
Abstract
Glucocorticoids (GCs) and estrogen can modulate neuron death and dysfunction during neurological insults. Glucocorticoids are adrenal steroids secreted during stress, and hypersecretion of GCs during cerebral ischemia compromises the ability of hippocampal and cortical neurons to survive. In contrast, estrogen can be neuroprotective after cerebral ischemia. Here we evaluate the protective potential of a herpes viral vector expressing a chimeric receptor (ER/GR), which is composed of the ligand-binding domain of the GC receptor (GR) and the DNA-binding domain of the estrogen receptor-α (ER). This novel receptor can transduce an endangering GC signal into a protective estrogenic one. Using an in vitro oxygen glucose deprivation model (OGD), GCs exacerbated neuron death in primary cortical cultures, and this worsening effect was completely blocked by ER/GR expression. Moreover, blocking GC actions with a vector expressing a dominant negative GC receptor promoted neuron survival during postischemia, but not preischemia. Thus, gene therapeutic strategies to modulate GC and estrogen signaling can be beneficial during an ischemic insult.
Introduction
Cerebral ischemia is a major neurological insult that disrupts brain function and causes neuron death. Two hormones, glucocorticoids (GCs) and estrogen, modulate ischemic injury in opposite ways (Merchenthaler et al, 2003; Sapolsky and Pulsinelli, 1985; Smith-Swintosky et al, 1996; Wise et al, 2000). Glucocorticoids are adrenal hormones secreted during stress, and hypersecretion of GCs can exacerbate ischemic neurotoxicity (Sapolsky and Pulsinelli, 1985; Smith-Swintosky et al, 1996). Studies of hippocampal and cortical cultures have shown that GCs can worsen an array of processes central to excitotoxic neuron death. This includes disruption of neuronal energetics by decreasing glucose uptake (Horner et al, 1990) and accelerating depletion of ATP (Tombaugh and Sapolsky, 1992). Glucocorticoids also exacerbate secondary effects such as accumulation of excitotoxic glutamate in the synapse (Stein-Behrens et al, 1992), increased mobilization of free cytosolic calcium (Elliott and Sapolsky, 1993), elevated levels of reactive oxygen species (McIntosh and Sapolsky, 1996) and cytoskeletal damage (Elliott et al, 1993; Smith-Swintosky et al, 1996). The adverse effects of GCs are primarily mediated by the low affinity glucocorticoid receptor (GR), which is heavily occupied during high-circulating GC levels, such as sustained stressors (McEwen et al, 1988; Sapolsky, 1996).
In contrast to these GC actions, estrogen protects against cerebral ischemia (Merchenthaler et al, 2003; Wise et al, 2001), as well as a number of other insults including excitotoxins (Turchan et al, 2001), hypoglycemia (Wise et al, 2001) and gp120 toxicity (Howard et al, 2001; Zemlyak et al, 2002) in primary cultures. The estrogen receptor-α (ERα) mediates the protective effects of estrogen against cerebral ischemia, as estrogen is no longer protective in ERa knockout mice (Dubal et al, 2001). The mechanisms underlying estrogenic protection include upregulation of the endothelial glucose transporter Glut-1, increased expression of anti-apoptotic proteins such as bcl-2, inhibition of expression of proapoptotic proteins such as Bax and Bad, and increased expression of neurotrophins such as brain-derived neurotrophic factor (BDNF; Wise et al, 2000, 2001).
Gene therapy approaches have showed the protective potential of a variety of genes against stroke (Sapolsky, 2003; Yenari et al, 2001a), including a glucose transporter, a calcium buffering protein, bcl-2, anti-oxidants and neurotrophins (Hoehn et al, 2003; Lawrence et al, 1996, 1997; Yenari et al, 2001b). Based on the contrasting effects of GCs and estrogen, we have designed a chimeric receptor (ER/GR) that is composed of the ligand-binding domain of GR and the DNA-binding domain of ERa (Kaufer et al, 2004). Because of this structure, ER/GR transduces an endangering GC signal into a protective estrogenic one, and decreases kainic acid damage to the hippocampus (Kaufer et al, 2004). It has been previously shown that upon activation, ER/GR translocates to the nucleus and regulates downstream gene expression mediated by ERa, such as N-methyl-D-aspartate receptor subunit 2D and BDNF (Kaufer et al, 2004). Here we used a herpes simplex virus amplicon system to express ER/GR and investigate its neuroprotective potential in the realm of cerebral ischemia. We examined whether ER/GR can offer protection in primary cultures under oxygen glucose deprivation, and in a transient middle cerebral artery occlusion (MCAO) model in rats. To dissociate the protective effects of ER/GR, we also tested whether blocking GCs alone with a dominant-negative GR (TdGR) can achieve similar neuroprotective potential offered by ER/GR.
