
Abstract
Development of neuronal and glial cells and their maintenance are under control of neurotrophic factors (NTFs). An exogenous administration of NTFs protects extremely sensitive brain tissue from ischemic damage. On the other hand, it is now known that neural stem cells are present in normal adult brain, and have a potential to compensate and recover neural functions that were lost due to ischemic stroke. These stem cells are also under control of NTFs to differentiate into a certain species of neural cells. Thus, the purpose of this review is to summarize the present understanding of the role of NTFs in normal and ischemic brain and the therapeutic potential of NTF protein itself or gene therapy, and then to summarize the role of NTFs in stem cell differentiation and a possible therapeutic potential with the neural stem cells against ischemic brain injury.
Among neural cells, neurons are particularly sensitive to various injuries such as ischemia, hypoxia, hypoglycemia, infection, and trauma. These vulnerabilities of neurons make it difficult to treat patients suffering from the above injuries in clinical situations. The vulnerability is different even within the neuronal populations (Abe et al., 1991, 1995; Abe and Kogure, 1993). Normal differentiation of neuronal and glial cells and their maintenance are under the control of neurotrophic factors (NTFs). In addition, an excessive administration of NTFs greatly protects sensitive brain tissue from injury. However, brain cells have long been believed not to differentiate again once they have matured with normal development. Recent advancements in molecular cell biology have discovered the presence of neural stem cells even in mature adult mammalian brain. It is now expected that neural stem cells have the potential to compensate and recover neural functions that were lost because of ischemic stroke. These stem cells are also under the control of NTFs to differentiate into a certain species of neural cells. Therefore, understanding the role of NTFs as a possible therapeutic potential and in a differentiation of neural stem cell may be essential to propel future development of the treatment for human stroke patients.
The purpose of this review is to summarize the present understanding of the role of NTFs in normal development and ischemic brain, and the therapeutic potential of NTF protein itself or of gene therapy, and then to summarize the role of NTFs in stem cell differentiation and as a possible therapeutic potential with neural stem cells.
DEVELOPMENTAL AND PHYSIOLOGIC ROLE OF NEUROTROPHIC FACTORS
A number of protein peptides affect target cells in different ways (Fig. 1). Endocrine hormones have a remote effect on their target cells through systemic circulation. In contrast, NTFs affect target cells by paracrine, autocrine, or juxtacrine (Fig. 1). This suggests a local role for NTFs in cell proliferation, migration, differentiation, and production of extracellular matrix in the nervous system. A study on epidermal growth factor (EGF) suggests that paracrine stimulation transiently activates intracellular signals and has a direct effect on proliferation, whereas juxtacrine stimulation continuously activates intracellular signals and has a greater effect on differentiation and survival (Higashiyama et al., 1995). Neurotrophic factors usually promote cell proliferation, but sometimes inhibit such a proliferation under certain circumstances or for a certain cell species. For example, fibroblast growth factor (FGF) inhibits proliferation of chondroblast, and a constant activation of FGF effect with FGF receptor mutation causes achondroplasia (Deng et al., 1996; Naski et al., 1996). Transforming growth factor (TGF)-β was originally found to promote transformation and growth of fibroblast. However, arecent study suggests that TGF-β usually inhibits cell growth for most epidermal cells (Royuela et al., 1998).
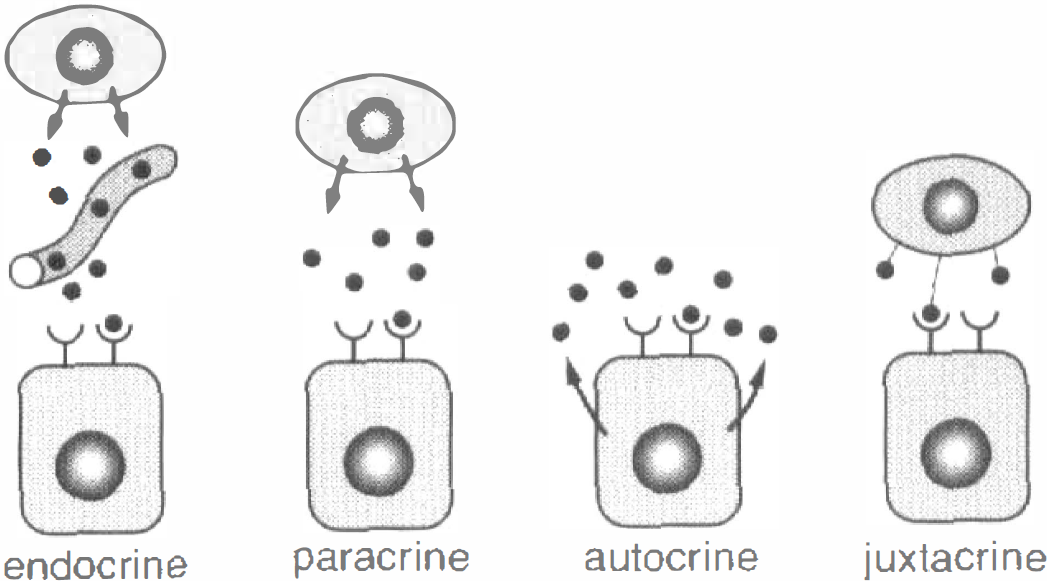
Ways of influencing target cells. Endocrine hormones produce a remote effect through the systemic circulation. However, neurotrophic factors (NTFs) affect target cells by paracrine, autocrine, or juxtacrine, suggesting a local role of NTFs in cell proliferation, migration, and differentiation.
Neurotrophic factors are usually classified into 3 or 4 subclasses based on their receptors (Table 1;Fig. 2). Neurotrophic factors that work with tyrosine kinase receptors are a major group consisting of EGF, FGF, hepatocyte growth factor (HGF), insulin-like growth factor (IGF-1), neurotrophins (nerve growth factor [NGF], brain-derived neurotrophic factor [BDNF], neurotrophin 3 [NT3], neurotrophin 4/5 [NT4/5]), platelet-derived growth factor (PDGF), vascular endothelial growth factor (VEGF), and so on. Once these proteins bind the receptors as monomer (FGF) or dimer (neurotrophins), the receptors form dimer and the intracellular tyrosine kinase domains are activated, those then transphosphorylate intracellular signal proteins that possess SH2 (Src homology 2), SH3, or PH (pleckstrin homology) domain (Table 1;Fig. 2A). Glial cell line-derived neurotrophic factor (GDNF) and colleague proteins—such as neurturin, artemin, and persephin—bind receptor complex of GFRα and c-ret as a monomer, then the intracellular tyrosine kinase domain is activated (Fig. 2B). Cytokines—such as ciliary neurotrophic factor (CNTF), leukemia inhibitory factor (LIF), and interleukin-2 (IL-2)—bind dimeric or trimeric receptors and activate intracellular tyrosine kinase proteins such as JAK (Table 1;Fig. 2C). The TGF-β super family consists of the activin family (activin-βA and -βB), bone morphogenetic protein (BMP) family (BMP2∼8, OP1∼2, GDF5∼7, and so on), and TGF-β family (TGF-β1∼3). Transforming growth factor-β ligands bind to types 1 and 2 receptors, and the receptors form tetrameric functional complex. Then type 2 receptors phosphorylate serine-threonine kinase domain of type 1 receptors so that the type 1 receptorsactivate subsequent signal systems such as Smad (Table 1;Fig. 2D).
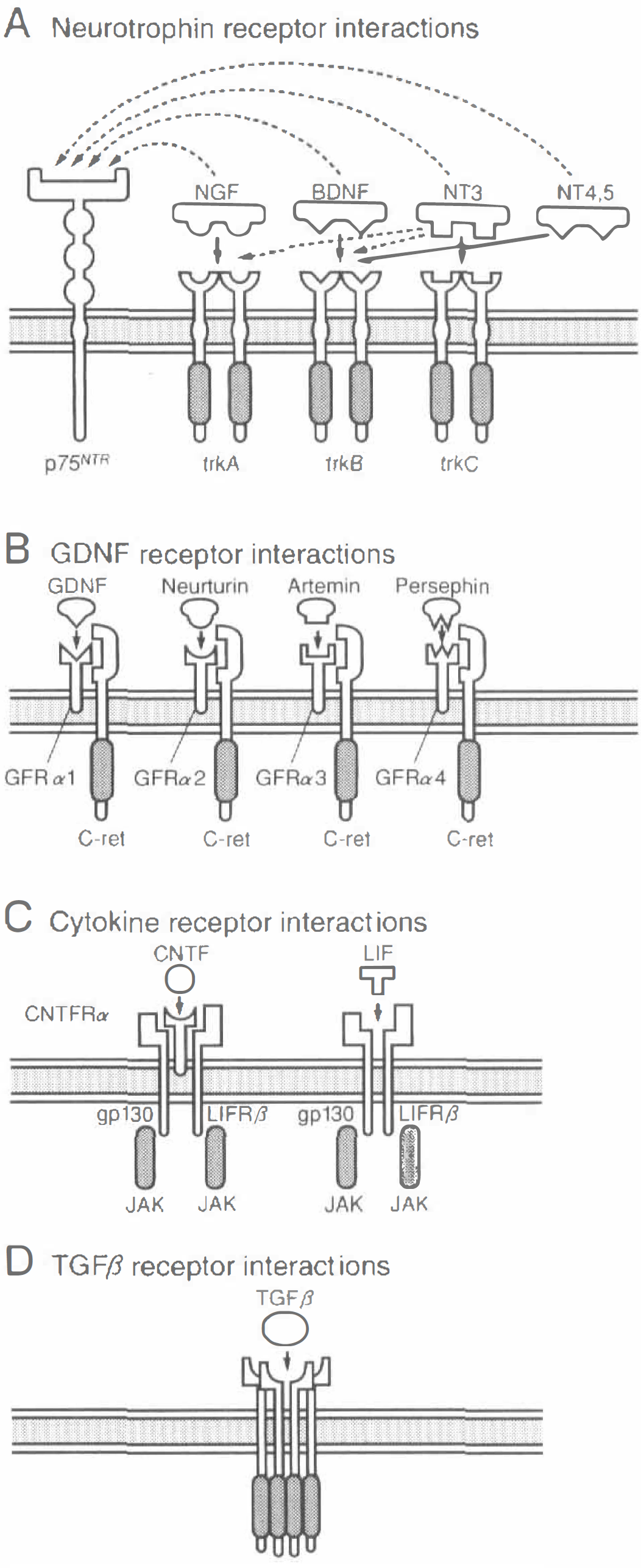
Different receptor types for neurotrophic factors (NTFs). Neurotrophins (NGF, BDNF, NT3, NT4/5) interact with tyrosine kinase receptors
Classification of neurotrophic factors based on their receptors

EGF, epidermal growth factor; TGF, transforming growth factor; FGF, fibroblast growth factor; HGF, hepatocyte growth factor; NGF, nerve growth factor; PDGF, platelet-derived growth factor; VEGF, vascular endothelial growth factor; CNTF, ciliary neurotrophic factor; LIF, leukemia inhibitory factor; BMP, bone morphogenetic protein.
