cannulation and after MCAO. Groups of untreated rats were killed at 20.40, and 80 minutes after MCAO. Rats received drug pretreatment 30 minutes before MCAO and were killed 40 minutes after MCAO. The following pretreatments were examined: MK-801 (0.5 mg/kg, intravenously), n = 5, NBQX (30 mg/kg, intravenously; 30 minutes before and 30 minutes after MCAO), n = 5; PBN (100 mg/kg, intraperitoneally), n = 5, MCAO, middle cerebral artery occlusion; MABP, mean arterial blood pressure; NBQX, 2,3-dihydroxy-6-nitro-7-sulfamoyl-benzo(f)quinoxaline; PBN, phenyl-tert-butyl-nitrone; pre, after femoral vessel cannulation; post, after MCAO.
Drug interventions
There were three treatment groups in this study: MK-801 (0.5 mg/kg, intravenous) (n = 5); 2,3-dihydroxy-6-nitro-7-sulfamoylbenzo(f)quinoxaline (NBQX) (30 mg/kg, intravenous) (n = 4), or α-phenyl-tert-butyl-nitrone (PBN) (100 mg/kg intraperitoneal) (n = 5). Vehicle solutions for these agents were: 0.9% saline; 5.5% glucose, and distilled water respectively. All drugs were administered 30 minutes before MCA occlusion and NBQX was administered again 30 minutes after MCAO. The doses for each drug used in this study have previously been shown to reduce the volume of infarct after permanent MCAO in the rat (Cao and Phillis, 1994; Gill et al., 1992; Park et al., 1988). For the drug-intervention studies animals were killed 40 minutes after permanent MCAO.
Immunohistochemistry
At the appropriate times after MCAO, the animals were killed by transcardial perfusion of 0.9% saline followed by 4% paraformaldehyde in 50 mmol/L phosphate buffer, pH 7.4. The brain was removed and post-fixed in 4% paraformaldehyde for a further 24 hours at 4°C and cryoprotected in 30% sucrose. The brains were then frozen for 2 minutes in isopentane chilled on dry-ice to −42°C and cut into 30-µm coronal sections in a cryostat. Sections were stored in cryoprotectant (30% glycerol/30% ethylene glycol in sodium phosphate buffer) at −20°C. For immunohistochemistry, free-floating tissue sections were rinsed in 50 mmol/L phosphate buffered saline pH 7.4 (2 × 5 minutes) and incubated in phosphate buffered saline containing 0.2% Triton X-100 for 30 minutes. Endogenous peroxidase activity was blocked by incubating in 3% hydrogen peroxide for 20 minutes. Sections were then incubated in phosphate buffered saline containing 10% normal horse serum/1% BSA (Tau 1, MAP2) or 10% normal goat serum/1% BSA (TP70, TP007) for 1 hour. Incubation with primary antibody (Tau 1, 1:1000; MAP2, 1:750; TP70, 1:2000; TP007, 1:500) was overnight at 4°C. Sections were then incubated with appropriate biotinylated secondary antibodies (horse anti-mouse 1:100, Vector Laboratories; goat anti-rabbit 1:400, DAKO) followed by ABC complex (Vector Laboratories, Peterborough, U.K.) with 3-3'-diaminobenzidine (Sigma, Poole, Dorset, U.K.) as the chromagen. Negative controls were included in each experiment, in which the primary antibody was omitted from the procedure. Sections were mounted onto poly-L-lysine coated slides then dehydrated and cleared before microscopic analysis.
Alkaline phosphatase pretreatment
Dephosphorylation of the tissue before staining with the Tau 1 antibody was performed using the method previously described by Papasozomenos and Binder (1987). Tissue sections were incubated with 130 µg/mL, of type VII-L alkaline phosphatase from bovine intestinal mucosa (Sigma) in 100 mmol/L Tris-HCl, pH 8 for 2.5 hours in a shaking water bath at 32°C. To inhibit proteolysis, 1 mmol/L phenylmethylsulfonyl fluoride, 10 µg/mL pepstatin and 10 µg/mL leupeptin were included in the incubation fluid. For controls, 100 mmol/L sodium pyrophosphate was included in the incubation solution as a competitive inhibitor of alkaline phosphatase. The dephosphorylation reaction was quenched by bathing the sections in ice-cold Tris saline (50 mmol/L Tris-HCl, 0.2 mol/L sodium chloride, pH 7.6), and immunostained immediately as described above.
Double-label immunohistochemistry
Sections were stained with Tau 1 (1:1000) as described above using Vector VIP (Vector Laboratories) as the chromagen. After rinses in distilled water and phosphate buffered saline, sections were incubated in phosphate buffered saline containing 10% normal goat serum/1% BSA for 1 hour and then for 1 hour at room temperature with either anti-transferrin (1:200) as a marker for oligodendrocytes (Conner and Finne, 1986; Martin et al., 1991) or anti-glial fibrillary acid protein (1:750) as a marker for astrocytes (Martin et al., 1991). Antibody binding was detected as described above for polyclonal antibodies, using Vector SG (Vector Laboratories) as the chromagen.
Antibodies
Tau was detected using three different antibodies: Tau 1, TP70, and TP007. The monoclonal antibody Tau 1 labels a dephosphorylated epitope between residues Ser 199 and 204 (Binder et al., 1985; Papasozomenos and Binder, 1987). TP70 is a polyclonal antibody directed against a synthetic peptide corresponding to residues 428 to 441 at the C-terminal of the protein (Brion et al., 1993), and TP007 is a polyclonal antibody raised to a synthetic peptide corresponding to the 16 most N-terminal residues of human tau protein (Davis et al., 1995). MAP2 was labelled using a monoclonal antibody which recognizes MAP2a, b and c (Sigma). Anti-transferrin and anti-glial fibrillary acid protein were polyclonal antibodies obtained from DAKO (High Lycombe, Bucks, U.K.).