Materials and methods
Tissue Culture
Primary cortical cultures were obtained from E18 rat embryos as described previously (Brooke et al, 2002). Experiments were performed on days 10 to 12. These mixed cultures are typically 20% to 30% neurons and 70% to 80% glia. Corticosterone (CORT; Sigma, St Louis, MO, USA) powder was dissolved in 100% ethanol (EtOH). CORT or EtOH (0.1 mmol/L) were applied 18 to 19 h before oxygen glucose deprivation (OGD), and these cultures were infected with viruses expressing the reporter gene, green fluorescent protein (GFP) or ER/GR receptor. During OGD, cultures were washed once with balanced salt solution with 0 mmol/L glucose to remove trace amounts of glucose, and hypoxia was induced by placing the cultures in a modular hypoxic chamber for 6 h. Control cultures were treated the same way except placement under normoxia conditions and maintenance of normal glucose levels (5.5 mmol/L) in the medium. After 6 h of OGD, cells were reperfused with 5.5 mmol/L glucose and returned to normoxic conditions. After cultures were reperfused for 24 h, cells were fixed with cold methanol, and the survival of cortical neurons was examined using an enzyme-linked immunosorbent assay with a neuron specific marker MAP2 (Sigma), as described previously (Brooke et al, 1999). As methanol disrupts GFP fluorescence, some of cells were fixed with 4% paraformaldehyde followed by examination under fluorescent microscope.
Generation of Herpes Simplex Virus Amplicons
We used herpes simplex virus (HSV)-based vector to express ER/GR or TdGR, with GFP as a reporter gene. Detailed construction of the ER/GR and TdGR has been previously described (Kaufer et al, 2004). Briefly, the chimeric ER/GR construct contains the C-terminal ligand-binding and dimerization domains of the rat GR, and the N-terminal transactivation, and DNA-binding domains of the primary human estrogen receptor (ERα), spliced together at a conserved hinge region. The TdGR construct contains the dominant negative b isoform of the human GR (Kaufer et al, 2004). Protocols used for the generation of HSV amplicons have been previously described (Ho et al, 1995). Briefly, E5 cells were transfected with plasmids expressing the chimeric ER/GR receptor with GFP (pa22-GFP-ERGR), the dominant negative rat GR with GFP (pa22-GFP-TdGR) or control vector expressing GFP alone (pa22-GFP), using lipofectamine. Twenty-hours after transfection, cells were superinfected with the helper virus d120 (multiplicity of infection = 0.03). The cells were harvested 48 h later, when the cytopathic effect reached 100%. The viruses were then extracted, concentrated and purified. The titer of the vector was quantified in Vero cells by counting GFP-expressing cells. The titer of the helper virus was determined by counting the plaques formed in the E5 cells. The titer of the viruses ranges from 7 to 9 × 106 viruses per milliliter. The ratio of vector to helper virus ratio was 1:1.
Focal Cerebral Ischemia
Male Sprague Dawley rats (270 to 300 g) were housed under 12:12 h light/dark cycle with food and water available ad libitum. All experiments were conducted in compliance with animal care laws and institutional guidelines, and approved by the Stanford Institutional Animal Care and Use Committee. Before experimental stroke, animals were first anesthetized with 5% isoflurane, then maintained on 2% to 3% isoflurane. Body temperature, heart rate and respiration were monitored every 15 mins and kept in physiologic ranges. Transient MCAO was performed as described (Zhao et al, 2003). The left common carotid, external carotid, pterygopalatine and internal carotid arteries were exposed. The left internal carotid artery was transiently occluded with a microsurgical clip, and a cut was made in the common carotid artery. A 3.0-monofilament suture with a rounded tip was inserted into the common carotid artery and advanced through the internal carotid artery to the ostium of the middle cerebral artery (MCA) to occlude the MCA. The intraluminal suture was left in place for 1 h and removed to allow reperfusion. Animals were killed 24 h after reperfusion and neuronal survival was quantitated.
