
Abstract
The purpose of this study was to examine the activation, topographic distribution, and cellular location of three mitogen-activated protein kinases (MAPKs) after permanent middle cerebral artery occlusion (MCAO) in mice. Phosphorylated MAPKs expression in the ischemic region was quantified using Western blot analysis and localized immunohistochemically using the diaminobenzide staining and double-labeled immunostaining. Extracellular signal-regulated kinases 1 and 2 (ERK1 and ERK2), p38 mitogen-activated protein (p38), and c-Jun NH2-terminal kinase or stress-activated protein kinase (SAPK/JNK) were initially activated at 30 minutes, 10 minutes, and 5 minutes, respectively, after focal cerebral ischemia. Peak expression represented a 2.7-fold, 3.7-fold, and 4.8-fold increase in each of these MAPKs, respectively. The immunohistochemical expressions of ERK1, ERK2, p38, and SAPK/JNK protein paralleled the Western blot analysis results. Double-labeled immunofluorescent staining demonstrated that the neurons and astrocytes expressed ERK1, ERK2, p38, and SAPK/JNK during the early time points after MCAO. The current results demonstrate that brain damage after ischemia rapidly triggers time-dependent ERK1, ERK2, p38, and SAPK/JNK phosphorylation, and reveals that neurons and astrocytes are involved in the activation of the MAPK pathway. This very early expression of MAPKs suggests that MAPKs may be closely involved in signal transduction during cerebral ischemia.
It is presently clear that certain signal transduction pathways that relay messages from the cell surface to the nucleus display a high level of evolutionary conservation. Stresses, including ischemia and reperfusion, may activate intracellular second messenger systems, and subsequently protein kinases and phosphatases (Wieloch, 1996). Mitogen-activated protein kinases (MAPKs) play a crucial role in the transduction of signals through protein kinases and protein phosphatases (Davis, 1993). The MAPK pathways are a ubiquitous group of protein serine and threonine kinases that regulate gene expression through transcription factor activity (Pages et al., 1995). The stimulus derived from ischemia may transduce to the nucleus to regulate gene expression through a distinct set of MAPK signal transduction cascades, including ERK1, ERK2, p38, and SAPK/JNK (Bogoyevitch et al., 1996). These pathways are important mediators of the signal transduction responsible for cell growth and proliferation (Seger and Krebs, 1995). In the central nervous system MAPKs are expressed at relatively high levels and are activated by stimulation of neurons (Fiore et al., 1993). Recently, three distinct MAPKs have been identified in vertebrates (Hunter, 1995). The most extensively characterized MAPKs are the 42 kDa and 44 kDa isoforms, also referred to as extracellular signal-regulated kinase 1 and 2 (ERK1 and ERK2), respectively. These serine and threonine kinases are unusual in that they require phosphorylation on tyrosine and threonine residues within the signature sequence T-E-Y (Yhr-183 and Yhr-185 in mammalian ERK2) for activity (Keyse, 1995). p38 mitogen-activated protein (p38) was the most recently identified member of the MAPK family (Davis, 1994). The c-Jun NH2-terminal kinase (JNK) protein kinases, also called stress-activated protein kinase (SAPK), were first identified as a protein kinase activity that phosphorylates the transcription factor c-Jun within its amino-terminal activation domain at Ser63 and Ser73. The nuclear targetsof these MAPK signaling pathways are transcriptional factors, such as transcriptional factor activator protein-1 (AP-1) and nuclear factor-kappa B (NFκB), which regulate the expression of various genes (Shimizu et al., 1998).
Focal cerebral ischemia produces biochemical changes in brain tissue. Increases in the expression of immediate early genes and late response genes can be detected (Akins et al., 1996). Alteration in gene expression may play a major role in the recovery process after cerebral ischemia or in further tissue injury by induction of genes involved in apoptotic cell death triggered by ischemia (Akins et al., 1996). The regulation of these gene expressions by the MAPK signal transduction pathway, especially ERK1 and ERK2, is likely to play an important role in focal cerebral ischemia. If this true, MAPK activation in the brain may provide fertile ground for the discovery of novel therapeutic agents for stroke. The purpose of the current study was to elucidate the signal-transduction pathway in the process of ischemic brain injury. Time courses for phosphorylated ERK1 and ERK2, p38, and SAPK/JNK expression during middle cerebral artery occlusion (MCAO) in mice were examined by Western blot analysis. The topographic distribution and cellular source of phosphorylated ERK1, ERK2, p38, and SAPK/JNK in the mouse brain after permanent MCAO were assessed using immunohistochemical study.
MATERIALS AND METHODS
Experimental model
Procedures using laboratory animals were approved by the institutional animal care and use committee. Adult male CD-1 mice (Charles River, Wilmington, MA, U.S.A.) weighing 30 to 35 g were anesthetized with an induction dose of 4% isoflurane in a 70%:30% N2O:O2 gas mixture and maintained at 1.5% isoflurane through a face mask. Body temperature was maintained at 37°C ± 0.5°C using a heated blanket with a feedback control system (73A; Yellow Springs, OH, U.S.A.).