Quantification of tau-positive oligodendrocytes
For each animal in this study, the total number of tau-positive oligodendrocytes in the entire subcortical white matter, extending from that underlying the perirhinal cortex to that underlying cingulate cortex was counted in one section from each of the two brain levels shown in Fig. 1. Sections were viewed at ×40 magnification. The investigator was blinded to the survival period and drug treatments of each animal. The area of subcortical white matter in each section counted was measured using an MCID image analyzer and the number of tau-positive oligodendrocytes/mm2 calculated for the white matter at each brain level. From this the average number of tau-positive oligodendrocytes/mm2 present in the 2 sections counted was calculated for each individual animal. The mean number of tau-positive oligodendrocytes/mm2 in each treatment group was calculated and data expressed as mean ± SD. The same quantitative analysis was performed in both hemispheres, ipsilateral and contralateral to the occluded MCA. Coefficient of variance for repeated measurements 1 week apart was 7%. Statistical analysis consisted of an ANOVA followed by Student's one-tailed t-test with appropriate Bonferroni correction. The a priori hypothesis tested was that the drugs would reduce the number of tau-positive oligodendrocytes.
Schematic representation of the sections in which the number of tau-positive oligodendrocytes/mm2 was determined in subcortical white matter. The sections corresponded to those at approximately 1.2 mm and 0.7 mm anterior to Bregma in the atlas of Paxinos and Watson. For each animal, the total number of tau-positive oligodendrocytes in the entire subcortical white matter was counted in one section from each of the two brain levels shown above. Sections were viewed at ×40 magnification and the investigator was blinded to the treatment of each animal. The area of subcortical white matter in each section was measured using an MCID image analyzer and the number