Stereotaxic Injection
Herpes simplex virus amplicons were delivered either 17 to 19 h before ischemia onset, or 1 to 2 h after ischemia. Rats were first anesthetized with 5% isoflurane, then maintained on 2% to 3% isoflurane. Body temperature, heart rate and respiration were monitored every 15 mins and kept in physiologic ranges. The rats were anchored in a stereotaxic station and an incision was made on the skin of the top of the head. A small hole was exposed with a drill without damaging the brain tissue. A 10 μL Hamilton syringe needle was stereotaxically inserted into the striatum (from bregma, AP = 0.2 mm, ML = 3.5 mm, DV = 5 and 4.5 mm). Herpes simplex virus amplicons overexpressing GFP (control), ER/GR or TdGR were infused with a microinfusion pump at 0.5 μL/min. The first injection (2 μL) was delivered at depth 5 μm, followed by a 1 min waiting period. The needle was then moved to depth 4.5 mm, and a second injection (2 μl) was delivered followed by a 5 min waiting period. Injections were administered on the ischemia side first, followed by the non-ischemic side 15 mins later. Wounds were then closed and animals were monitored for recovery and return to homecages.
Histopathology and Data Analysis
Animals were killed 24 h after cerebral ischemia by an overdose of isoflurane, and transcardially perfused with cold 1 × phosphate-buffered saline followed by 4% paraformaldehyde and postfixed in 20% sucrose in 4% paraformaldehyde for 24 h. Sections of 30 mm were collected onto superfrosted slides using a cryostat and several duplicates were collected. One of the set was used for cresyl violet staining to assess infarct size. Another set was used for quantifying neuron survival in the striatum. The study focused on the striatum because it is the most