Examples of the roles of NTFs in normal cellular development and differentiation are shown in Fig. 3. Many different classes of neurons, glial cells, and other cell types develop from the multipotent neural crest cells (Fig. 3A). Specific growth factors control the fate of certain neural crest cells. Bone morphogenetic proteins (BMPs) 2 and 4 promote the differentiation of sympathetic neurons, glial growth factor (GGF) represses neurogenesis and promotes Schwann cell differentiation, and TGF-β promotes smooth muscle cell differentiation (Fig. 3A). In optic nerve (Fig. 3B), PDGF secreted by astrocytes maintains the proliferation of oligodendrocyte progenitor cells. Postnatally, these progenitor cells begin to differentiate into oligodendrocytes or astrocytes with or without CNTF secreted by astrocytes (Fig. 3B;Lillien and Raff, 1990). In sympathoadrenal lineage of the neural crest (Fig. 3C), sympathoadrenal precursor cells differentiate into adrenal chromaffin cells, noradrenergic and cholinergic sympathetic neurons with glucocorticoids, FGF/NGF, or LIF/CNTF, respectively. Thus, NTFs are actively involved in many steps of normal cell development and differentiation (Fig. 3;Jessell and Sanes, 2000).
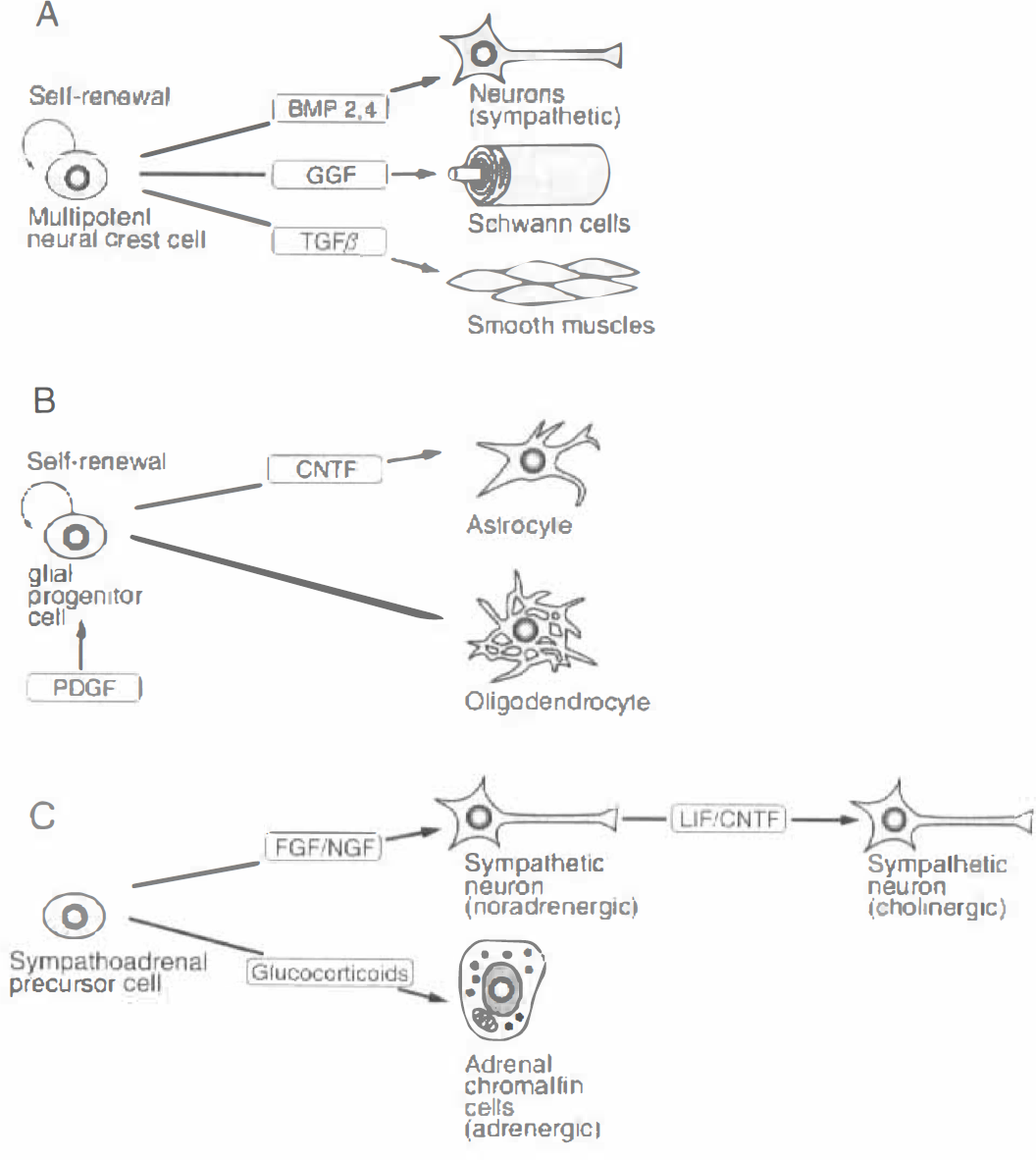
Role of neurotrophic factors (NTFs) in the differentiation of neural crest cell
Neurotrophic factors are also involved in normal maintenance and survival of neuronal cells after differentiation. Constant secretion of NTFs keep cell survival signals activated and death signals inactive (Fig. 4). For example, the presence of nerve growth factor maintains cell survival by activating cell survival Bcl-2 and inhibiting death signals such as Apaf-1 and caspases (Jessell and Sanes, 2000). However, once NTFs are deprived for some reason, such as ischemia, the balance between survival and death signals is tapped in favor of death signals that finally result in neuronal cell death. Many other stimuli—such as reactive oxygen species (ROS), FAS ligand, and tumor necrosis factor (TNF)—activate a distinct biochemical pathway that triggers a family of caspases and leads to cell death.
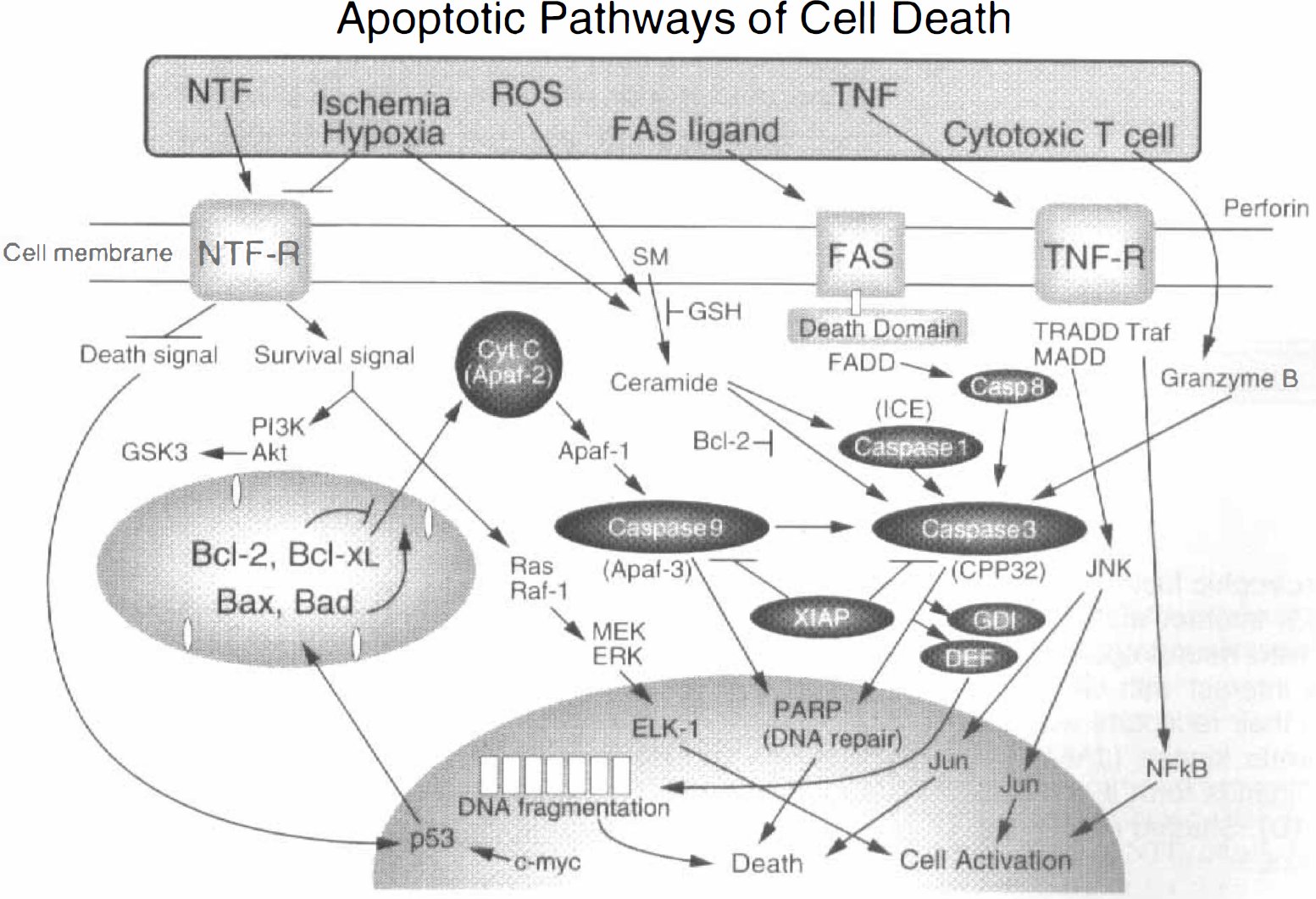
Schematic illustration of the cascade involving cell survival and death signals. Constant secretion of neurotrophic factors (NTFs) keep cell survival signals activated and death signals inactive. However, with a pathologic condition such as ischemia or reactive oxygen species, the balance of survival and death signals could be inverted. Large amounts of exogenously added NTFs may keep survival signals activated and death signals inactive, resulting in cell protection.