of tau-positive oligodendrocytes/mm2 calculated for the white matter of each brain level. From this, the average number of positive cells/mm2 in each treatment group was calculated (mean ± SD). The same quantitative analysis was performed in both hemispheres, ipsilateral and contralateral to the occluded MCA. () Area of contralateral white matter where tau-positive oligodendrocytes were counted. ( ) Area of ipsilateral white matter where tau-positive oligodendrocytes were counted. (
) Area of ipsilateral white matter where tau-positive oligodendrocytes were counted. ( ) Area of ischemic damage as detected with loss of MAP2 immunoreactivity. The arrow indicates the region of subcortical white matter from which photographs shown in Figs. 2, 4 and 5 were taken.
) Area of ischemic damage as detected with loss of MAP2 immunoreactivity. The arrow indicates the region of subcortical white matter from which photographs shown in Figs. 2, 4 and 5 were taken.
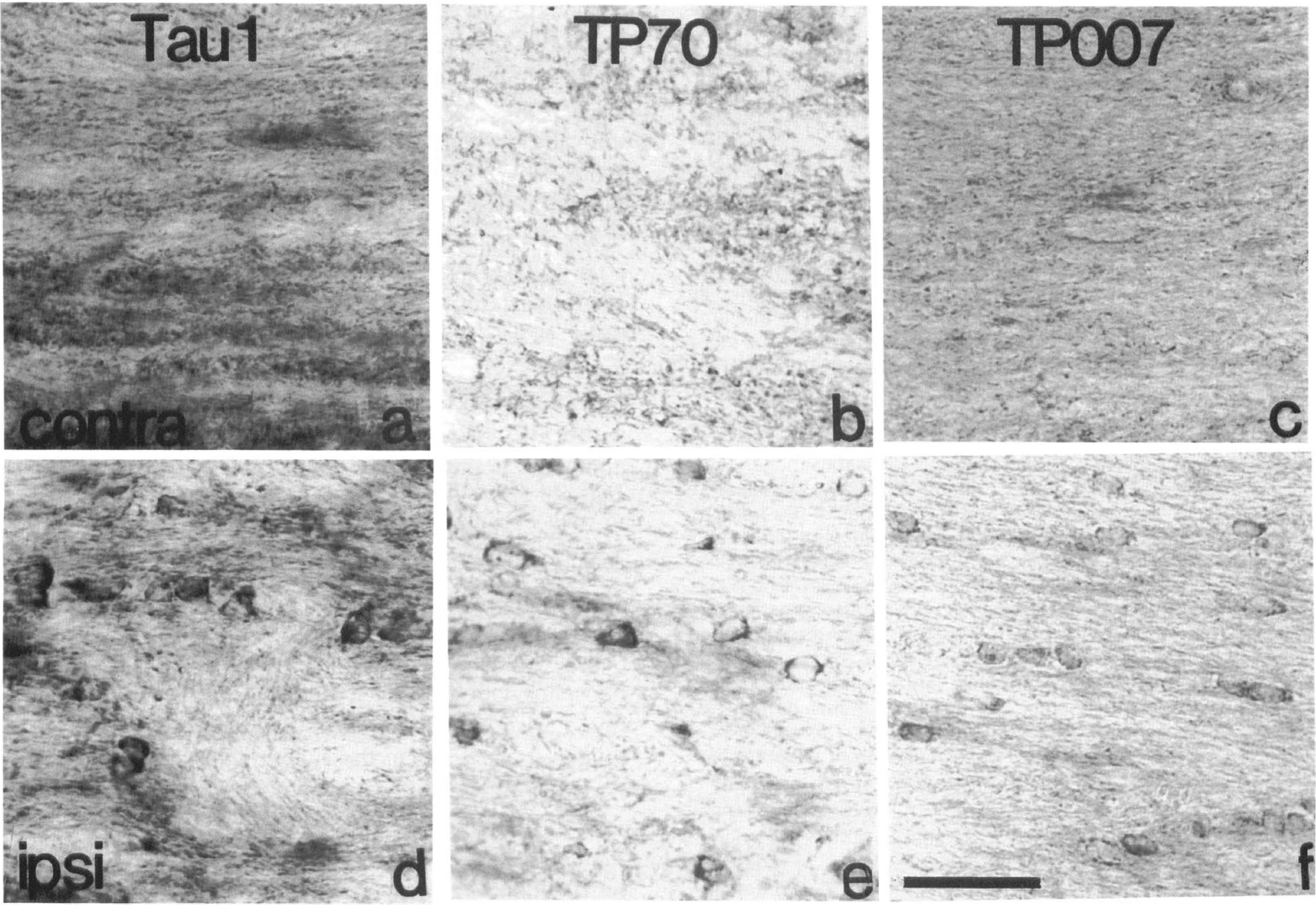
Tau immunostaining of subcortial white matter 40 minutes after MCAO in the rat. Tau immunostaining was localized mainly within the axons of the contralateral subcortical white matter 40 minutes after MCAO (
RESULTS
MAP2 immunoreactivity
MAP2 immunostaining has been shown to be a sensitive marker of ischemic damage in animal models of cerebral ischemia (Dawson and Hallenbeck, 1996; Kitagawa et al., 1989; Yamamoto et al., 1986; Yanagihara et al., 1990). Although traditional histological stains such as hematoxylin and eosin have been used within 4 hours of permanent focal ischemia at shorter survival times, these stains do not permit definitive conclusions to be drawn about neuronal viability. Therefore, in this study involving very short survival times after MCAO, the anatomical extent of ischemic damage was detected using MAP2 immunostaining. Twenty minutes after MCAO, an area in which MAP2 immunostaining was decreased was detected within the caudate nucleus in the ipsilateral hemisphere. MAP2 immunostaining