consistent site of damage in our stroke model. Neuron death in the striatum was determined by counting the number of GFP-positive striatal neurons; these were counted only if they are contained within the striatum and possessed characteristics of neuronal morphology. Neuron survival was expressed as percentage of GFP-positive cells in the ischemic striatum as compared with contralateral non-ischemic striatum. Brains without infarctions were excluded from the analysis. Student's t-test was used to compare between two groups, and one-way ANOVA was used for comparisons between three or more groups, followed Bonferroni's multiple comparison post hoc test.
Results
Expression of ER/GR Promotes Neuron Survival in Cortical Cultures During Oxygen Glucose Deprivation
We first examined the effects of CORT (the GC of rats) on neuron survival during OGD in cortical cultures. Six hours of OGD caused a significant 25% neuron death, when comparing between EtOH normal and EtOH OGD groups (Figure 1). Pretreatment of CORT at 0.01 to 1 μmol/L exacerbated neuron loss during OGD, relative to EtOH OGD control (Figure 1). These CORT treatment increased neuron death from ~25% to 35%, which is a 40% increase in neuron death (CORT versus EtOH OGD).
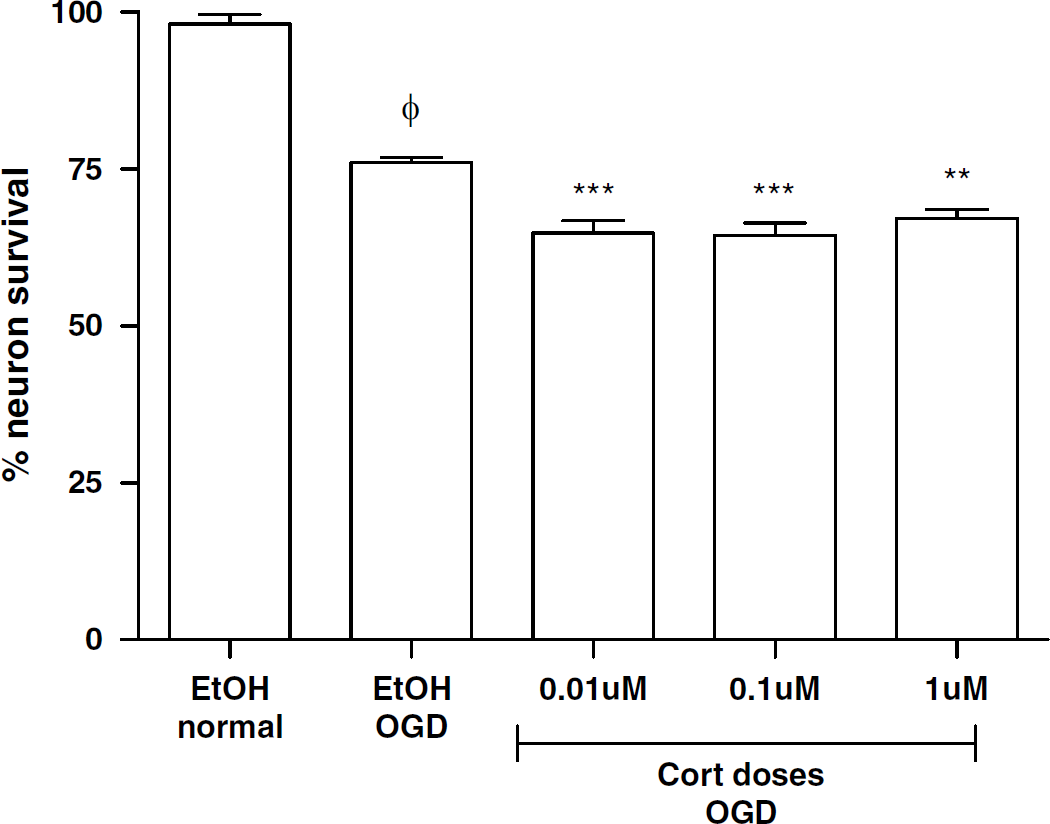
Corticosterone exacerbated neuron death during OGD. Pretreatment of corticosterone (CORT, 0.01 to 10 μmol/L) decreased neuron survival during OGD, when compared with EtOH OGD group (n = 12 to 15 per group). ϕ < 0.001 indicates significant difference from EtOH normal. ** p < 0.01, *** P < 0.001 indicates significant difference from EtOH OGD group, using one-way ANOVA with Bonferroni's post hoc test. Data are expressed as percentage over its own group during normal condition±s.e.m.