INDUCTION OF NEUROTROPHIC FACTORS AFTER ISCHEMIA
Under pathologic conditions such as ischemia and kinate-induced seizures, NTFs—such as BDNF, bFGF, GDNF, NGF, and TGF-β1—are induced in brain cells (Kato et al., 1992; Logan et al., 1992; Comelli et al., 1993; Humpel et al., 1994; Lee et al., 1994). However, each NTF species shows a different temporal and cellular distribution. Glial cell line-derived neurotrophic factor was originally associated with survival and neurite outgrowth of dopaminergic and motor neurons as well as peripheral sensory and sympathetic neurons (Henderson et al., 1994; Li et al., 1995; Tomac et al., 1995). Although immunoreactive GDNF was not detected in sham-control brain (Fig. 5a), it was expressed in neuronal cytoplasm of the cerebral cortex (Fig. 5b, arrowheads) at 1 hour of reperfusion after 90 minutes of transient middle cerebral artery occlusion (MCAO). The induction reached a maximum at 3 hours (Figs. 5c and 5d, arrowheads) with a gradual decrease by day 1 (Fig. 5e, arrowheads). Brain cells positive for GDNF-like immunoreactivity were primarily large pyramidal neurons in layers 4 to 6 of cerebral cortex (Fig. 5f), and no glial cell was stained in the brain sections (Abe and Hayashi, 1997).
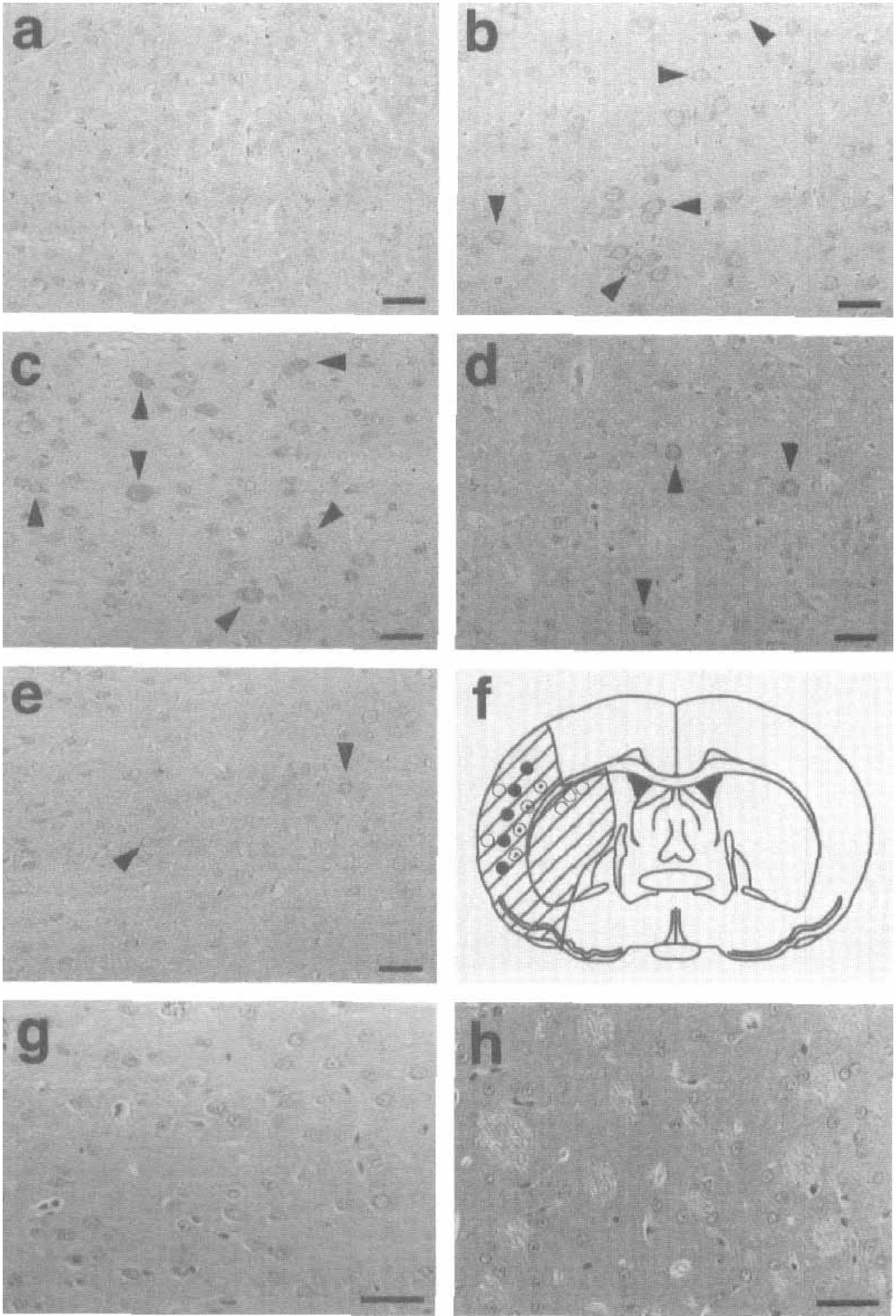
Changes of glial cell line-derived neurotrophic factor (GDNF) immunohistochemistry after 90 minutes of transient middle cerebral artery occlusion at 1 hour
Ciliary neurotrophic factor abundantly expressed in astroglial and Schwann cells was identified as a neurotrophic molecule for parasympathetic ciliary ganglion neurons, but also affected the survival of other neuronal cells (Barbin et al., 1984; Arakawa et al., 1990, Ip et al., 1991). Ciliary neurotrophic factor levels were significantly higher in the frontal, parieto-temporal, and occipital lobes at 2 days after 120 minutes of transient MCAO (Hu et al., 1997). With subsequent immunohistochemical analysis, astrocytes were predominantly stained in the marginal region of the infarct from 1 day to 7 days of reperfusion after 90 minutes of transient MCAO (Fig. 6).
Ciliary neurotrophic factor (CNTF) immunohistochemistry after 90 minutes of transient middle cerebral artery occlusion at 3 days of reperfusion. Infarction is obvious in the right caudate with CNTF staining in the marginal region
Vascular endothelial growth factor, also known as vascular permeability factor, is mitogenic for endothelial cells. By alternative splicing four different isoforms exist in vivo: VEGF206, VEGF189, VEGF165, and VEGF121; VEGF165 and VEGF121 act as diffusible factors. A recent report (Hayashi et al., 1997) showed that immunoreactive VEGF was normally present in the ependymal cells, but was not in neurons (Fig. 7a), glial cells, vascular endothelial cells, or pial cells (Fig. 7e) in control brains. However, neurons of the ischemic cerebral cortex stained positively for VEGF from 1 hour (Fig. 7b) to 1 day (Fig. 7d) of reperfusion after 90 minutes of transient MCAO with a peak at 3 hours (Fig 7c). Pial cells of the MCA area became positive for VEGF from 1 hour with a peak at 3 hours (Fig. 7f), which was sustained until 3 to 7 days of reperfusion (Fig. 7g, arrowhead).
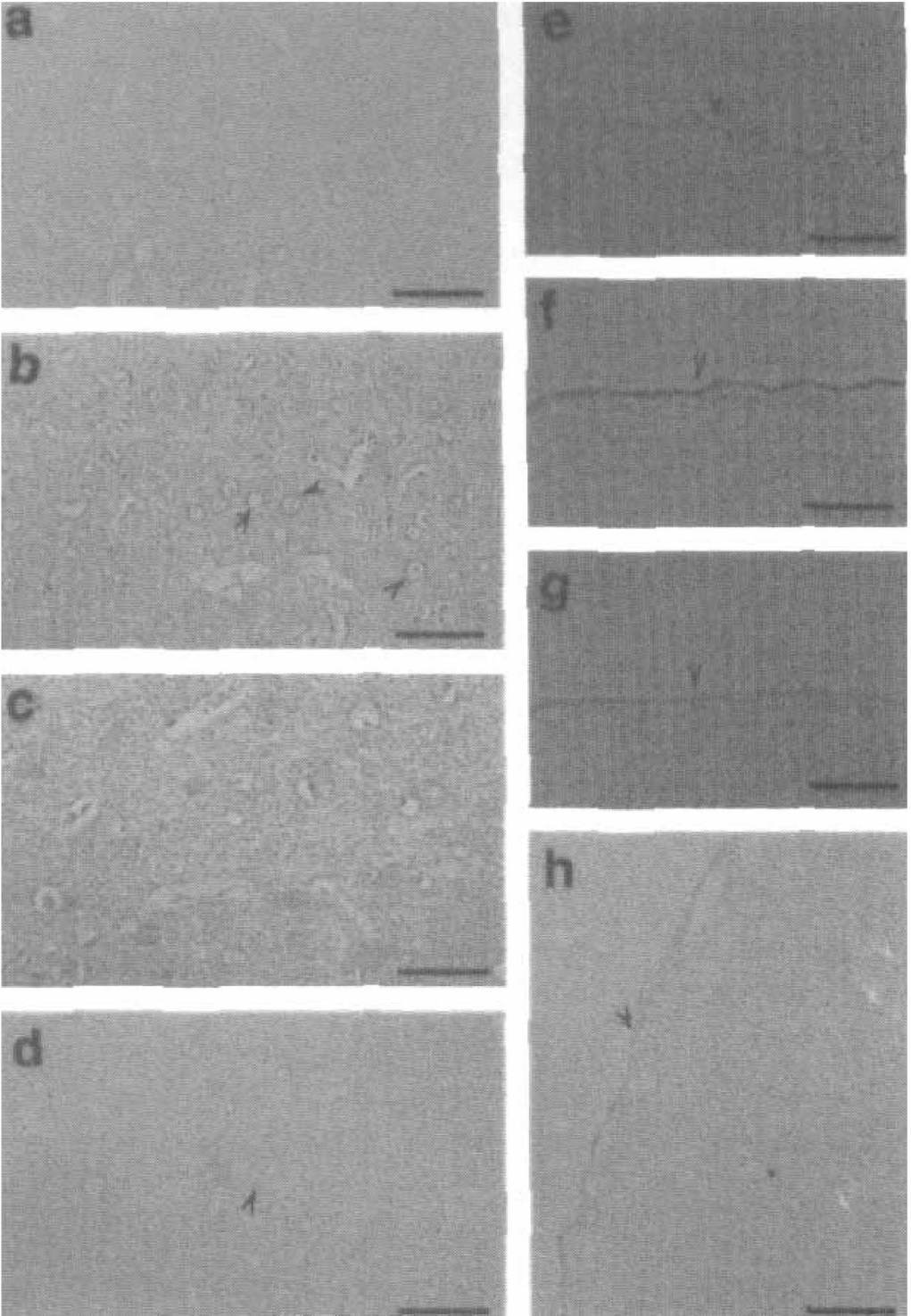
Vascular endothelial growth factor (VEGF) immunohistochemistry in cerebral cortex
Although GDNF was originally isolated from cultured glial cells, the gene was expressed only in neurons in the current study (Fig. 5). Because the induction of CNTF was predominantly observed in the astroglia of the marginal region (Fig. 6), injured neurons may stimulate the synthesis of CNTF in astroglia, which may in turn support survival and regeneration of neurons in the injured regions. Because the VEGF gene was also induced in other brain cells under hypoxic conditions or during continuous MCAO (Fischer et al., 1995; Ijichi et al., 1995; Nomura et al., 1995; Kovacs et al., 1996), sensitivity for inducing the VEGF gene may be different in each neural cell type under conditions of hypoxia–ischemia or continuous ischemia–reperfusion. Neovascularization usually begins at 2 to 5 days after the ischemic insult and continues for months (Krupinski et al., 1994). Therefore, the sustained expression of the VEGF gene in the pia mater after ischemia (Fig. 7e to 7h) suggests its role in new vessel formation in the affected area. However, the rapid and transient induction of VEGF in neurons (Fig. 7a to 7d) may be related to the protection of the vascular system against ischemic damage.