was decreased within the caudate nucleus within the territory ipsilateral to the occluded MCA 40 and 80 minutes after occlusion. In addition, areas of decreased immunoreactivity were also detected within the ipsilateral cortex, however this was variable and patchy in its distribution. Decreased MAP2 immunostaining in the hemisphere ipsilateral to the occluded MCA was detected in all animals used in this study.
Time course of increased tau-immunoreactivity in oligodendrocytes
In the hemisphere contralateral to the occluded MCA of all animals in this study Tau 1, TP70, and TP007 immunoreactivity within the cortex and caudate nucleus was predominantly located within neurons, similar to that previously described (Binder et al., 1985; Irving et al., 1996a). Occasional tau-positive cells were detected in the subcortical white matter of the contralateral hemisphere (Figs. 2A–C and 3) with Tau 1, TP70, and TP007. The number of Tau 1-positive oligodendrocytes detected in the contralateral subcortical white matter 20 minutes after MCAO was not significantly increased after dephosphorylation of the tissue (+alkaline phosphatase [AP]: 19 ± 7/mm2; -AP: 23 ± 22/mm2). Twenty minutes after MCAO (Fig. 3) occasional tau-positive cells with the morphological appearance of glia were detected within the ipsilateral subcortical white matter and the number was greater than that in the contralateral hemisphere.
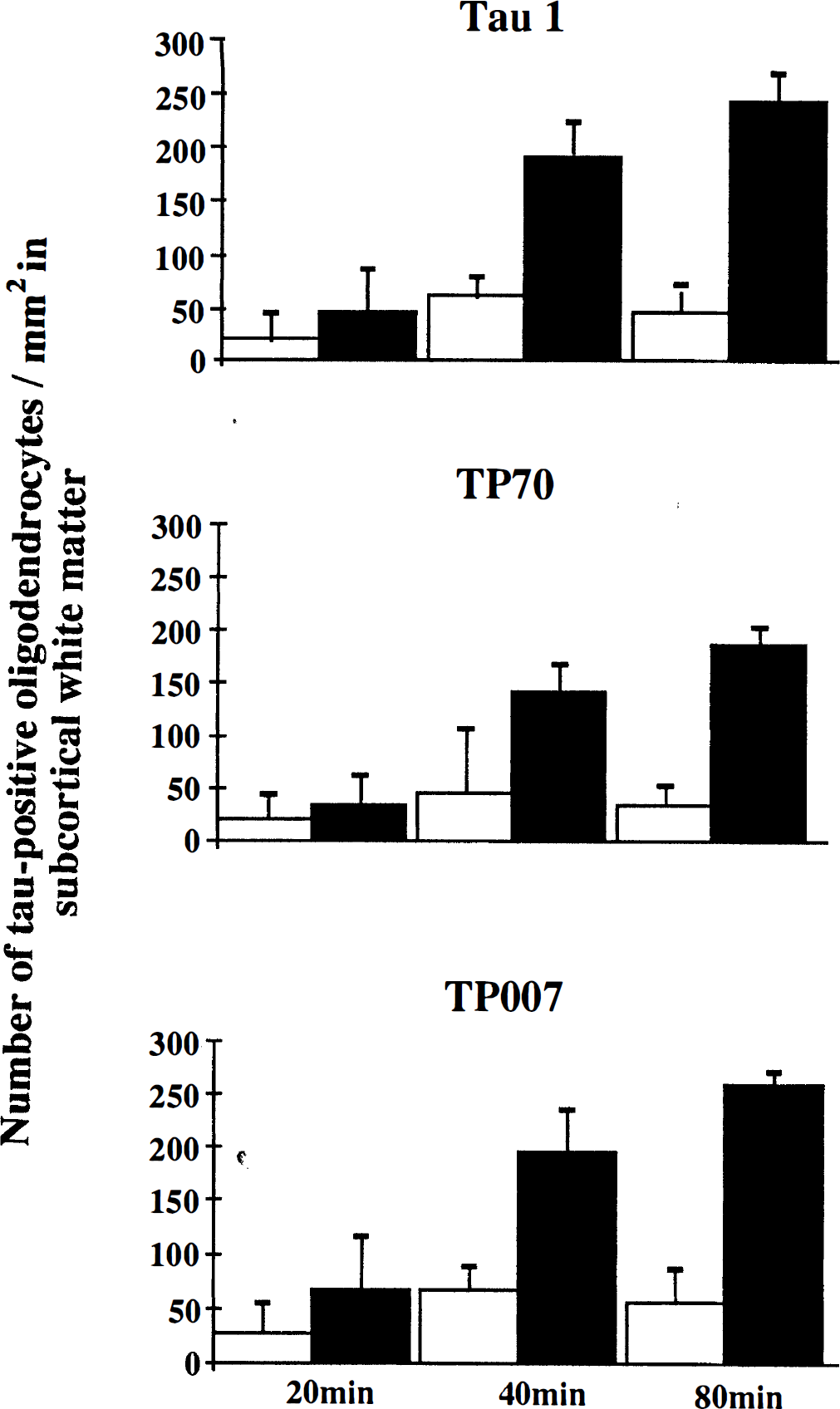
The number of tau-positive oligodendrocytes increased with time following MCAO. Twenty minutes after MCAO the number of tau-positive oligodendrocytes/mm2 in the ipsilateral subcortical white matter was increased slightly as compared with contralateral white matter. The number of tau-positive oligodendrocytes/mm2 increased significantly 40 and 80 minutes after MCAO in the white matter ipsilateral to the occluded MCA. This increase was detected with all three tau antibodies, Tau 1, TP70, and TP007. Data are presented as mean ± SD. Hollow bar = contralateral subcortical white matter. Solid bar = ipsilateral white matter.