The MCAO method has been described previously (Yang et al., 1994). Briefly, under an operating microscope (12×; Carl Zeiss, Oberkochen, Germany), the left common carotid artery was exposed through a midline incision. The internal carotid artery was then isolated and its branch, the pterygopalatine artery, was ligated close to its origin. A 2-cm length of 5-0 rounded tip nylon suture was advanced from the external carotid artery through the common carotid artery and into the internal carotid artery for a distance of 11.0 ± 0.5 mm. Sham-operated mice underwent standard exposure of the common carotid artery; the pterygopalatine artery and distal branches of the external carotid artery were cauterized and cut, but no suture was introduced. During the occlusion procedure, surface blood flow was measured by laser–Doppler flowmetry to confirm the occlusion. Nine groups of mice (n = 3 to 5 in each group) underwent 0, 5, 10, 20, 30 minutes, 1, 2, 6, and 24 hours of MCAO. The animals were killed and brains were removed immediately after the MCAO for Western blot analysis. Five other groups of mice (n = 6 to 8 per group) underwent 0, 10, 20, 30, and 120 minutes of MCAO. These animals were perfusion-fixed and brains were removed, postfixed, and immersed in 25% sucrose solution for immunohistochemistry.
Tissue preparation
Brains were quickly removed after MCAO for Western blot analysis. Brain tissue from the basal ganglia of the ischemic hemisphere was immediately removed and sonicated in a sodium dodecyl sulfate (SDS) sample buffer (containing 62.5 mmol/L Tris-HCl, pH 6.8, 25°C, 2% w/v SDS, 50 mmol/L DTT, and deionized water). Twenty μL of sample was taken for protein assay (Bio-Rad, Hercules, CA, U.S.A.), and the remaining solution was frozen and stored at −80°C for Western blot analysis.
For immunohistochemistry, mice were anesthetized while perfused transcardially with 2% paraformaldehyde in 0.1 mol/L phosphate buffer solution (PBS, pH 7.4) for 10 minutes at 4°C. Brains were removed and immersed in 25% sucrose solution until they sunk to the bottom and were then embedded in a 60%:40% sucrose: OCT (Sakura Finetek USA, Torrance, CA, U.S.A.) mixed solution. Coronal sections (20 μm in thickness) were cut on a cryostat (CM1800; Leica, Bensheim, Germany) and mounted on microscope slides (precleaned; Fisher Scientific, Pittsburgh, PA, U.S.A.).
Western blot analysis
Thirty-five micrograms of protein were boiled at 95°C for 5 minutes and run on 10% polyacrylamide gels with a 4% stacking gel (SDS-PAGE) for 2 to 4 hours. The protein was then transferred to hybond-C pure nitrocellulose membrane (Amersham Pharmacai Biotech, Piscataway, NJ, U.S.A.) overnight at 4°C. The membrane was blocked in 1% Carnation nonfat dry milk and 1% bovine serum albumin in TBST (0.05% Tween, 150 mmol/L NaCl, 60 mmol/L Tris base, pH 7.6) for 1 hour at room temperature and washed in TBST 3 times for 10 minutes in each. The membrane was incubated with either monoclonal phospho-specific ERK1 and ERK2 antibody (1:2000; New England BioLabs, Beverly, MA, U.S.A.), or polyclonal phospho-specific anti-p38 (1:1000), or polyclonal phospho-specific anti-SAPK/JNK antibodies (1:1000; New England BioLabs) in blocking solution overnight at 4°C. The membranes were washed with TBST in each, 3 times for 10 minutes, then incubated with either peroxidase-conjugated goat anti-mouse (1:2000) or goat anti-rabbit antibody (1:2000) in blocking solution. Finally, the membranes were incubated in ECL detection reagent (Amersham) for 1 minute and then exposed to Kodak X-OMAT film (Kodak, Rochester, NY, U.S.A.). The relative densities of the ERK1, ERK2, p38, and SAPK/JNK protein bands were analyzed using National Institutes of Health Image Analysis Version 1.62.
Immunohistochemistry
Sections were incubated in 5% normal goat serum for 30 minutes at room temperature, rinsed with 50 mmol/L Tris buffer saline (TBS), then incubated in polyclonal phospho-specific ERK1 and ERK2 antibody, (1:300) or polyclonal phospho-specific anti-p38 (1:300), or polyclonal phospho-specific anti-SAPK/JNK antibodies (1:300) overnight at 4°C. An equivalent dilution of rabbit IgG was used as a primary antibody for the negative control. Sections were immersed in 0.3% H2O2 in 30%:70% methanol:PBS solution for 30 minutes. The sections were incubated in biotinylated goat anti-mouse or goat anti-rabbit secondary antibodies at a 1:300 ∼500 dilution for 90 minutes at room temperature followed by an ABC (Vector Laboratories, Burlingame, CA, U.S.A.) process. Finally, the sections were developed with stable DAB (Research Genetics, Huntsville, AL, U.S.A.) and counterstained with hematoxylin and eosin.
Double-labeled fluorescence staining
After washing in TBS-0.1% Triton solution, the sections were incubated with 2% normal donkey serum, 2% bovine serum albumin, 0.02% Saponin, and 0.1% Triton in TBS solution. Sections were incubated with two primary antibodies: polyclonal rabbit anti-ERK1 and anti-ERK2 (1:50), or rabbit anti-p38, or rabbit anti-SAPK/JNK (1:50), and either sheep anti-neuron specific enolase antibody (1:100; Capricorn, Scarborough, ME, U.S.A.) or goat anti-glial fibrillary acidic protein antibody (1:100; Santa Cruz Biotechnology, Santa Cruz, CA, U.S.A.) with 2% donkey and 2% bovine serum albumin in 0.02% saponin in TBS for 48 hours at 4°C. The sections were incubated with two secondary antibodies: donkey anti-rabbit IgG-fluorescence and either donkey anti-sheep IgG-rhodamine (Cortex Biochem, San Leandroca, CA, U.S.A.) or donkey anti-goat IgG-rhodamine (Santa Cruz) for 90 minutes at room temperature. Double-labeled immunostained sections were evaluated using a fluorescence microscope (Leitz, W. Nuhsbaum, McHenry, IL, U.S.A.) with a filter cube (excitation filter, 450 to 490 nm; suppression filter, 515 to 560 nm) for fluorescent isothiocyanate labeling and another filter cube (excitation filter, 515 to 560 nm; suppression filter, 590 nm) for rhodamine. Photomicrographs for double-labeling illustrations were obtained by changing the filter cube without altering the position of the section or focus.