Thus, NTFs are induced in different neural cell types (neuronal, astroglial, and pial) with different temporal profiles, suggesting a different role of each NTF for each neural cell species at a particular stage after ischemia (Table 2).
Temporal and cellular profiles of neurotrophic factor expression after 90 minutes of transient MCAO, in rat brain

MCAO, middle cerebral artery occlusion; GDNF, glial cell line-derived neurotrophic factor; CNTF, ciliary neurotrophic factor; VEGF, vascular endothelial growth factor.
PROTECTIVE EFFECTS AND THE MECHANISM OF ACTION OF NEUROTROPHIC FACTORS IN ISCHEMIC BRAIN INJURY
Neurotrophic factors are essential for the survival and differentiation of normally developing neurons, but they also play important roles in the protection and recovery of mature neurons under pathologic conditions (Lin et al., 1993). Galectin-1 regulates initial axonal growth in peripheral nerves after axotomy (Horie et al., 1999). Protective effects of GDNF in various injuries for central and peripheral nervous tissue have been reported (Schaar et al., 1993; Krieglstein et al., 1995). Ciliary neurotrophic factor infusion prevented the learning disability and neuronal loss induced by ischemia in gerbils (Wen et al., 1995).
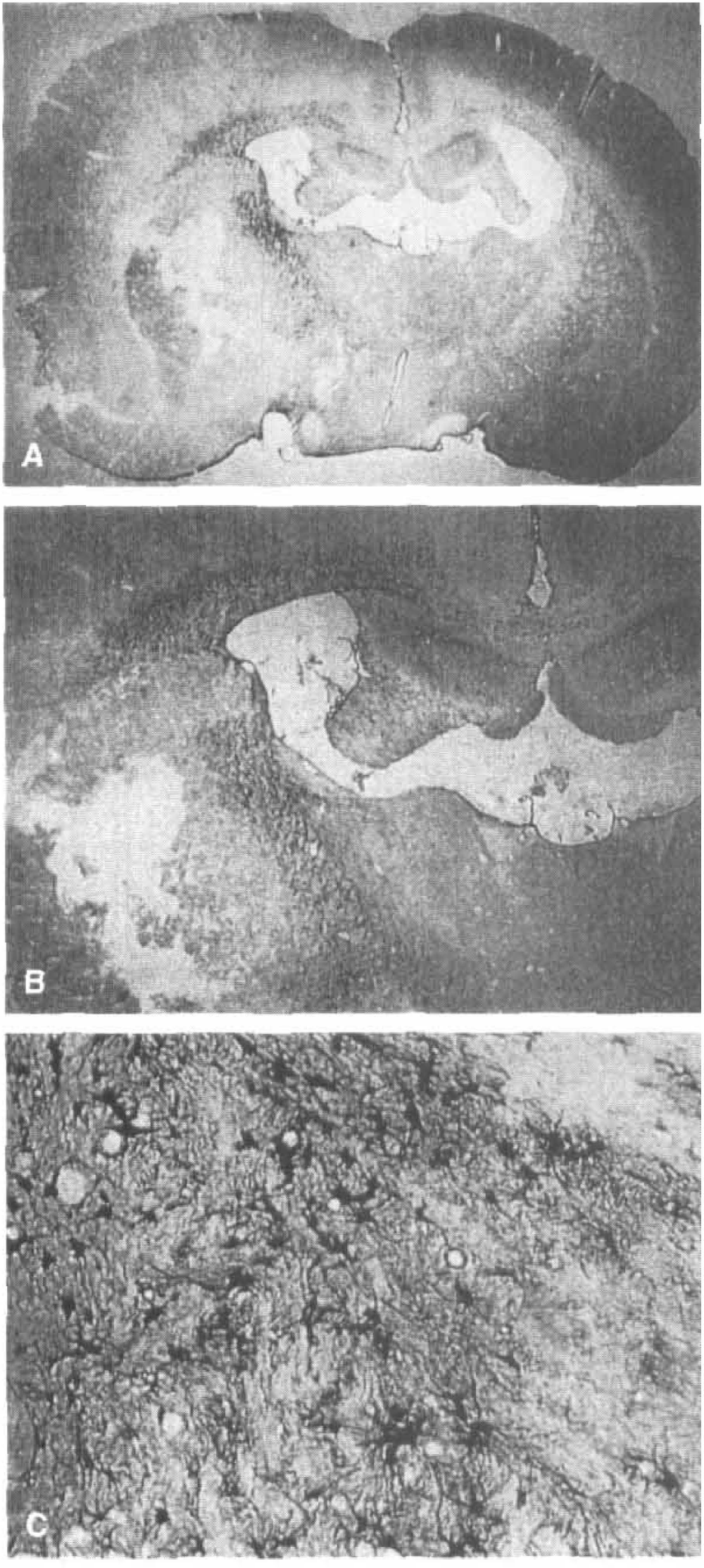
To examine possible direct protective effect of NTF proteins against ischemic brain damage, topical administration of NTFs such as GDNF, NT-3, VEGF, IGF-1, and BDNF onto the ischemic brain surface have recently been tried in continuous and transient focal cerebral ischemia models. Kitagawa et al. (1998b) reported that topical application of GDNF greatly reduced the infarct size (48%) and brain edema (30%) at 24 hours of continuous MCAO in rats. The reduction of the infarct size was not related to a change of cerebral blood flow (CBF), but was accompanied by marked reduction in TUNEL and caspase positive cells in the affected area (Fig. 8). Thus, GDNF showed a direct protective effect against ischemic brain damage, but not by improving CBF (Fig. 8D). Induction of TUNEL was located primarily in the ischemic core, and induction of caspases 1 to 3 was mainly in the penumbra, suggesting that the main mechanism of ischemic neuronal cell death is necrosis in the core and apoptosis in the penumbra. Although the infarct size became smaller with GDNF treatment, the dissociative spatial distribution among TUNEL and caspases 1 to 3 positive cells was similar to vehicle treatment (Fig. 8E). These data suggest that treatment with GDNF was protective not only by reducing the apoptotic process, but also by reducing the necrotic process.
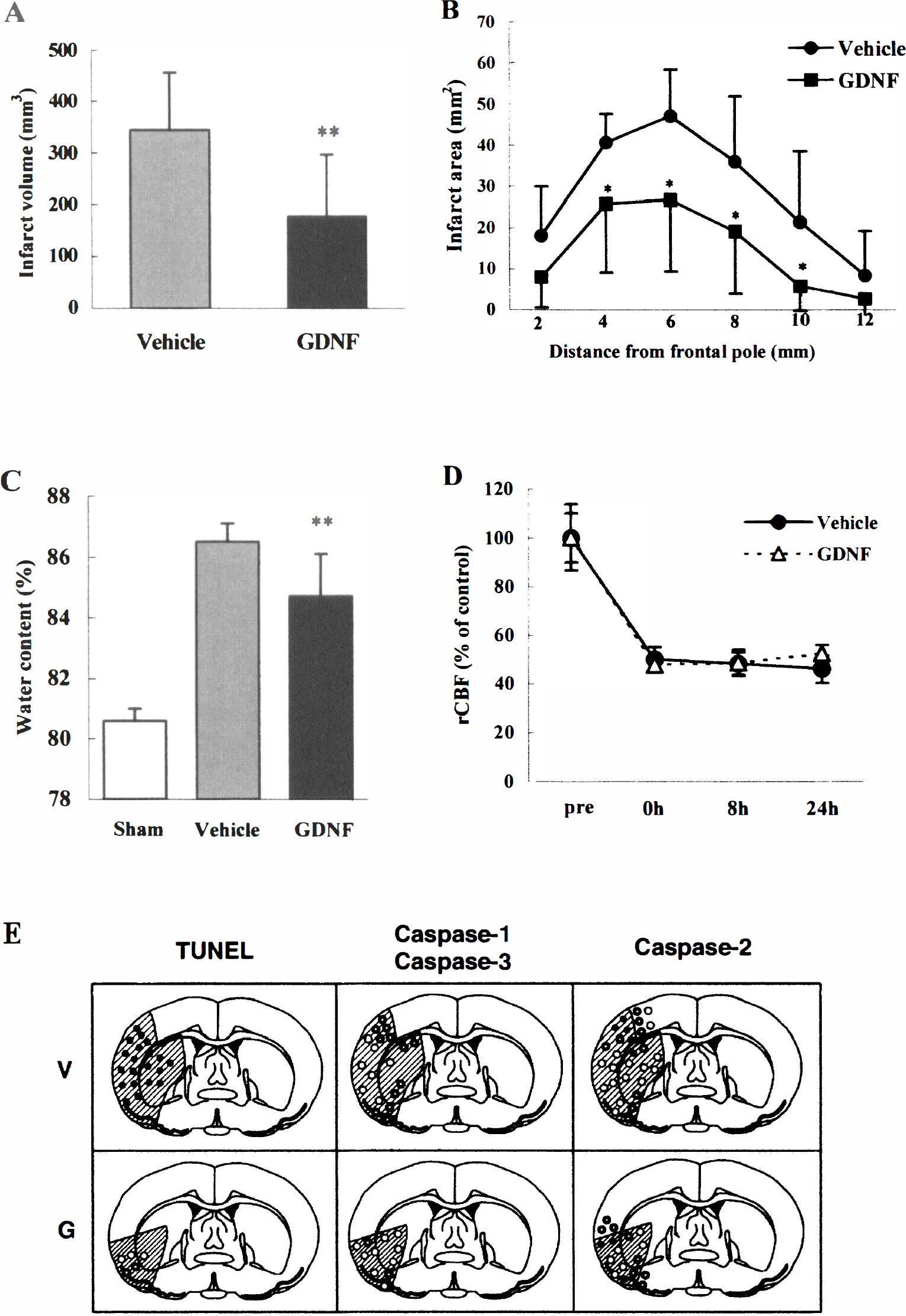
Topical application of glial cell line-derived neurotrophic factor (GDNF) reduces infarct volume
Similar results were obtained with different NTFs and animal models, but ischemic injury was smaller with shorter periods of transient focal ischemia than with continuous ischemia. In fact, reduction of brain edema by GDNF was greater with transient MCAO (Abe et al., 1997a) than with continuous MCAO (Kitagawa et al., 1998a) in rats. In transient MCAO models, treatment of ischemic rats with topical application of NT-3, VEGF, and IGF-1 ameliorated infarct sizes and was associated with a reduction in the number of TUNEL and caspase positive cells (Hayashi et al., 1998; Zhang et al., 1999, Wang et al., 2000). However, the pharmacologic potency was strongest for GDNF (that is, GDNF > NT-3 > VEGF > IGF-1 in this order). Yamashita et al. (1997) delivered BDNF into the territory of MCA through an osmotic mini-pump (1 μ/hr) and found a 33% reduction in total infarct volume.