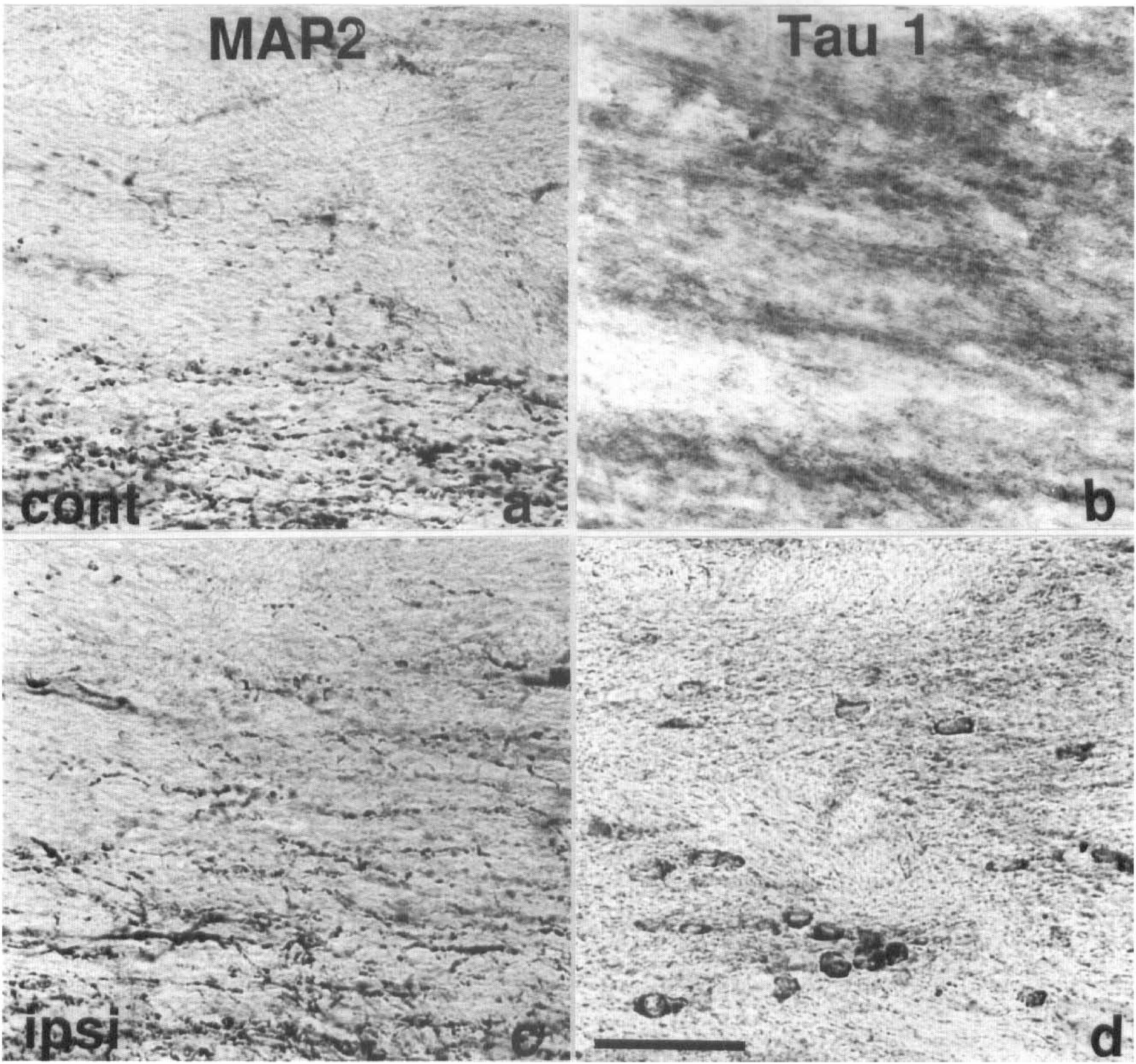
MAP2 and Tau 1 immunostaining of subcortical white matter 40 minutes after MCAO in the rat. MAP2 immunostaining was localized predominantly within dendrites of the contralateral (
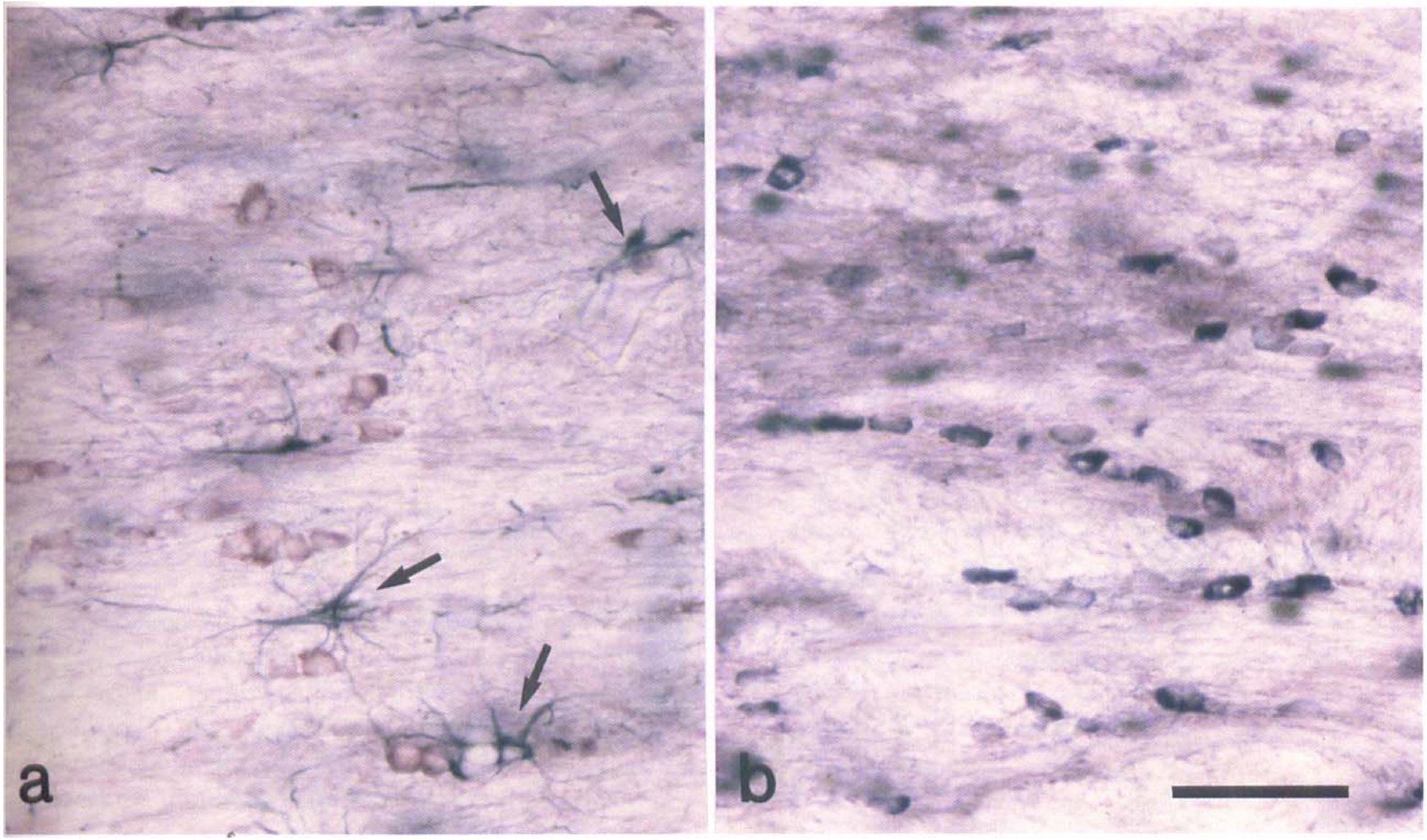
Identification of tau-positive glia as oligodendrocytes. (
In rats subjected to 40 minutes (Fig. 2D–F) or 80 minutes of focal ischemia, the number of tau-positive glial cells in the ipsilateral subcortical white matter, detected with all three tau antibodies, was increased compared with the contralateral white matter (Fig. 3). Dephosphorylation of tissue before Tau 1 immunostaining failed to significantly alter the number of Tau 1-positive glial cells detected in the subcortical white matter ipsilateral to the occluded MCA 20 (+AP: 26 ± 21/mm2; −AP: 46 ± 38/mm2), 40 (+AP: 163 ± 44/mm2; −AP: 192 ± 33/mm2), or 80 (+AP: 243 ± 132/mm2; −AP: 243 ± 26/mm2) minutes after the induction of focal cerebral ischemia. It should be noted that the number of tau-positive oligodendrocytes detected with TP007 seemed to be higher in all animals as compared with Tau 1 and TP70. This may possibly be explained by the small degree of cross reactivity of this antibody with neurofilament proteins. Whereas increased tau immunoreactivity was most prominent within glial cells located in the subcortical white matter, cells with a similar morphological appearance were also present to a lesser extent in the gray matter of the caudate nucleus and the neocortex ipsilateral to the occluded MCA. These cells were detected using all three tau antibodies and were present in gray matter only within the boundary of ischemic damage as determined by MAP2 immunostaining. However, in contrast to tau immunoreactivity, MAP2 immunoreactivity was not detected in glial cells within the subcortical white matter contralateral or ipsilateral to the occluded MCA, 20, 40, or 80 minutes after the induction of focal cerebral ischemia (Fig. 4).