Statistical analysis
Phosphorylated ERK1 and ERK2, p38, and SAPK/JNK expression were semiquantified as mean ± SD. Parametric data among the time points were compared using analysis of variance followed by Scheffe F-test between group comparisons. P < 5% was considered significant.
RESULTS
Surface cerebral blood flow (CBF) was measured before and after MCAO to verify that the MCAO was successful in experiments and constant in each group. Surface CBF was reduced to 10% to 20% of baseline CBF in the ipsilateral hemisphere and maintained at ∼100% of baseline CBF in the contralateral hemisphere in all groups of mice. There was no statistical significance among groups (data not shown).
Western blot analysis of MAPK proteins
Fig. 1 to Fig 3 illustrate the changes of MAPK protein levels in the mouse brain after 0, 5, 10, 20, 30 minutes, 1, 2, 6, and 24 hours of MCAO. Very low levels of phosphorylated ERK1, ERK2, p38, and SAPK/JNK expression were detected in the contralateral hemisphere after different time points of MCAO (data not shown). Western blot analysis revealed a double band at 42 and 44 kDa, indicating phosphorylated ERK1 and ERK2 was expressed in the ischemic hemisphere of the mouse brain. Phosphorylated ERK1 and ERK2 expression was increased as early as 30 minutes (1.8-fold, P < 0.05) after permanent MCAO, peaked at 2 hours (2.7-fold), and decreased to the control levels by 6 hours (Fig. 1). Western blot analysis revealed a band at 38 kDa, indicating that phosphorylated p38 was expressed in the ischemic hemisphere of the mouse brain. Phosphorylated p38 expression was increased at a very early MCAO time point (10 minutes, 3.0-fold, P < 0.05), peaked at 20 minutes, and returned to control level by 60 minutes (Fig. 2). SAPK/JNK showed a band at 46 kDa. Phosphorylated SAPK/JNK expression was increased earlier than both ERK1 and ERK2 and p38 groups (5 minutes after MCAO, 3.2-fold, P < 0.05); it peaked at 30 minutes, maintained the increase for 120 minutes, then decreased to the control levels by 6 hours.
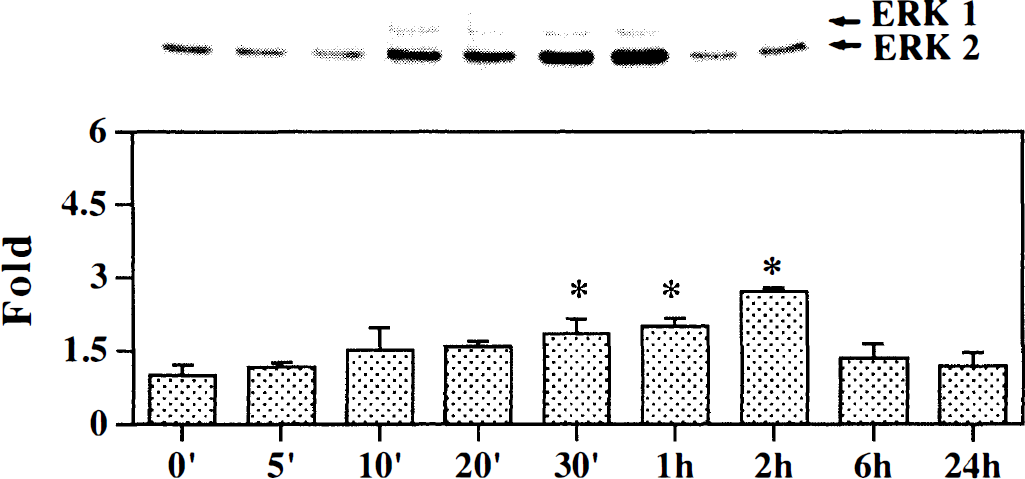
Time course of phospho-ERK1 and ERK2 expression during ischemia in the mouse brain. Total soluble protein extracts were prepared from the ischemic hemisphere in mouse brain. The upper panel shows equal amounts of extracts (35 μg) separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and immunoblotted with monoclonal ERK1 and ERK2 antibody. The 42 and 44 kDa MAP kinase bands are clearly indicated with an arrow. The bar graph shows optical density with equal amounts of isolated protein from the ipsilateral hemisphere in mice after 0, 5, 10, 20, 30 minutes, 1, 2, 6, and 24 hours of middle cerebral artery occlusion. The mean value of sham-operated mice is represented as 1. Values of ischemic samples are corrected relative to sham-operated mice and represent mean ± SD. * P < 0.01, compared with sham-operated mice; n = 3 to 5 for each time point.