Next we tested whether the ER/GR receptor can block the worsening effect of CORT during OGD. Vectors expressing control GFP or ER/GR mainly infect neurons in cortical cultures, as indicated by the MAP2 staining (Figure 2A). Twenty-four hours before OGD, cortical cultures were treated with vectors expressing ER/GR or control GFP. In the presence of viral vectors, the endangering effect of CORT is greater (Figure 2B). During OGD, pretreatment of 0.1 μmol/L CORT increased neuron death from 25% to 60% neuron death in the control GFP group, which is a ~ 140% increase in neuron death. Expression of ER/GR completely blocked the exacerbating effects of CORT during OGD (P < 0.001; Figure 2B).
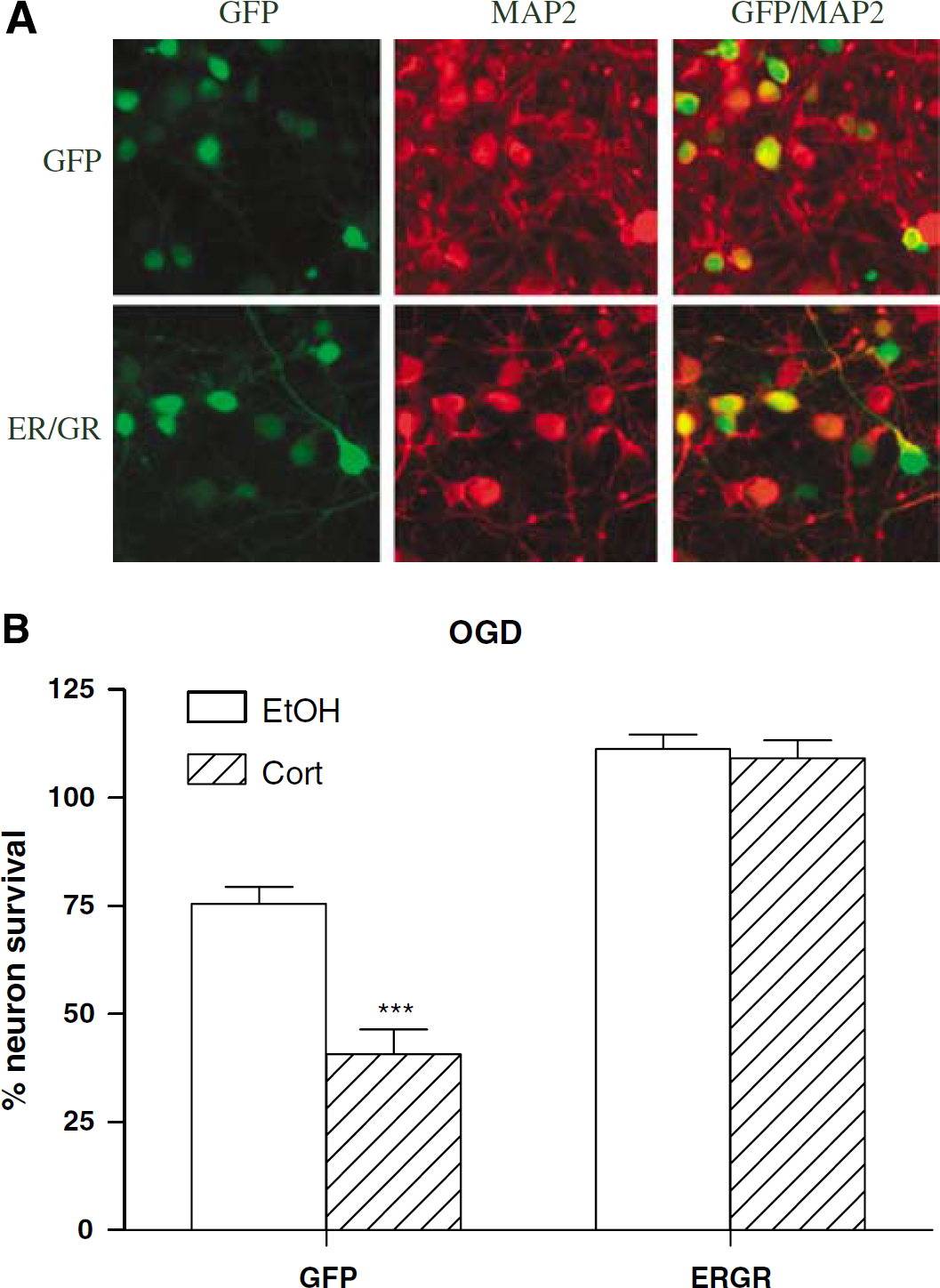
ER/GR promotes neuron survival in cortical cultures during OGD. (
Expression of ER/GR Before or After Ischemia Promotes Striatal Neuron Survival
To evaluate the neuroprotective potential of ER/GR in vivo, we expressed ER/GR in the striatum before ischemia (MCAO) onset in rats. Green fluorescent protein or ER/GR vector was infused into the striatum and infarct size was visualized by cresylviolet staining (Figure 3A). Twenty-four hours after ischemia, there were approximately twice the number of ER/GR + neurons, as compared with GFP + neuron (P = 0.029; Figures 3B and 3C). This protective effect could be due to ER/GR blocking GC actions and/or mimicking estrogen actions. To address this, we also tested the neuroprotective potential of a TdGR, which only blocks GC actions without mimicking any estrogenic signaling. If expressing TdGR did not protect against neuron death, this suggests that the protective effects of ER/GR was due to the estrogenic signaling. TdGR expression caused only a trend toward protection when infused before ischemia (P = 0.115; Figure 3C).