Double-label immunohistochemistry showed that tau-positive cells in the subcortical white matter ipsilateral to the occluded MCA were glial fibrillary acid protein negative and morphologically distinct from astrocytes (Fig. 5A). In addition, these cells were transferrin positive (Fig. 5B) and had a morphological appearance consistent with that of oligodendrocytes. Thus, we conclude that the tau-positive glial cells were oligodendrocytes.
Effect of drug intervention on tau-positive oligodendrocytes
Pretreatment with the spin-trap agent PBN (100 mg/kg) significantly reduced the number of tau-positive oligodendrocytes within the subcortical white matter ipsilateral to the occluded MCA of animals subjected to 40 minutes cerebral ischemia as compared with untreated animals (Fig. 6). Pretreatment with the glutamate receptor antagonists MK-801 (0.5 mg/kg) or NBQX (2 × 30 mg/kg) failed to decrease the number of tau-positive oligodendrocytes present in the subcortical white matter ipsilateral to the occluded MCA compared with untreated animals (Fig. 6). Dephosphorylation of tissue before Tau 1 immunostaining failed to significantly alter the number of tau-positive glial cells detected in the subcortical white matter ipsilateral to the occluded MCA 40 minutes after the induction of focal cerebral ischemia in animals pretreated with PBN (+AP: 81 ± 26/mm2; -AP: 103 ± 46/mm2, NBQX (+AP: 213 ± 83/mm2; -AP: 233 ± 124), or MK-801 (+AP: 288 ± 97/mm2; -AP: 279 ± 100/mm2.
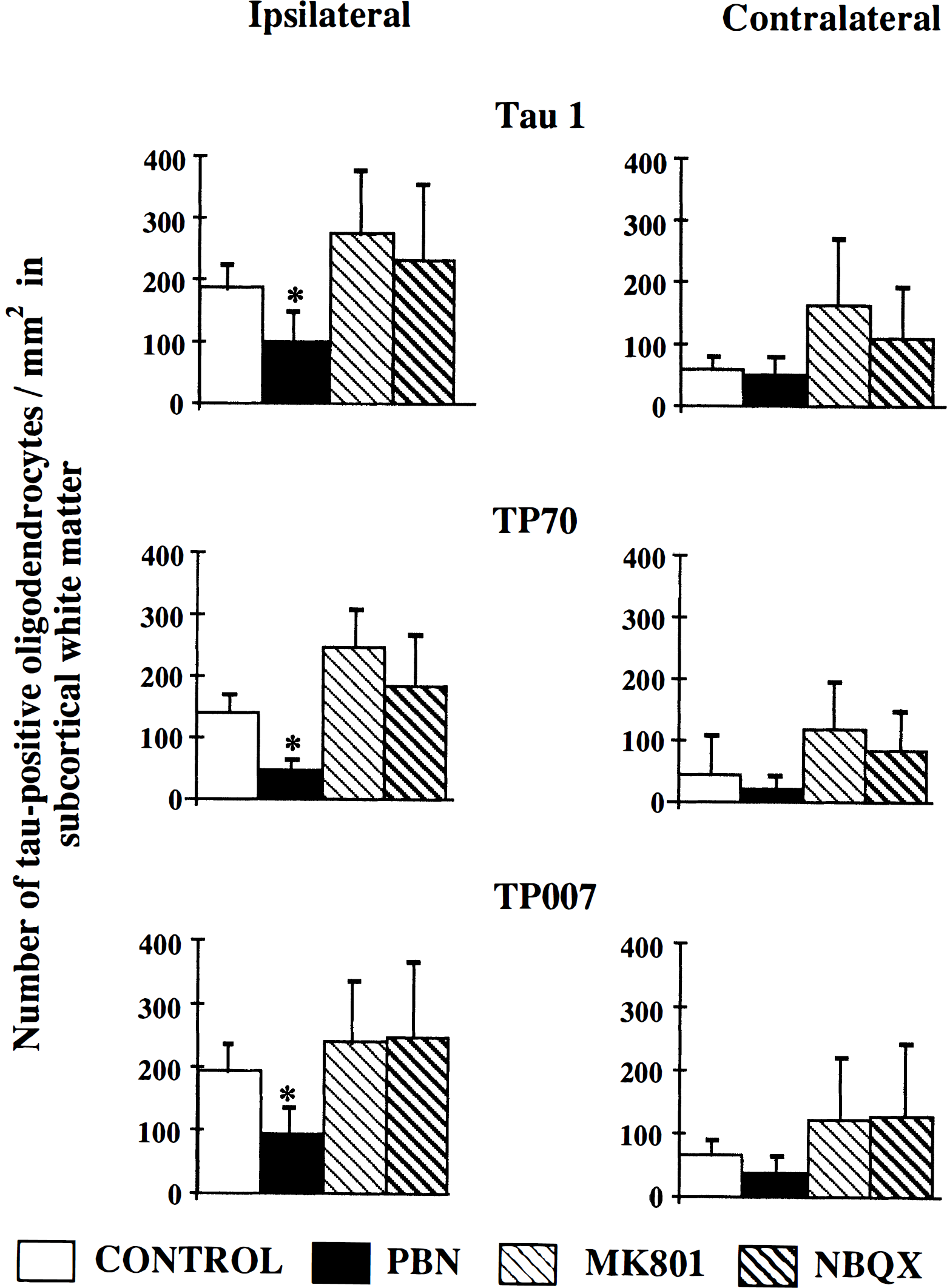
Effect of drug intervention on the number of tau-positive oligodendrocytes 40 minutes after MCAO. Pretreatment with the spin trap agent PBN (100 mg/kg) significantly reduced the number of tau-positive oligodendrocytes/mm2, as detected with all three tau antibodies, Tau 1, TP70, and TP007, in the ipsilateral subcortical white matter as compared to untreated animals (*P < 0.05). In contrast, pretreatment with the glutamate receptor antagonists MK-801 (0.5 mg/kg) or NBQX (30 mg/kg × 2) failed to significantly reduce the number of tau-positive oligodendrocytes/mm2 in the subcortical white matter ipsilateral to the occluded MCA. Data is presented as mean ± SD. Statistical analysis consisted of analysis of variance followed by Student's unpaired t-test with Bonferroni correction.