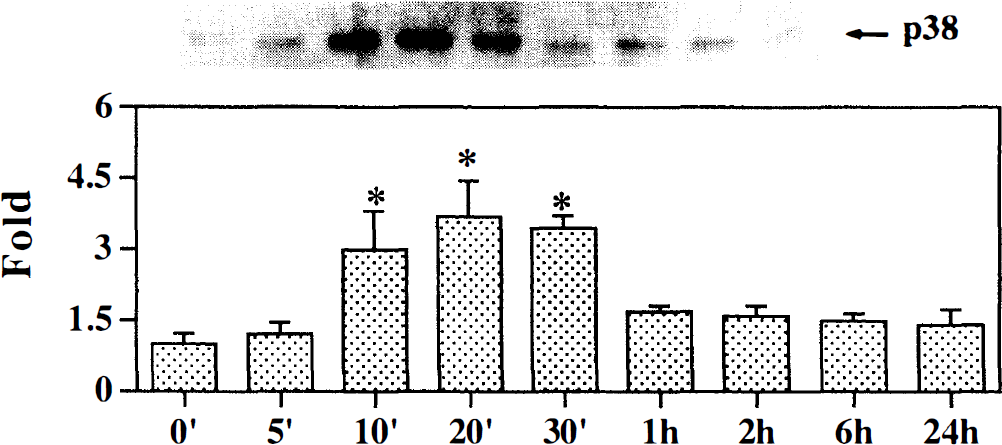
Time course of phospho-p38 expression during ischemia in the mouse brain. Total soluble protein extracts were prepared from the ischemic hemisphere in mouse brain. The upper panel shows equal amounts of extracts (35 μg) separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and immunoblotted with monoclonal p38 antibody. The 38 kDa MAP kinase band is indicated with an arrow. The bar graph shows optical density with equal amounts of isolated protein from the ipsilateral hemisphere in mice after 0, 5, 10, 20, 30 minutes, 1, 2, 6, and 24 hours of middle cerebral artery occlusion. The mean value of sham-operated mice is represented as 1. Values of ischemic samples are corrected relative to sham-operated mice and represent mean ± SD. * P < 0.01, compared with sham-operated mice; n = 3 to 5 for each time point.
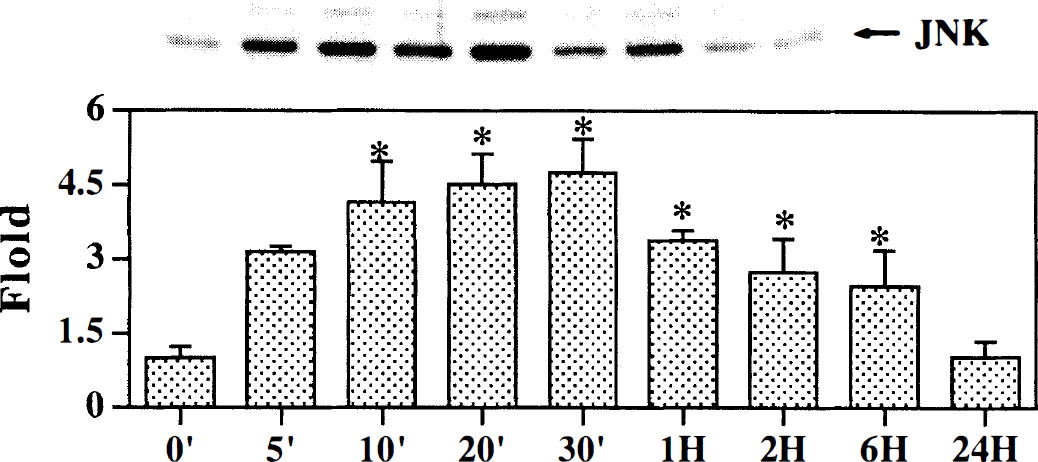
Time course of phospho-JNK expression during ischemia in the mouse brain. Total soluble protein extracts were prepared from the ischemic hemisphere in mouse brain. The upper panel shows equal amounts of extracts (35 μg) separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and immunoblotted with monoclonal SAPK/JNK antibody. The 46 kDa SAPK/JNK band is indicated with an arrow. The bar graph shows optical density with equal amounts of isolated protein from the ipsilateral hemisphere in mice after 0, 5, 10, 20, 30 minutes, and 1, 2, 6, and 24 hours of middle cerebral artery occlusion. The mean value of sham-operated mice is represented as 1. Values of ischemic samples are corrected relative to sham-operated mice and represent mean ± SD. * P < 0.01, compared with sham-operated mice; n = 3 to 5 for each time point.
Immunohistochemistry of MAPK proteins
Immunohistochemical results were consistent with Western blot analysis in the current study. Few phosphorylated ERK1 and ERK2, p38, and SAPK/JNK immunopositive stained cells were detected in normal brain sections and in sham control mice (data not shown). The negative control showed no immunopositive staining without phospho-specific ERK1, ERK2, p38, and SAPK/JNK antibodies (Figs 4A, 5A, and 6A). ERK1, ERK2, p38, and SAPK/JNK phosphorylation were detected as early as 5 minutes of MCAO in the ischemic core and perifocal regions. ERK1 and ERK2 phosphorylation increased at 2 hours of MCAO (Fig. 4), p38 phosphorylation increased at 20 minutes of MCAO (Fig. 5), and SAPK/JNK phosphorylation increased at 30 minutes of MCAO (Fig. 6). Comparison of the phosphorylated ERK1, ERK2, p38, and SAPK/JNK expression with hematoxylin counterstaining indicated that the expression of these proteins was primarily localized to the morphologically relatively intact cells or dark shrunken cells. The most immunopositive neuronlike cells were seen in the ipsilateral cortical neuronal layer and subcortical regions, including the molecular layer of hippocampus, the outermost layer of the core, and the corpus callosum of the ischemic hemisphere. These proteins were expressed in the neuronal cell bodies, neurofibrils, and extensive dendritic arborizations. In addition, clearly immunopositive staining was seen in the axon hillocks and proximal axons of pyramidal neurons.