In terms of the protective mechanism of NTFs on ischemic brain injury, exogenously administered excessive amounts of NTFs could partially inhibit activation of the apoptotic signal cascade, including caspases, and could in turn potentiate survival signal pathways (Fig. 4). However, GDNF was also protective in the cases of continuous ischemia in which the necrotic mechanism was more important than the apoptotic mechanism (Kitagawa et al., 1998b). Thus, treatment of NTFs may have the potential to be protective against apoptotic and necrotic mechanisms of cell death. These potent effects of NTFs against ischemic brain injury could have applications for improvement in clinical outcome for human stroke, or even to prolong the therapeutic time window for an alternative effective therapy.
GENE THERAPY FOR THE ISCHEMIC BRAIN USING NEUROTROPHIC FACTOR GENES
Gene delivery systems using virus vectors have been reported in many fields including not only genetic diseases but also in some acquired diseases such as cerebrovascular diseases and cancer (Heistad and Faraci, 1996; Karpati et al., 1996; Abe et al., 1997b). Initial experimental gene transfer into the brain was performed with replication-defective herpes simplex virus (HSV) type 1 (Pallela et al., 1989; Fink et al., 1992; Wolfe et al., 1992). Although HSV vectors can carry a relatively large gene of up to 36 kb, cytopathogenicity such as neuronal death may be a major disadvantage (Breakfield, 1993; Culotta, 1993; Mulligan, 1993). The advantages and disadvantages of each vector is shown in Table 3. Retroviral vectors also have the potential to deliver genes into the brain (Price et al., 1987; Walsh and Cepko, 1988). However, the retroviral vectors do not infect nondividing cells such as neurons, and in vivo risks of mutagenesis and tumorinogenesis are also indicated (Mulligan, 1993; Karpati et al., 1996). Lentivirus can transduce a gene to nondividing cells and has long-term expression, but has the potential to cause insertional mutagenesis (Kafri et al., 1997). Direct injection of plasmid vector with or without a conjugation of liposome also carries genes into the brain and muscle (Ono et al., 1990; Danko et al., 1993). However, the efficacy of gene expression is low (Ono et al., 1990; Culotta, 1993).
Characteristics of vectors of gene therapy for ischemic stroke

Usually considered as a disadvantage in gene therapy, but is an advantage for acute stroke. HSV, herpes simplex virus; AAV, adeno-associated virus.
In an experiment with replication-defective adenoviral vector, the E. coli lacZ gene was already expressed at 8 hours after the inoculation in control brain. The cell number of expression and staining gradually increased with a peak at 7 days (Abe et al., 1997b). In brains exposed to continuous MCAO, the lacZ gene was expressed at 8 hours of ischemia in the cerebral cortex around the injection needle (Fig. 9a, arrows indicate needle route). With reperfusion after transient 90 minutes of MCAO, no or only a few cells expressed the lacZ gene at 8 hours (Fig. 9e) and 2 days of reperfusion. However, many brain cells expressed the gene at 7 days (Figs. 9f and 9g). Cells expressing the lacZ gene were also found in the contralateral cerebral cortex (Fig. 9h). At 21 day of reperfusion, the majority of brain cells were no longer positively stained for the β-gal blue color. Double staining showed only a small percentage (5% to 10%) of cells that were double positive for glial fibrillary acidic protein and β-gal (Fig. 9d, filled arrowheads). There was only minimal traumatic injury and leukocyte infiltration (Fig. 9b, arrow). Chronological and spatial changes of the staining are illustrated in Fig. 10. Transient expression of an adenoviral vector-mediated gene may be advantageous in situations in which it is appropriate for the foreign gene to disappear from the tissue after it has fulfilled its role.
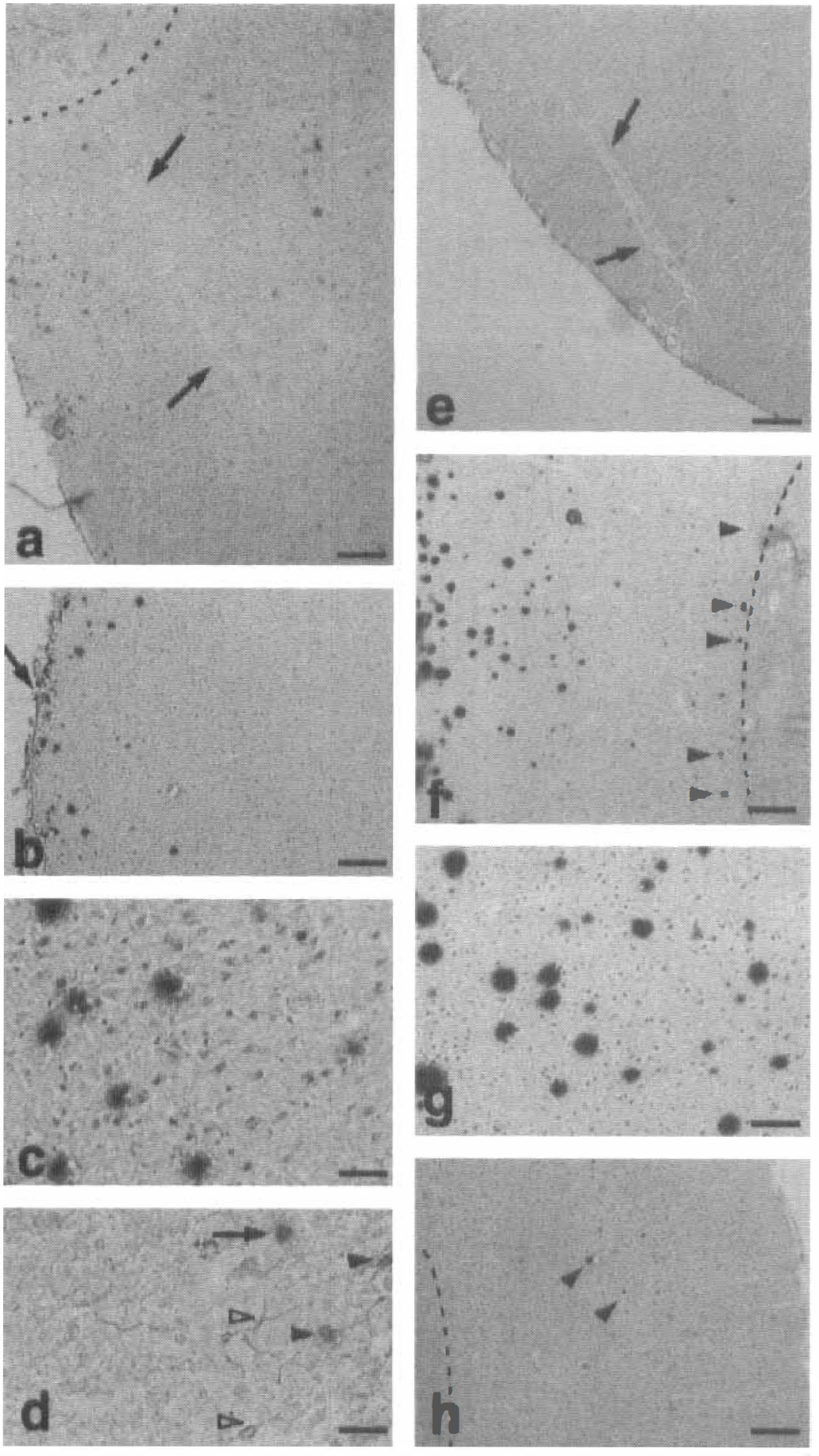
Representative microphotographs of brain sections from continuous middle cerebral artery occlusion (MCAO;
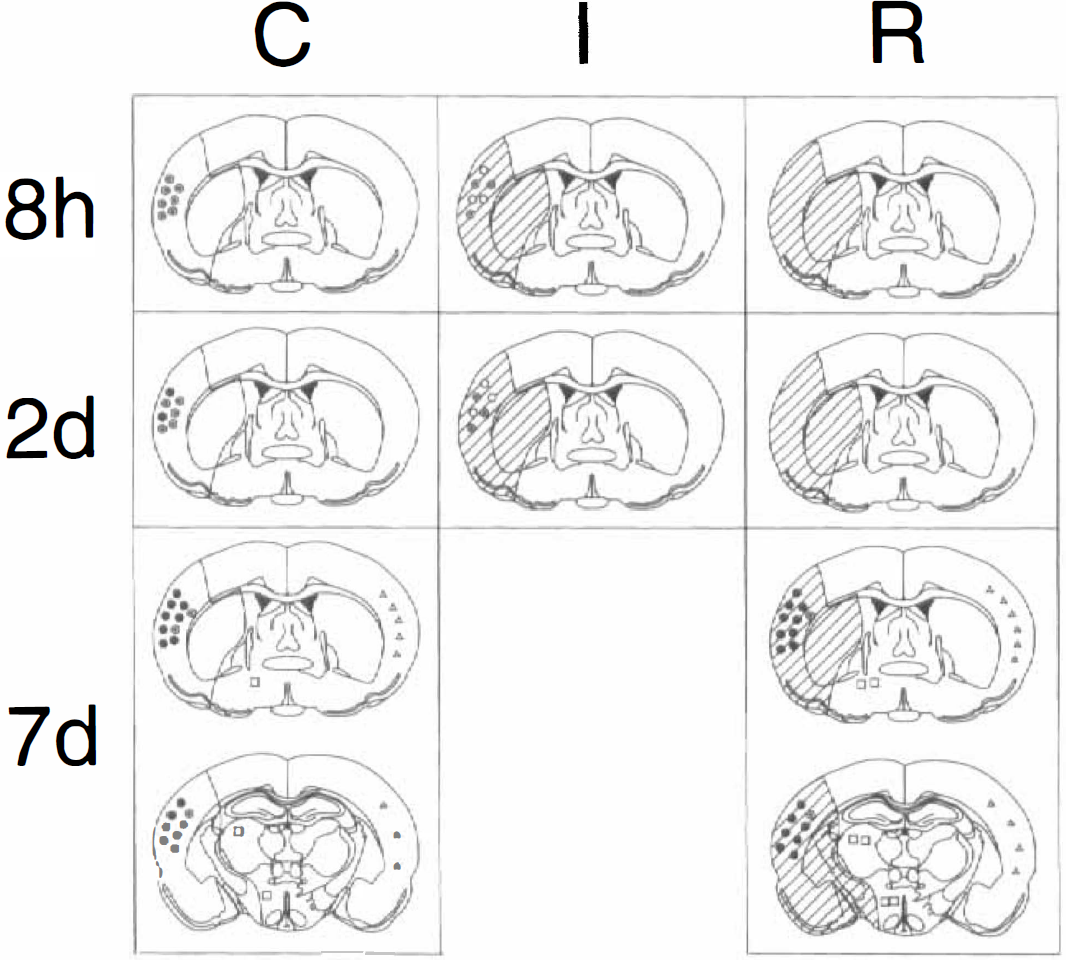
Schematic illustrations showing representative chronological and spatial distributions of X-Gal positive cells in the control (C), continuous ischemic (I), and ischemic–reperfused (R) brains at 8 hours, 2 days, or 7 days after the injection of adenoviral vector into the cerebral cortex. Shaded areas show the territory of right middle cerebral arteries. Open, dotted, and filled circles represent staining grades of (±), (+) to (2+), or (3+), respectively (as in Fig. 5), in the ipsilateral cerebral cortex. Open squares and open triangles represent the staining grade of (±) in the ipsilateral and contralateral hemisphere, respectively. Referred from Abe et al. (1997b).