Although the mean number of tau-positive oligondendrocytes in the contralateral compared with the ipsilateral hemisphere was greater in animals that received MK-801 and NBQX (Fig. 6), the statistical significance of this was not determined because of the a priori hypothesis that was being tested: drug interventions reduced the response in the ischemic hemisphere.
DISCUSSION
The present study shows that the microtubule-associated protein tau undergoes rapid alteration in oligodendrocytes in response to permanent focal cerebral ischemia in the rat. Although tau was originally believed to be neuron specific, Lopresti et al. (1995) recently described the presence of tau within primary cultures of ovine oligodendrocytes in vitro. However, in our study using adult rat brain, tau immunoreactivity was only detected in a few oligodendrocytes within normal tissue using the three antibodies Tau 1, TP70, and TP007 before or after dephosphorylation. The discrepancy between our study and that of Lopresti et al. may reflect low levels of the protein in mature oligodendrocytes in rat brain in vivo.
Twenty minutes after MCAO the number of tau-positive oligodendrocytes was not significantly different in the ipsilateral hemisphere compared with the contralateral hemisphere. However, 40 and 80 minutes after MCAO, the number of tau-positive oligodendrocytes in the ischemic hemisphere, as detected with all three antibodies, was increased compared with the contralateral hemisphere. Tau-positive oligodendrocytes were located predominantly within the subcortical white matter ipsilateral to the occluded MCA, although they were also detected scattered throughout the caudate nucleus and cortex within the area of ischemic tissue defined by decreased MAP2 immunostaining.
Although Tau 1 immunoreactivity was reported to increase in neurons after ischemic brain injury in the rat (Dewar and Dawson, 1995; Geddes et al., 1994), TP70 immunoreactivity was shown to decrease (Dewar and Dawson, 1995). This suggests that tau present in oligodendrocytes and neurons may be affected differently after ischemic brain injury and may reflect different functional roles of tau protein in these two cell types. Tau and MAP2 share homologous C-terminal repeat regions thought to represent the microtubule binding domain. However, MAP2 immunoreactivity was not detected in oligodendrocytes in the absence of or after ischemic brain injury. This suggests that tau is specifically altered in oligodendrocytes after focal cerebral ischemia and that this is not a generalized microtubule-associated protein response.
The source of increased tau immunoreactivity in ischemic oligodendrocytes at present remains unclear. Increased Tau 1 immunoreactivity was detected in oligodendrocytes after focal cerebral ischemia, suggesting that tau may undergo dephosphorylation within these cells in response to ischemic brain injury. However, in the absence of cerebral ischemia, dephosphorylation of tissue before immunostaining failed to induce Tau 1 immunoreactivity in significant numbers of oligodendrocytes, suggesting that mechanisms other than dephosphorylation of existing protein may be involved. In support of this, increased tau immunoreactivity was detected using TP70 and TP007 antibodies that detect tau independent of the phosphorylation state of the protein. Detection of oligodendrocytes with all three tau antibodies suggests the accumulation of full-length protein within these cells. Tau protein has been shown previously to be sensitive to fixation conditions, however tau immunoreactivity has previously been reported in oligodendrocytes in situ using 4% paraformaldehyde fixative as was used in this study (Migheli et al., 1988). The possibility that in non-ischemic tissue, tau present in oligodendrocytes may be masked by fixation, and that the presence of tau immunoreactivity in oligodendrocytes after ischemic brain injury may reflect changes in existing tau, resulting in increased accessibility of the antibodies to the protein can not be ruled out. However, changes in epitope accessibility may reflect conformational changes in the protein and such structural alterations could lead to the disruption of normal tau function within oligodendrocytes. Alternatively, increased tau immunoreactivity may reflect increased synthesis of tau protein, however the increase in tau immunoreactivity occurred rapidly after MCAO and therefore de novo synthesis of tau protein seems unlikely. Ongoing studies in our laboratory are currently investigating changes in tau synthesis in response to a focal ischemic challenge.