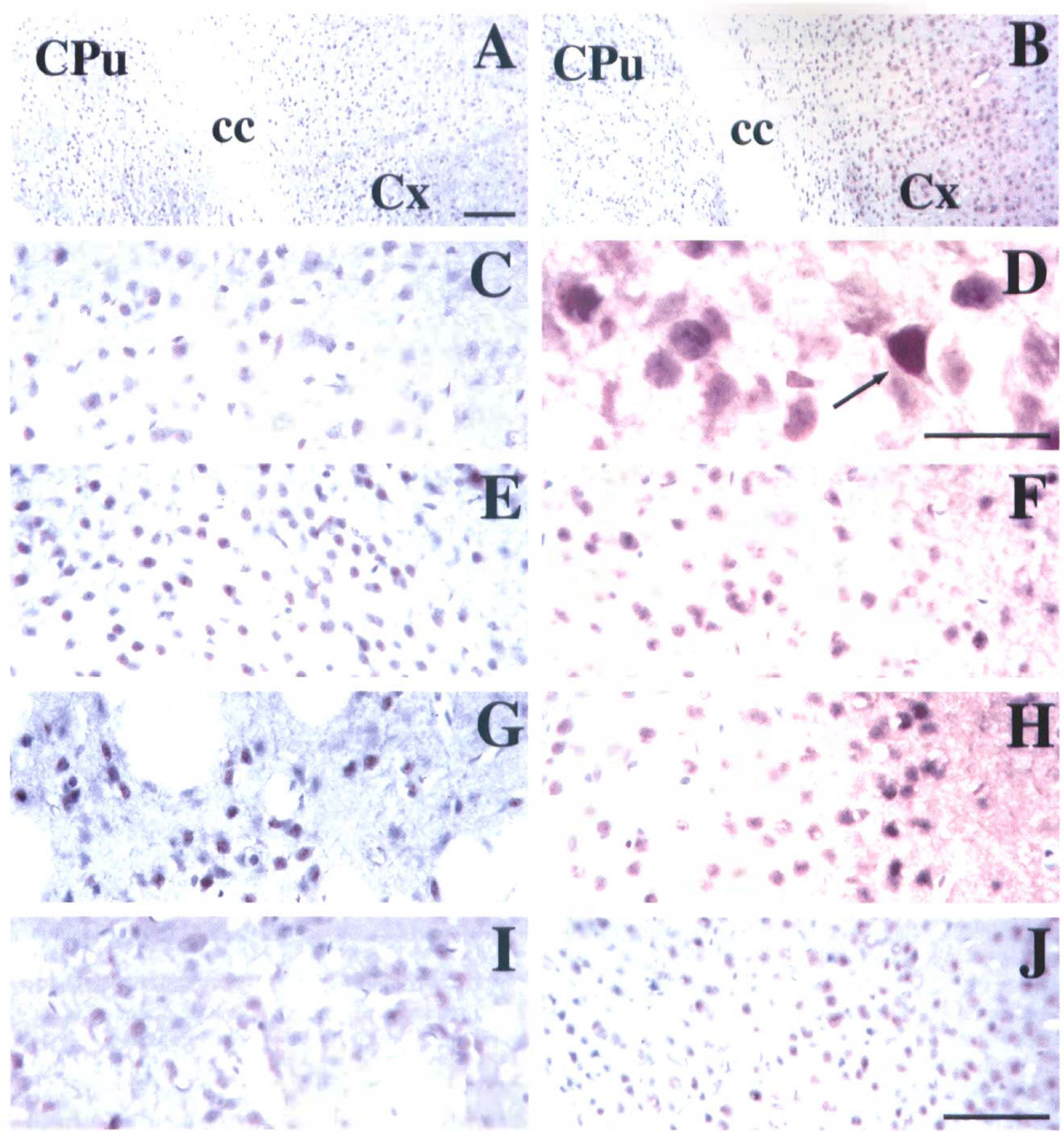
Photomicrographs show the activated ERK1 and ERK2 expression in the mouse brain after permanent middle cerebral artery occlusion (MCAO). There is no dark brown staining in the negative control sections
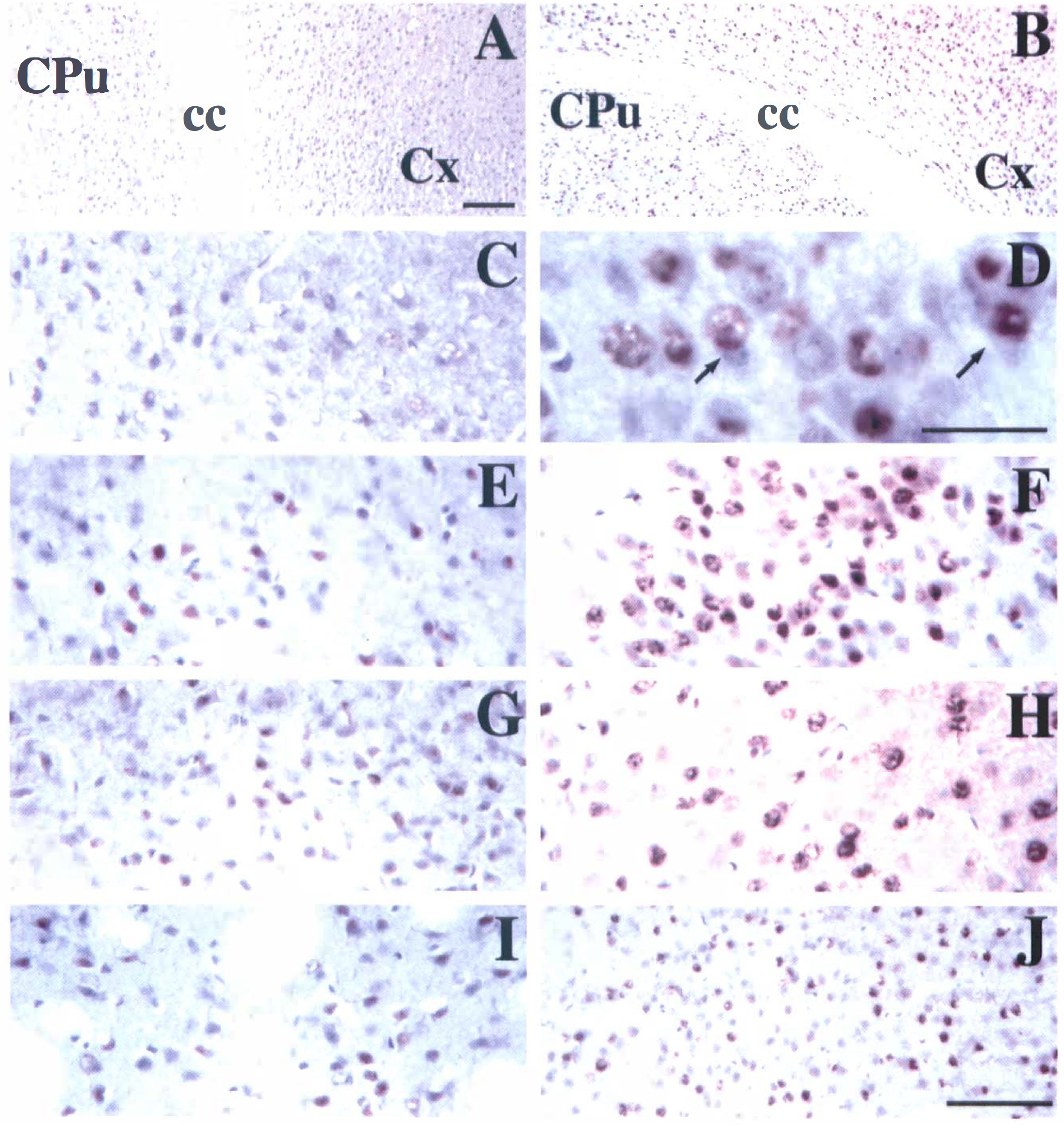
Photomicrographs show phosphorylated p38 expression in the mouse brain after middle cerebral artery occlusion (MCAO). There is no dark brown staining in the negative control sections
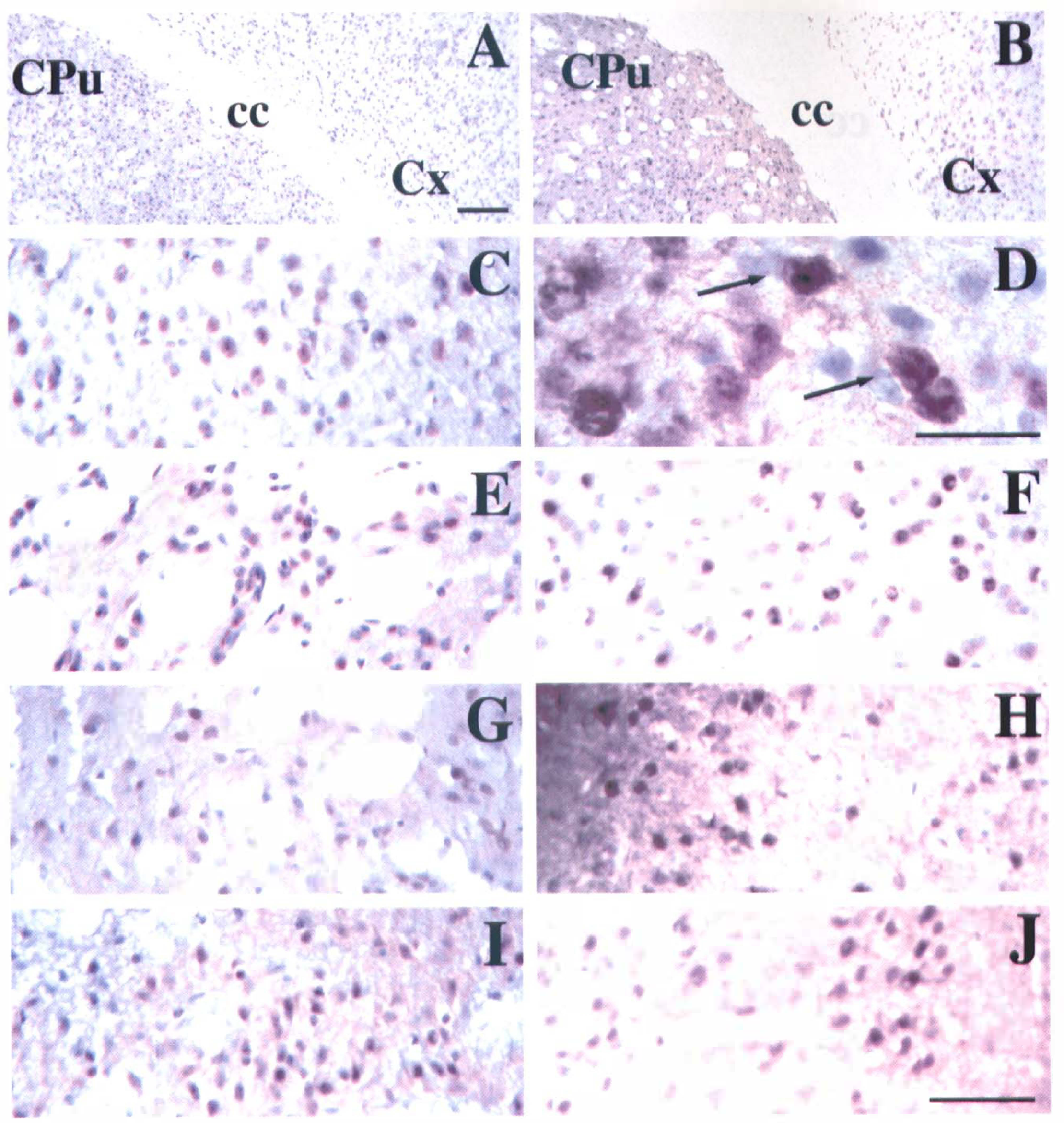