Based on the potent protective effect of NTF protein in ischemic brain damage (Fig. 8) and the considerable transfer of a foreign gene into ischemic brain (Fig. 9), an adenovirus vector containing the GDNF gene (Ad-GDNF) was prepared, and a possible protective effect of the Ad-GDNF transfer was examined after transient MCAO in the rat. Pretreatment of animals with Ad-GDNF 24 hours before a subsequent 90 minutes of transient MCAO effectively reduced infarct volume and area (Figs. 11A and 11B) without affecting regional CBF as compared with the vehicle or Ad-LacZ animal groups (Kitagawa et al., 1999). With immunohistological analyses, treatment with Ad-GDNF greatly reduced the number of TUNEL, caspase-3, and cytochrome c positive cells (Fig. 11C). No leukocyte infiltration existed in Ad-GDNF treated rats. Inhibition of the cytosolic release of cytochrome c by Ad-GDNF (Fig. 11Ci) suggests a target of protection in an apoptotic pathway through cytochrome c and caspase-3 in the penumbra (Fujimura et al., 1998).
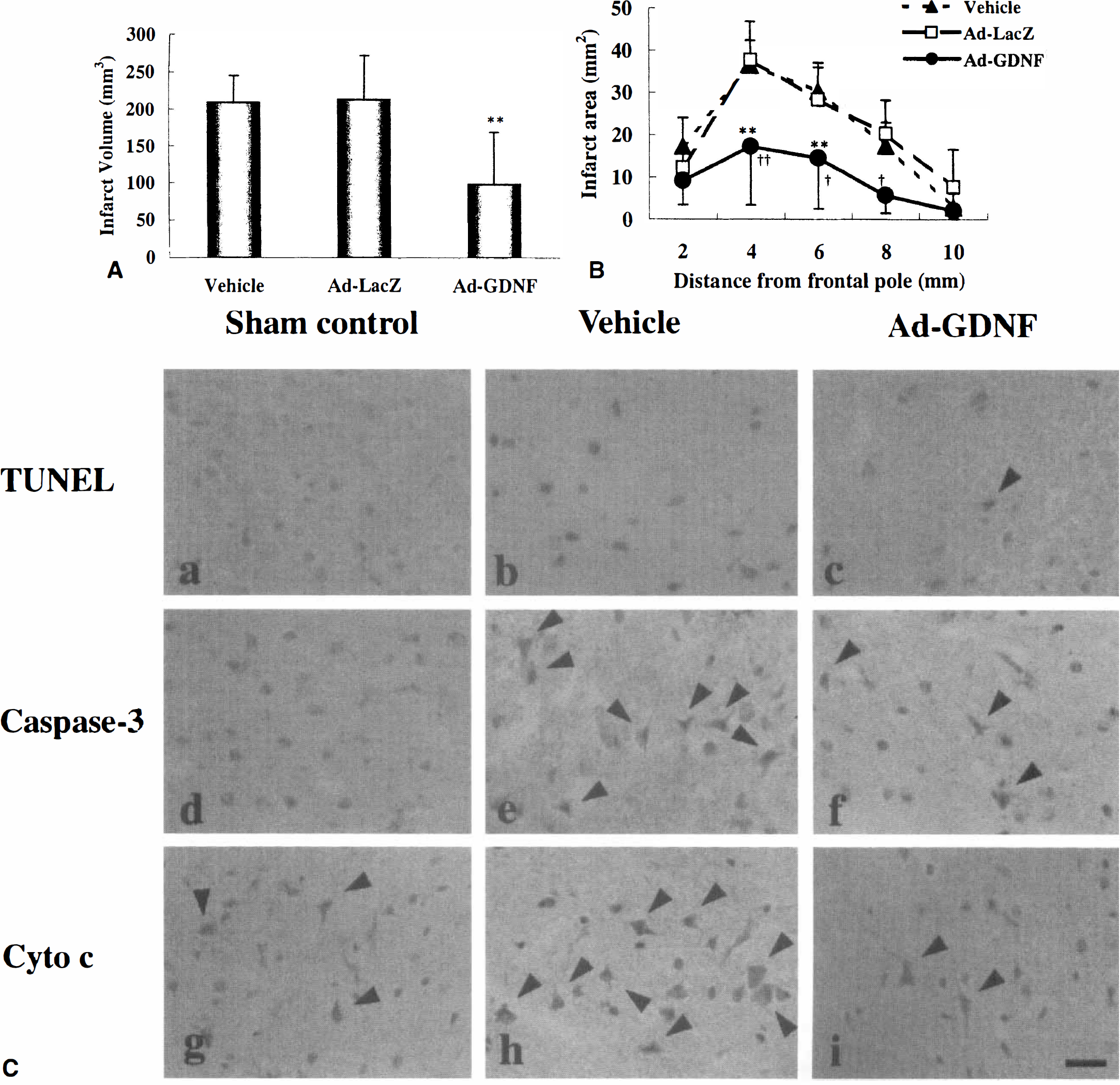
Effect of Ad-GDNF on infarct volume
Many other therapeutic gene studies for cerebral ischemia have been successful in animal models. Adenovirus-mediated neuronal apoptosis inhibitory protein and an interleukin-1 receptor antagonist had protective effects against ischemic brain injury (Betz et al., 1995; Xu et al., 1997). Herpes simplex virus-mediated HSP72 or Bcl-2 gene transfer improved brain damage in focal ischemia (Linnik et al., 1995; Lawrence et al., 1997; Yang et al., 1997; Yenari et al., 1998). Each vector has some disadvantages such as toxicity or efficacy of geneexpression, and each experiment, including the current data, has other problems to solve such as timing of gene transfer (pretreatment), spatial limitation of the exogenous gene expression around the injection site or periventricular ependymal cells, and gene delivery. Development of a more effective and safer vector may be necessary for future practical gene therapy (Ferrari et al., 1997; Verma and Somia, 1997). Finally, the route of administration may be another important aspect for the gene therapy of stroke. Direct injection of the vector into brain parenchyma or the ventricle may not be practical for future clinical applications. A development of ischemic tissue-specific targeted gene transfer using intraarterial or venous administration may be expected (Abe et al., 1997b). A few additional approaches of gene therapy for stroke could be possible as shown in Fig. 12 (Heistad and Faraci, 1996; Abe et al., 1997b). Stabilization of atherosclerotic plaque and restenosis (Morishita et al., 1997, 1998) or stimulation of new collateral vessels may also be feasible. When these problems are successfully resolved, gene therapy could have great promise for stroke therapy in the future.
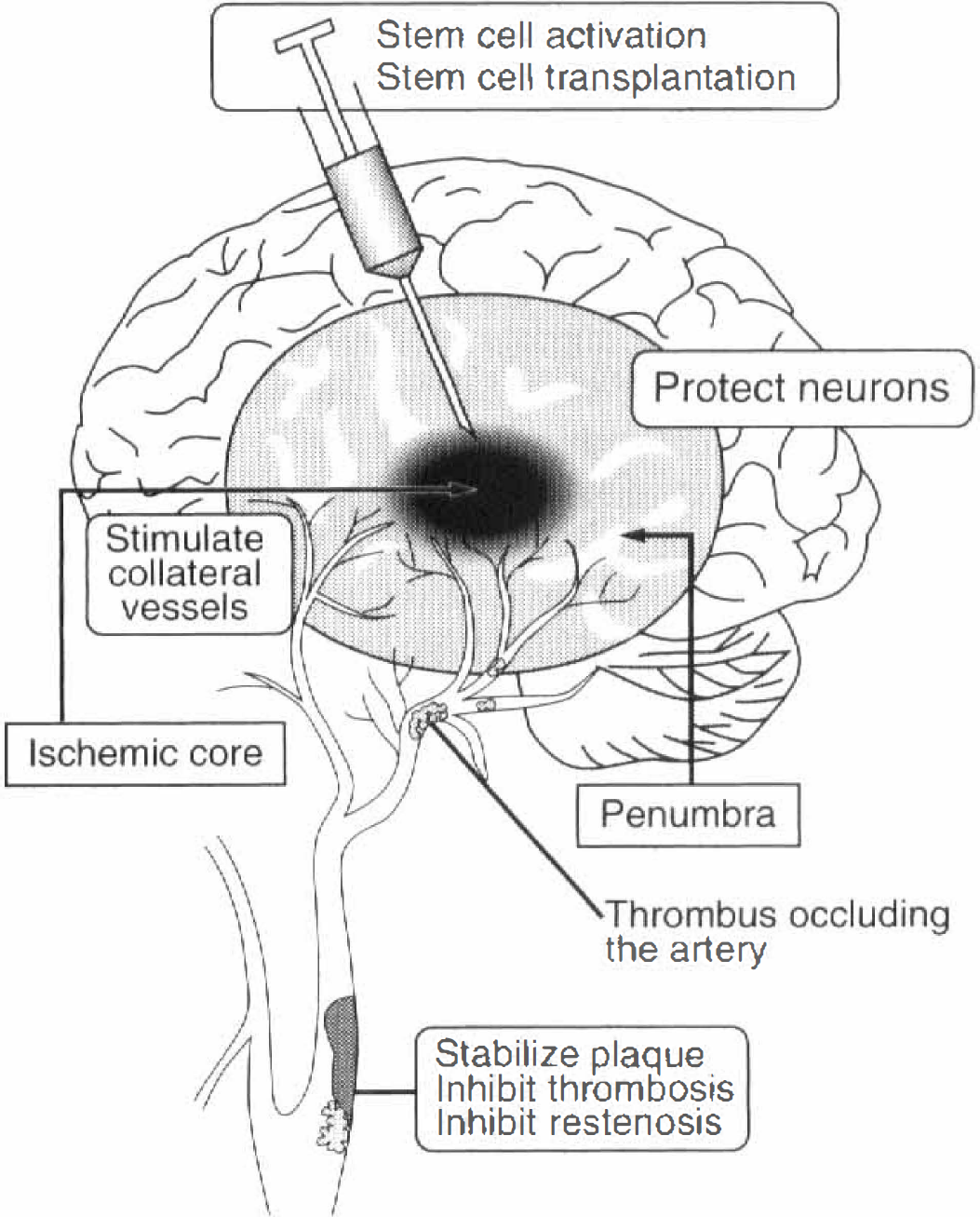
A possible strategy for gene therapy in ischemic stroke. Neurons may be protected by vector-mediated neurotrophic factor (NTF) gene expression. Collateral circulation may be improved or newly constructed by angiogenic genes such as VEGF or bFGF. Stem cells that are genetically manipulated could be transplanted into the ischemic lesion or NTFs may be directly injected to activate intrinsic stem cells. To avoid the risk of a cerebrovascular accident, arteriosclerotic plaques may be stabilized, or arterial thrombosis may be inhibited in advance. Modified from Heistad and Faraci, 1996).