In contrast to tau present in GFT (Abe et al., 1992; Iwatsubo et al., 1994; Nishimura et al., 1992; Yamada et al., 1992), tau present in oligodendrocytes after ischemia is dephosphorylated at the Tau 1 epitope. Presently, the consequence of increased tau immunoreactivity in oligodendrocytes is unknown. However, it is possible that this may form part of a protective response within these glial cells because dephosphorylated tau binds to and stabilizes microtubules (Brandt and Lee, 1994; Kosik, 1993; Trinczek et al., 1995). It is interesting to note that Petito (1986) showed the accumulation of microtubules in the cytoplasm of a population of oligodendrocytes 30 minutes to 2 hours after ischemia in the rat. Oligodendrocytes have also been shown to proliferate after trauma in mice (Ludwin et al., 1984) and in acute demyelination in humans (Morris et al., 1994), showing that these glial cells do respond to brain injury. Growing evidence suggests that the oligodendrocyte cytoskeleton may play a role in process extension and myelination within these cells (Wilson and Brophy, 1989); therefore it is possible to speculate that increased tau protein may be required for process extension after injury. Alternatively, accumulation of tau may represent an early stage of GFT formation; tau becoming abnormally phosphorylated over time thus leading to GFT formation.
Whether the presence of tau immunoreactivity in oligodendrocytes is part of a protective response or constitutes an early marker of oligodendrocyte degeneration is not clear presently. However, the results of our study indicate that the initiating stimulus for the response involves events mediated by free radicals. Primary cultures of bovine oligodendrocytes were shown to be susceptible to oxygen radicals derived from glucose/glucose oxidase and hypoxanthine/xanthine oxidase reactions (Kim and Kim, 1991). In that study, cytotoxicity was measured by tryptan blue exclusion and lactate dehydrogenase leakage, and the data showed that oxygen radicals induced significant membrane damage to oligodendrocytes within 2 hours of exposure.
In addition, Oka et al. (1993) showed in rat primary oligodendrocyte cultures that glutamate-induced cell death was mediated, not through glutamate receptor activation, but through free radical-mediated mechanisms. Excessive extracellular glutamate concentrations led to the efflux of cystine from these cells through the actions of the glutamate/cystine transporter which in turn resulted in glutathione depletion. Glutathione is an important protective agent against oxidative stress (Bast et al., 1991) and its depletion renders the cell vulnerable to oxidative stress. PBN is a nitrone compound that reacts with many types of free radicals resulting in the formation of the stable nitroxide radical adducts (Oliver et al., 1990). Extracellular glutamate concentrations peak 40 minutes after MCAO in the rat (Butcher et al., 1990), and it is possible to speculate that treatment with PBN protects oligodendrocytes indirectly from glutamate toxicity and directly from free radical-mediated damage resulting from ischemic injury thus preventing accumulation of tau within these cells.
In contrast to PBN, neither the NMDA antagonist MK-801 nor the AMPA antagonist NBQX prevented increased tau immunoreactivity in oligodendrocytes after 40 minutes of cerebral ischemia indicating that glutamate receptor activation is not involved in the response. However, both primary oligodendrocyte cultures and immortalized cultures derived from rat brain express non-NMDA receptor subtypes (Patneau et al., 1994; Puchalski et al., 1994; Yoshioka et al., 1995). Moreover, damage to these cells was mediated by non-NMDA receptor activation. Thus in our in vivo experimental paradigm, while it is possible that activation of AMPA receptors by excessive extracellular glutamate concentrations may damage oligodendrocytes, this signalling pathway does not seem to be involved in the tau protein response. Alternatively, the rapid desensitization of AMPA receptors in oligodendrocytes in response to glutamate (Yoshioka et al., 1995) may have masked any effect of receptor blockade by NBQX.
Finally, although the selection of the drug doses were based on those previously shown to reduce infarct volume after permanent focal cerebral ischemia in the rat (Gill et al., 1992), the possibility that the concentration of NBQX used in this study may be too low to prevent ischemic-induced accumulation of tau in oligodendrocytes cannot be ruled out. Mature oligodendrocytes have been reported to contain non-NMDA receptors; however mRNA for NMDA receptors was not detected (Patneau et al., 1994; Puchalski et al., 1994; Yoshioka et al., 1995) and therefore it is not surprising that MK-801 failed to prevent increased tau immunoreactivity in these cells after ischemic injury. However, the mean value of tau-positive oligodendrocytes present in both ipsilateral and contralateral white matter was greater after MK-801 treatment compared with controls. This suggests that MK-801 alone may be able to induce increased tau in oligodendrocytes. In the absence of a direct effect on oligodendrocytes themselves, increased tau immunoreactivity within these cells may arise as an indirect consequence of neuronal changes both in proximity and distant to ischemia. MK-801 and other NMDA ion channel blockers induce a range of neurochemical alterations within neurons that include glucose use and heat shock protein gene expression (Kurumaji et al., 1988; Sharp et al., 1992). In the light of the growing literature on neuronal-glia communication it may be envisaged that MK-801-induced changes in neuronal activity in turn induce responses in associated oligodendrocytes. Alterations in the cytoskeletal protein tau may represent such a response.
In conclusion, the microtubule-associated protein tau undergoes rapid alteration in oligodendrocytes after ischemic injury that is initiated through free radical-mediated mechanisms. This shows that oligodendrocytes respond rapidly to cerebral ischemia. Presently, it is not clear if increased tau immunoreactivity in oligodendrocytes represents a protective response or is a marker for a degenerative process within these cells. However, the observations in this study may have important implications for the development of neuroprotective strategies aimed at ameliorating acute brain damage.
Footnotes
Abbreviations used
Acknowledgements
We thank Professor B.H. Anderton and Mr. D. Davies (Institute of Psychiatry, London) for their kind gift of Tau 1, TP007, and TP70 antibodies. We thank the staff at the Wellcome Surgical Institute for their technical assistance. E.A. Irving was a Wellcome Trust Prize Scholar.