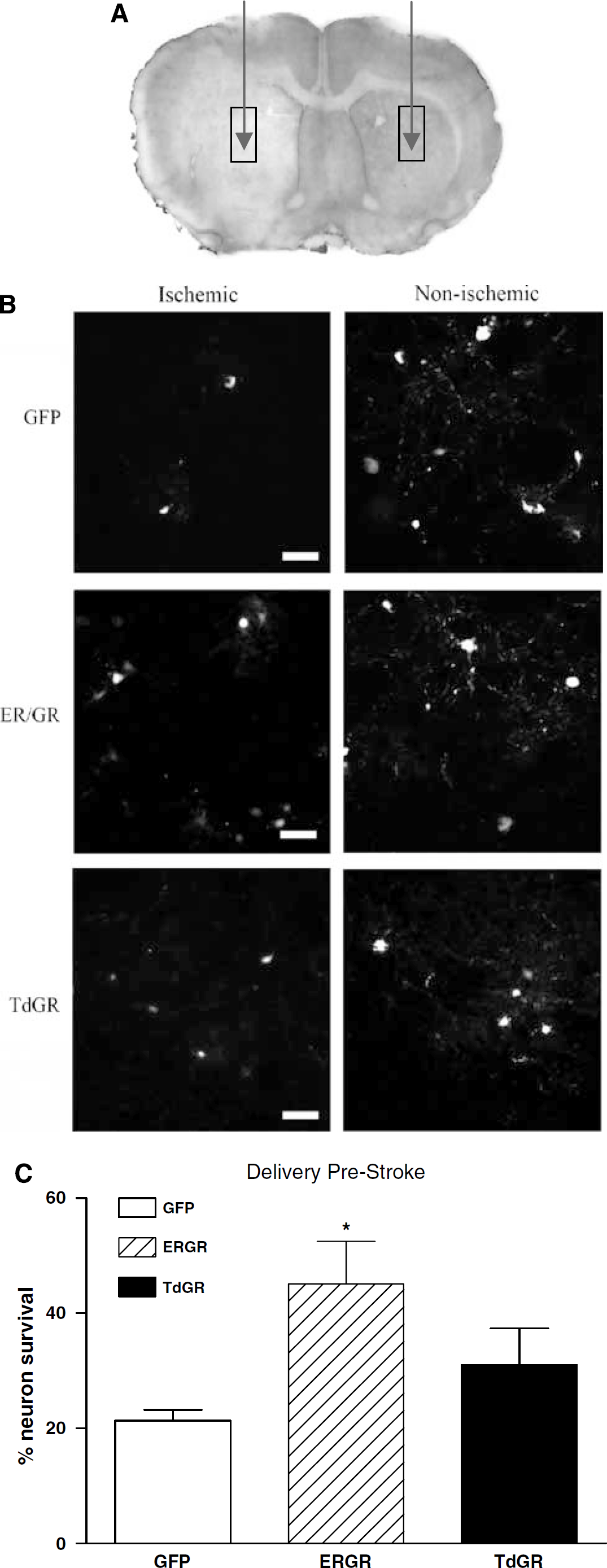
Expression of ER/GR prior to ischemia promotes striatal neuron survival in vivo. (
Although expression of ER/GR before an ischemic insult can promote striatal neuron survival, it is also important to evaluate whether ER/GR can still afford neuroprotection. Thus, we infused ER/GR 1 to 2 h after reperfusion and examined its neuroprotective potential. The number of ER/GR+ striatal neurons was double that of GFP + ones 24h after ischemia (P = 0.022). Interestingly, delivery of TdGR after ischemia also promoted striatal neuron survival (P = 0.013) (Figure 4).
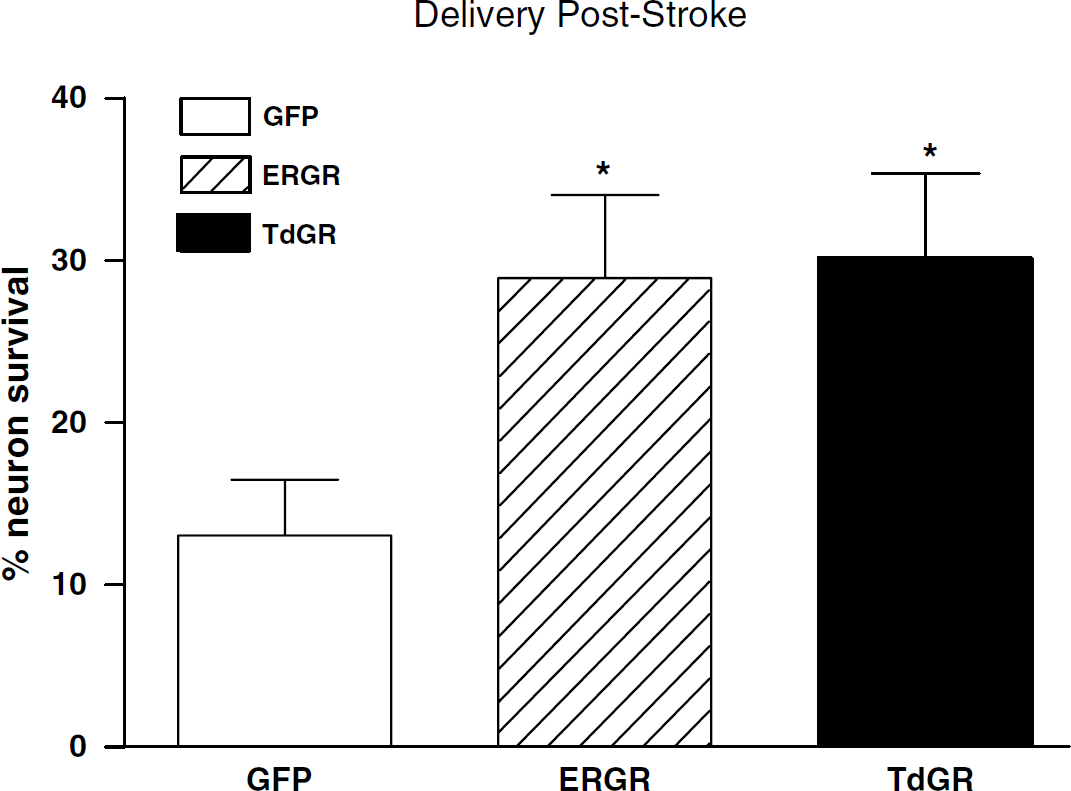
Expression of ER/GR or TdGR postischemia promotes striatal neuron survival in vivo. Neuron survival was higher in rats expressing ER/GR or TdGR, when compared with GFP control (n = 9 to 11 per group). Data are expressed as percentage of neuron survival by quantifying GFP-positive cells in the ischemic striatum versus contralateral non-ischemic striatum. * P < 0.05 indicates significant difference from GFP control, using one-way ANOVA with Bonferroni's post hoc test.
Discussion
Our results show that modulating GC and estrogenic signaling can promote neuron survival during an ischemic insult. Using a chimeric receptor, ER/GR, that can turn an endangering GC effect into beneficial estrogenic signaling, we show that expression of ER/GR completely blocked the worsening effects of CORT in primary cortical cultures during OGD (Figure 2B). Furthermore, in vivo expression of ER/GR also promoted striatal neuron survival in a transient MCAO model, whether administered before or after ischemia (Figures 3 and 4).