DIFFERENTIATION OF NEURAL STEM CELLS IN NORMAL DEVELOPMENT
Recent advancements in molecular cell biology have discovered the presence of neural stem cells, even in adult human and monkey brains (Eriksson et al., 1998; Gould et al., 1999). Various neurons and glial cells originate from a common stem cell (Fig. 13). The stem cell possesses two potentials such as self-renewal and multidirectional differentiation. Development and differentiation of the nervous system depends on the expression of particular genes at particular places and times. This spatial and temporal pattern of gene expression is regulated by internal molecular programs and external epigenetic environments. Internal influences include transcription factors that act at the DNA level to control gene expression, and cell surface and secreted molecules that control the fate of neighboring cells. External factors include secreted factors, nutrients, sensory stimuli, and social experience that mediate patterned changes in the activity of nerve cells. The interaction of these intrinsic and environmental factors is critical for the proper differentiation of each nerve cell. At each step of differentiation, particular intrinsic determinants (for example, transcription factors) are involved (Fig. 13). The stem cell is positive for nestin at an initial stage, but secondary progenitor cells become negative for nestin, and turn positive for Mash1, Ngn1/2, and Prox1. This cell then differentiates into a neuronal progenitor cell that is positive for Prox1 and Bm4, or into a glial progenitor cell that is positive for repo in the presence of gcm (Nakafuku and Nakamura, 1995; Nakagawa et al., 1996; Sakakibara and Okano, 1997; Torii et al., 1997; Shimazaki et al., 1999). From these neuronal or glial progenitor cells, mature neurons or glial cells are finally produced (Fig. 13).
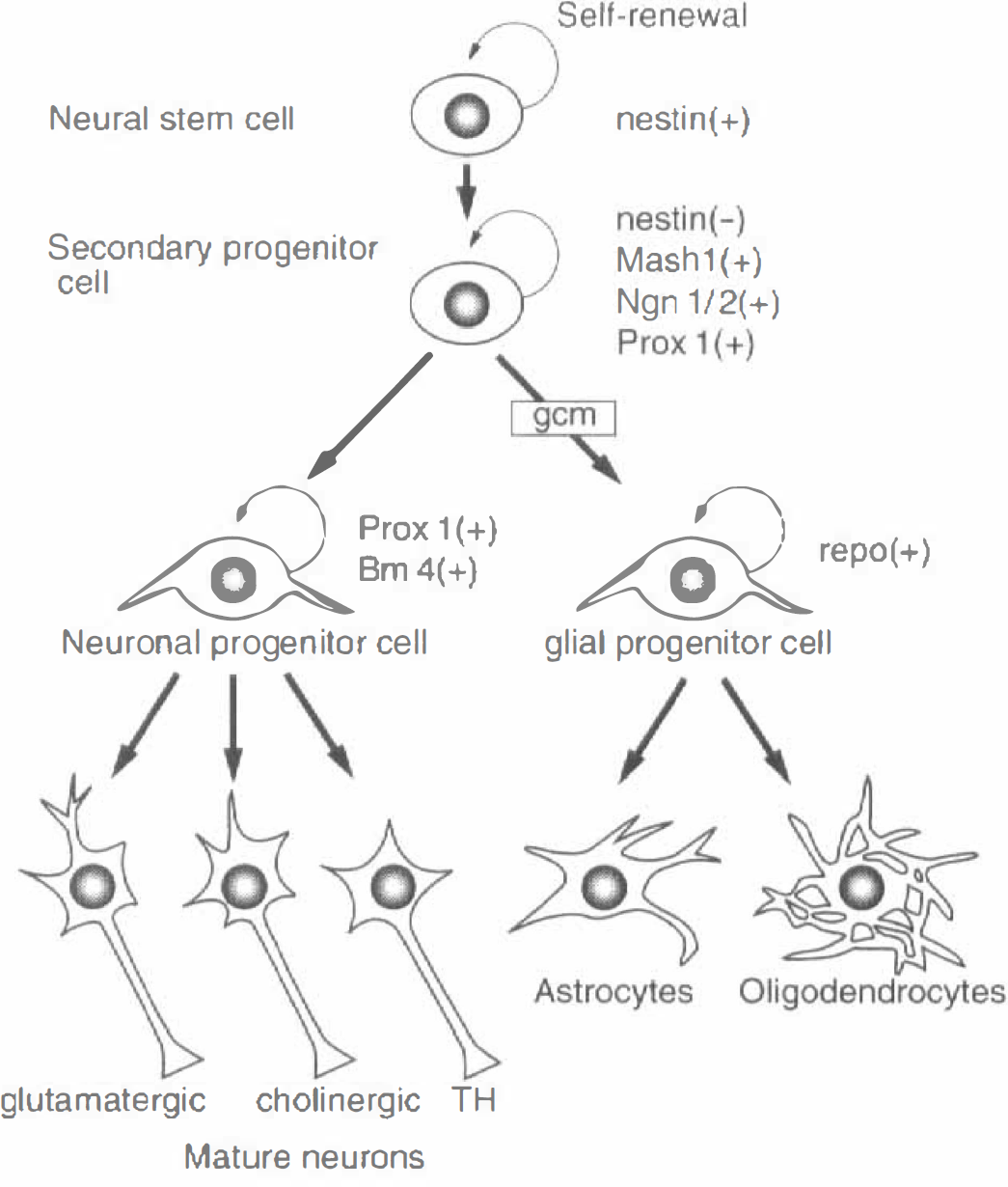
Neural stem cells differentiate into neurons or glial cells with certain transcription markers such as nestin, Mash1, Ngn1/2, Prox1, Bm4, and repo at certain stages.
The central nervous system originates from the neural crest or neural tube that forms a simple epithelial monolayer. In terms of the spinal cord and peripheral nerves, neural crest cells migrate from the dorsal neural tube along different paths. Many gene factors and NTFs play pivotal roles in the differentiation of neural crest cells into neuron, glia, or muscle cells, and further differentiation (Figs. 3 and 13) (Takahashi et al., 1999). As for the cerebral cortex, the neurons are generated from an epithelial layer of progenitor cells that lines the lateral ventricles. Once they have left the cell cycle, the immature neurons migrate out of the ventricular zone to form the cortical plate (future gray matter). Neurons born at early stages within the ventricular zone migrate along with radial glial cells to the deepest layers of the cortical plate. Neurons born at later stages migrate past the earlier-generated neurons to form the more superficial layers of the cortex. Thus, cortical neurons in a deep layer are generated first, and those in an outer layer are generated later. Early in normal development, the progenitor cells expand the population of neuronal precursor cells by rapid proliferation, thus giving rise to neurons and additional progenitor cells, finally generating only neurons (Jessell and Sanes, 2000).
ACTIVATION OF NEURAL STEM CELL AFTER CEREBRAL ISCHEMIA
It has been well known that stem cells are present in the bone marrow that generates peripheral blood cells throughout life. However, stem cells in adult mammalian brain are now believed to be present in the hippocampal dentate gyrus (DG) and subventricular zone (SVZ) of the striatum (Gould et al., 1999; Kempermann and Gage, 1999). Three DG layers such as hilus, subgranular layer, and granular cell layer generate stem cells for the hippocampus. The SVZ of the striatum provides new neurons to the olfactory bulb. Although the exact origin of the neuronal progenitor cell is still debated as being either subventricular cells, ependymal cells, or subventricular astrocytes (Johansson et al., 1999; Doetsch et al., 1999), it is interesting that differentiated cells such as ependymal cells or astrocytes could be neuronal progenitors. These data suggest the possibility of brain cells reverting back to undifferentiated cells and redifferentiating into neuronal progenitor cells. Thus, these cells have the potential for recovery and rearrangement of neuronal networks after destruction of the brain by ischemia.
Dividing cells can be detected by the incorporation of BrdU after its administration intraperitoneally. With double staining for the neuronal marker NeuN and calbindin-D28k, neurogenesis and age-related decreases were found in the hippocampal DG of adult rats (Kuhn et al., 1996). These data explain in part the decreased plasticity of the aged brain. Based on such a study of normal brain, preliminary studies on a possible activation of neural stem cells has been examined in ischemic brains. Proliferation of neuronal precursor cells in the DG was accelerated after 15 minutes of transient forebrain ischemia in mice (Takagi et al., 1999). The number of BrdU-labeled cells increased in DG and paraventricle regions at 3, 7, and 10 days after reperfusion (Fig. 14). Liu and Sharp (1999) found a 12-fold increase of BrdU incorporation at the subgranular zone of DG between 1 and 2 weeks after 10 minutes of global ischemia in gerbils. A 2-or 4-minute occlusion time was the threshold for ischemia-induced stem cell proliferation in the DG. Confocal microscopy confirmed that 60% of the BrdU positive cells in the granule cell layer expressed neuronal markers NeuN, Calbindin, and MAP-2 at 1 month after the BrdU labeling, and that many cells expressed glial marker glial fibrillary acidic protein in the dentate hilus between 2 weeks and 1 month after the BrdU injection (Liu et al., 1998). Activation of neurogenesis has also been reported in a systemic chemoconvulsant model (Parent et al., 1997), where hippocampal network plasticity associated with epileptogenesis may arise from aberrant connections formed by newly born dentate granule cells.