As reviewed, high levels of GCs released during acute neurological insults such as cerebral ischemia and seizure can worsen neuron survival, by affecting a number of cellular processes central to excitotoxic death (Chou et al, 1994; Lawrence and Sapolsky, 1994; Sapolsky and Pulsinelli, 1985; Smith-Swintosky et al, 1996; Stein-Behrens et al, 1992; Tombaugh and Sapolsky, 1992; Virgin et al, 1991). The adverse effects of GCs can be reduced by inhibitor of GC production (e.g., metyrapone; Smith-Swintosky et al, 1996) or in adrenalectomized animals that cannot produce GCs (Stein and Sapolsky, 1988). Blocking GC actions by using the GR antagonist RU486 has also been shown to protect against oxygen glucose deprivation and oxidative stress (Behl et al, 1997; Mulholland et al, 2005). In contrast, estrogen protects against excitotoxins, hypoglycemia and cerebral ischemia (Merchenthaler et al, 2003; Wise et al, 2001). In a transient cerebral ischemia model that causes cortical and striatal infarct, estradiol treatment is effective in reducing total infarct volume (Wise et al, 2000, 2001); however, it appears that a pretreatment period is required for its protection, suggesting a possible genomic mechanism. The genomic effect of estrogen is primarily mediated by ERa, and its downstream effects include upregulation of the endothelial glucose transporter Glut-1, increased expression of antiapoptotic proteins such as bcl-2, inhibition of expression of proapoptotic proteins such as Bax and Bad, and increased expression of neurotrophins such as BDNF (Wise et al, 2000, 2001). We have previously shown that activation of ER/GR increases expression of BDNF (Kaufer et al, 2004), which could be one of the mechanisms by which ER/GR exerts its protection. Nongenomic rapid mechanisms also exist for estrogen protection (Otto et al, 2006), such as its effects on membrane fluidity and its antioxidant effects; however, our chimeric vector uses the genomic ERa receptor, which indicate that the protective effects of ER/GR is likely to involve genomic mechanisms.