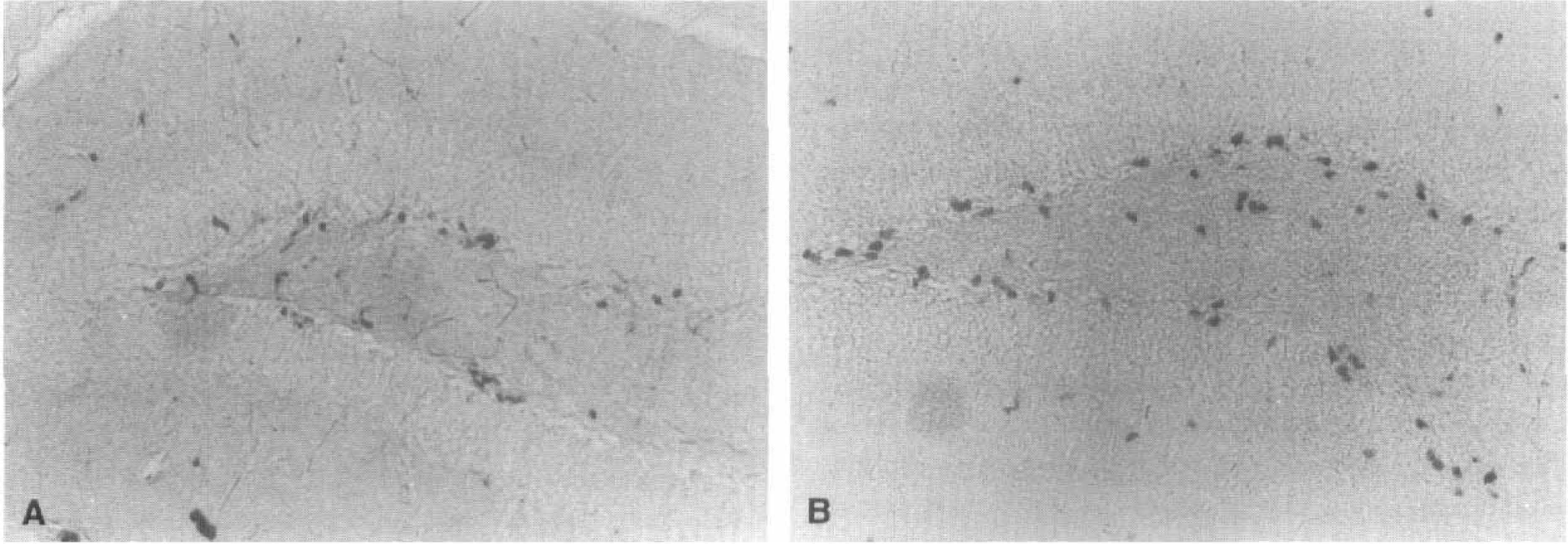
Intrinsic stem cell activation after ischemia. Compared with sham control brain
These pioneering works suggest that neural stem cells in the DG respond to ischemic injury by increasing the rate of proliferation in the subgranular zone in mice and gerbils. The neuronal and glial progeny derived from the stem cells migrated into different anatomical locations such as the granule cell layer and the dentate hilus. Enhanced neurogenesis in the DG may be a compensatory adaptive response to ischemic injury and could promote functional recovery after ischemia. Expression of highly polysialylated neural cell adhesion molecule in mossy fiber of granule cells in adult rat DG could be important in the migration of stem cells (Seki and Arai, 1993, 1999).
POTENTIAL OF NEURAL STEM CELL TRANSPLANTATION FOR THE ISCHEMIC BRAIN
Several trials have been performed to transplant brain cells into ischemic brain. For example, fetal rat hippocampal neurons were stereotaxically transplanted into 5-day-old ischemic hippocampal CA1 lesions of adult rat (Aoki et al., 1993). In the recipient brain, the transplanted cells survived well and formed clusters in the host CA1 subfield at 14 or 100 days after the transplantation and 67% of animals exhibited supragranular mossy fiber sprouting in DG. Hodges et al. (1997) reported that fetal CA1 grafts promoted recovery from cognitive deficits in marmosets induced by excitotoxic damage to the CA1 field.
Novel challenges have begun using various stem cells and modern techniques. The hematopoietic myeloid progenitor cells (1 × 105 per μL) cultured from adult mouse bone marrow were implanted into the penumbra of the ischemic striatum at 4 or 24 hours after 3 hours of transient MCAO in mice (Li et al., 1999). In another experiment, fresh bone marrow cells (1 × 106 per 10μL, including the supportive tissue (stroma) obtained directly from adult rats) were grafted into the penumbra of the ischemic striatum at 4 or 24 hours after 2 hours of transient MCAO in rats. The data demonstrated that adult hematopoietic bone marrow cells that were labeled with green fluorescent protein (GFP) survived in the adult mouse and rat brains after ischemia. Migration of SVZ cells was more prominent in the rat model than in the mouse, suggesting that a grafting microenvironment that includes stroma increased the potential for stem cell migration. Colocalization of mitotic and apoptotic cells in the graft suggests that ischemic injured brain and the graft cells may revert to an early stage of development to promote repair.
In another experiment, neuroepithelial stem cell lines (MHP36), which are genetically engineered to replicate into neurons or glia at 33°C, but cease replication and differentiate at body temperature of 37°C, were transplanted (75,000 cells per 3 μL) at 2 weeks after 60 minutes of transient MCAO into 8 sites of the striatum and cortex (Veizovic et al., 1999). The grafted animals recovered from sensorimotor deficit to a similar level as sham control throughout 18 weeks of testing. With this cell line (MHP36), Sinden et al. (1997) showed recovery from functional deficit in adult rats after 15 minutes of global ischemia, and found an extensive repopulation of the grafted cells into neuronal and glial phenotypes in the hippocampus. Further study showed that MHP36 stem cell restored cognitive function in marmosets after NMDA-induced excitotoxic hippocampal CA1 lesions as effectively as fetal homografts (Hodges et al., 1999; Virley et al., 1999).
There could be two ways to use stem cells as a therapy for ischemic brain. One way is to activate intrinsic stem cells located in the hippocampal DG or the SVZ of the caudate (Fig. 15A). Craig et al. (1996) showed that infusion of EGF into adult mouse ventricles induced migration of subependymal cells away from the lateral ventricle walls into adjacent parenchyma. Kuhn et al. (1997) found a neurogenesis in olfactory bulb after injection of bFGF or EGF into the ventricle. As mentioned above, these stem cells are also activated after ischemic injury. Addition of NTFs could further potentiate such a spontaneous activation of stem cells after ischemia. A second approach is to transplant manipulated stem cells originated from fetal or adult brain, or even embryonic stem cells (Flax et al., 1998; Vescovi et al., 1999) into the injured lesion of the brain (Fig. 15B). However, it is not possible at the current time to make these stem cells divide, differentiate, and migrate to fulfill the space lost by ischemia and to form functionally normal neuron networks. To develop future therapy for human stroke, further experimental studies and a better understanding of the role of each NTF, genetic markers at each stage of stem cell differentiation, and ways of genetic direction to a certain species of brain cells will be essential. Even though it may take time, the prospect of successful research in the future looks promising.
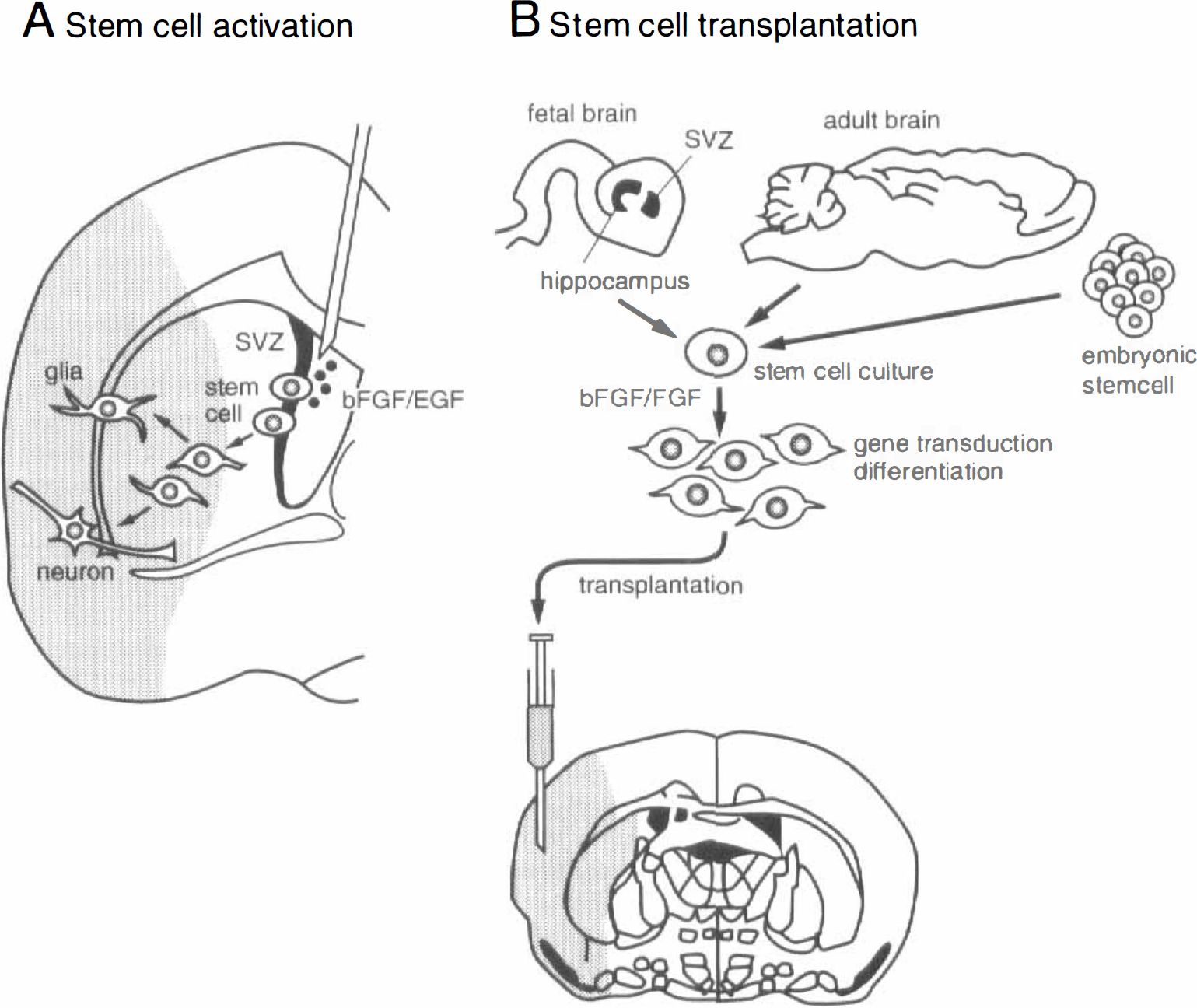
Strategy for neuroregenesis with stem cells. With injection of neurotrophic factors, such as bFGF or EGF, intrinsic neural stem cells may be activated, migrate to the shaded infarcted area, and differentiate into neurons or glia
Footnotes
Abbreviations used
Acknowledgements
The author thanks his colleagues, Drs. T. Hayashi, H. Kitagawa, H. Warita, and Y. Manabe, and Miss M. Endo for their help in writing this review article.