In primary cortical cultures, we show that CORT treatment worsens neuron survival during OGD (Figure 1). Although the exacerbating effect of CORT during OGD appeared small in Figure 1 (i.e., a maximum of ~40% increase in neuron death versus EtOH control), this worsening effect by CORT is greatly enhanced when cultures were infected with HSV (~ 140% increase in neuron death versus EtOH control; Figure 2B). This increased worsening is likely due to the HSV infection per se, as this is a challenge itself that makes the cells more vulnerable to the exacerbating effect of CORT. However, HSV expressing ER/GR reverses this CORT effect and promotes neuron survival during OGD (Figure 2B). Using MCAO, we show that delivery of ER/GR before ischemia can promote striatal neuron survival against cerebral ischemia (Figure 3C). As HSV infection area is local and limited, neuron survival is measured by counting GFP reporter positive cells in the infected area. Although HSV gene therapy is local and has no effect on infarction volume, its localized expression can still spare function in some cases (Dumas and Sapolsky, 2001). In our studies, ER/GR is mainly expressed in neurons as HSV is a neurotrophic virus (Figure 2A); however, some glia are still infected. Glucocorticoid levels can have adverse effects in glia (Virgin et al, 1991), and the neuroprotective effects of estrogen could also be partly mediated through astrocytes (Dhandapani and Brann, 2007). Thus it is possible that ER/GR protection involves both cell types.
We have tested whether ER/GR expression is protective in vivo, both before and after insult. It is important to determine whether delivering a gene-therapy vector after insult is protective, as such therapies in a clinical setting are likely to occur after insult. We show that expression of ER/GR, before or 2 h after, can promote neuron survival in cerebral ischemia. This protective time window is similar to previously reported gene therapy studies, such as overexpression of calbindin, bcl2, HSP72 and Gpx (Hoehn et al, 2003; Yenari et al, 1998, 2001b; Zhao et al, 2003).
To dissociate whether ER/GR's protection is due to blocking GCs and/or enhancing estrogenic signaling, we expressed a TdGR; it reduces GC signaling by competing with wild-type GR for GCs, as well as by heterodimerizing with wild-type GR, thereby blocking its genomic actions (Kaufer et al, 2004). Previous studies have shown that both TdGR and ER/GR protect against excitotoxicity in the hippocampus (Kaufer et al, 2004). We observe that although TdGR is not protective when introduced before insult, it is as protective as ER/GR when either was introduced after insult; this implies that the protective effects of ER/GR before insult are mediated by the estrogenic actions of the gene, whereas postinsult protection can be attributed to ER/GR's anti-GC actions.
The lack of additional estrogenic protection by ER/GR after insult may be due to several reasons. First, most estrogen protection studies require pretreatment of estrogen to be effective (Merchenthaler et al, 2003), which suggests a possible genomic involvement. Our chimeric vector uses the genomic ERa receptor, which would require a period of time to activate genomic events. Second, it is possible that the protection offered by TdGR after insult may have reached maximum protection, such that an additional ERa component cannot exceed this ceiling effect. Expression of an ERa-alone vector may further clarify this issue. Third, protein synthesis is inhibited after stroke, and thus, although ER/ GR can be expressed after stroke, it's likely that ER/GR's downstream genes may not be activated and synthesized at full potential. Unlike TdGR, which only needs to be expressed to block GCs and do not require transcriptional activation for its protection. Fourth, this discrepancy could also reflect the time course of vector expression, which begins 4 to 6 h after injection, peaks at 12 to 24 h and declines thereafter. Thus, when vector is introduced 17 to 19 h before insult, expression is at peak levels at the time of the ischemia; in contrast, when introduced 2 h after insult, less than half the survival period (of 24 h) is spent without maximal expression, and such low expression may be insufficient for estrogenic actions to be protective. As such, our results may reflect the limitations of this vector system, rather than the biology of estrogenic actions. Given the same time course of expression of the TdGR vector, our data suggest that postinsult events are more sensitive to the adverse effects of GCs than to the protective effects of estrogens. These ideas require further testing.
Our studies show that using gene therapy that turns an endangering GC signal into beneficial estrogenic signaling, we can promote neuron survival in cerebral ischemia, whether initiated before or after stroke. However, the limitations of gene therapy, such as the number of infected neurons and the route of administration, still need to be improved to provide an efficient and feasible way for clinical treatment. Nevertheless, modulating GC and estrogenic signaling can be beneficial during acute neurological insult, and pharmacologic treatment of GC inhibitor and estrogen may be a promising treatment for stroke.
Footnotes
Acknowledgements
The authors thank I-Ping Lee for technical assistance, and Nathan Manley, Rupshi Mitra, and Carolina Souza for helpful discussions.
None for all authors.