Photomicrographs show phosphorylated SAPK/JNK expression in the mouse brain after middle cerebral artery occlusion (MCAO). There is no dark brown staining in the negative control sections
Double-labeled immunofluorescence studies revealed that the ERK1, ERK2, p38, and SAPK/JNK phosphorylation were colocalized with NSE in cell bodies and extensive dendritic arborizations (Fig. 7 to Fig 9). The ERK1, ERK2, p38, and SAPK/JNK immunopositive cells were often located at the subcortical regions in the ischemic hemisphere. This showed that ERK1, ERK2, p38, and SAPK/JNK phosphorylation occurred in the neurons and dendritic arborizations after MCAO in mouse brain. Double-labeled immunofluorescence for phospho-specific ERK1, ERK2, p38, SAPK/JNK, and glial fibrillary acidic protein demonstrated that the phosphorylated ERK1, ERK2, p38, and SAPK/JNK immunoreactivities colocalized with glial fibrillary acidic protein (Fig. 7 to Fig 9), indicating that astrocytes and their processes were ERK1, ERK2, p38, and SAPK/JNK immunopositive.
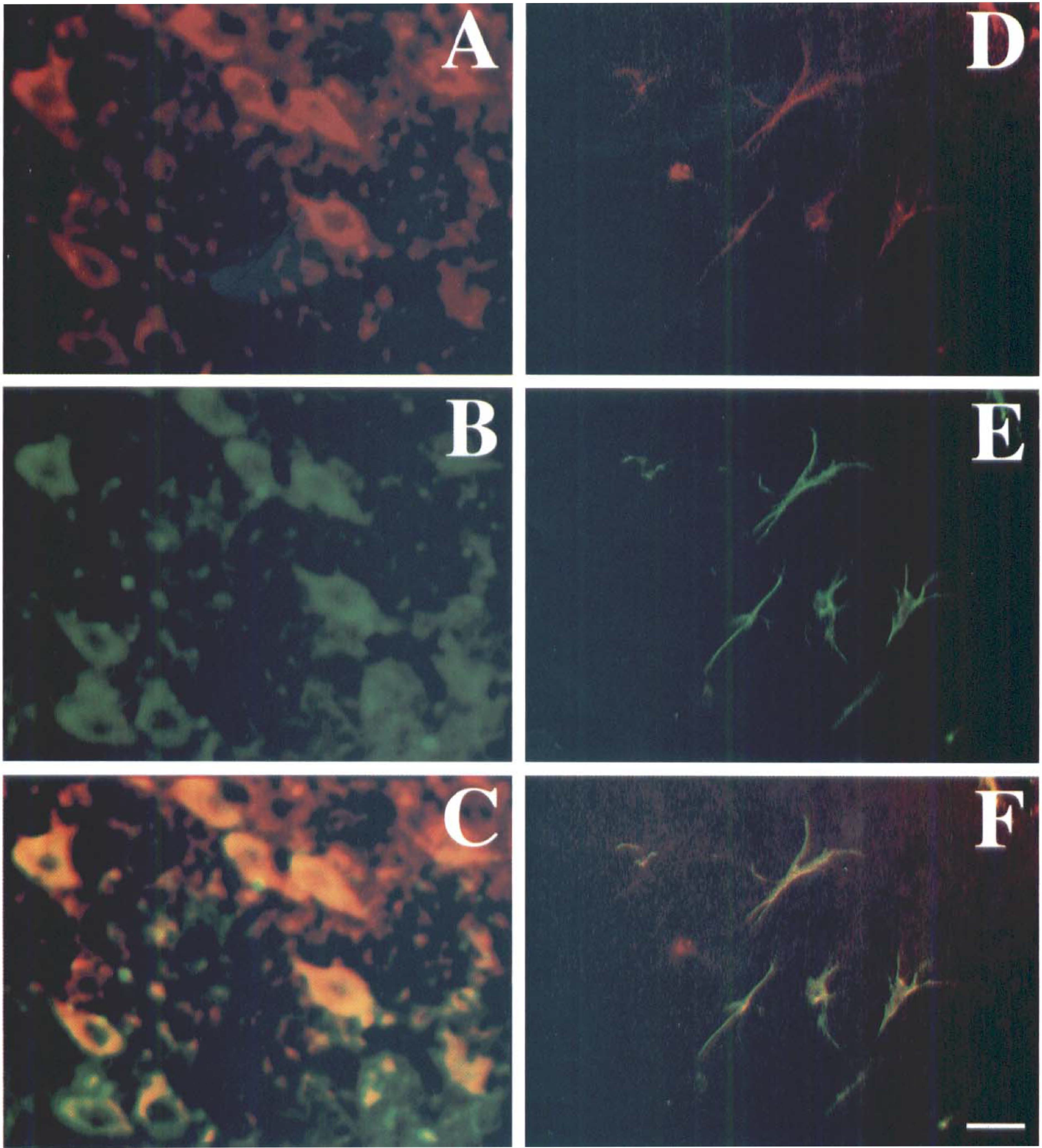
Photomicrographs showing double-labeled immunofluorescence staining in the mouse brain after 2 hours of middle cerebral artery occlusion (MCAO). Phosphorylated ERK1 and ERK2 positive stained cells (
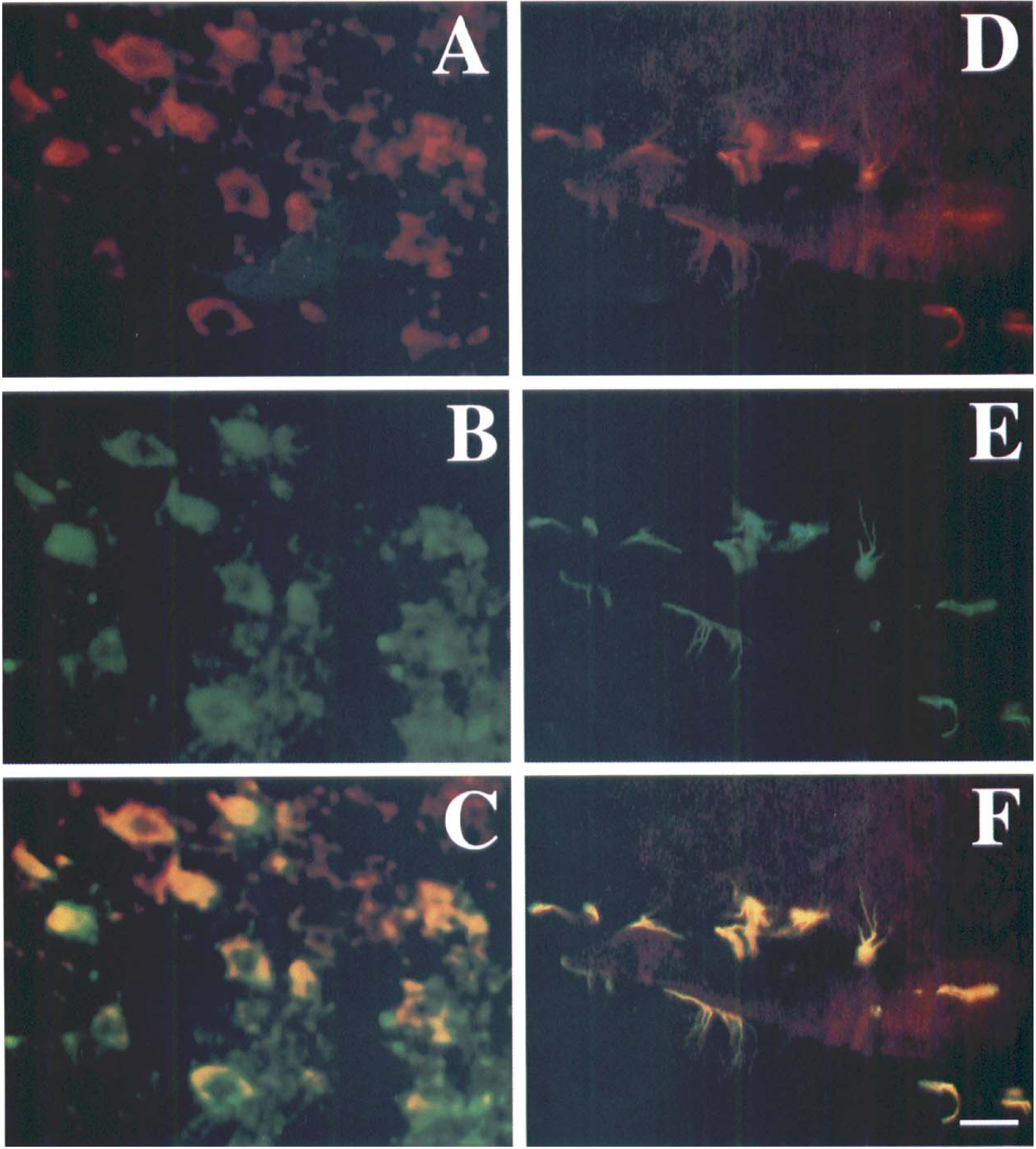
Photomicrographs showing double-labeled immunofluorescence staining in the mouse brain after 20 minutes of middle cerebral artery occlusion (MCAO). Phosphorylated p38 stained cells (
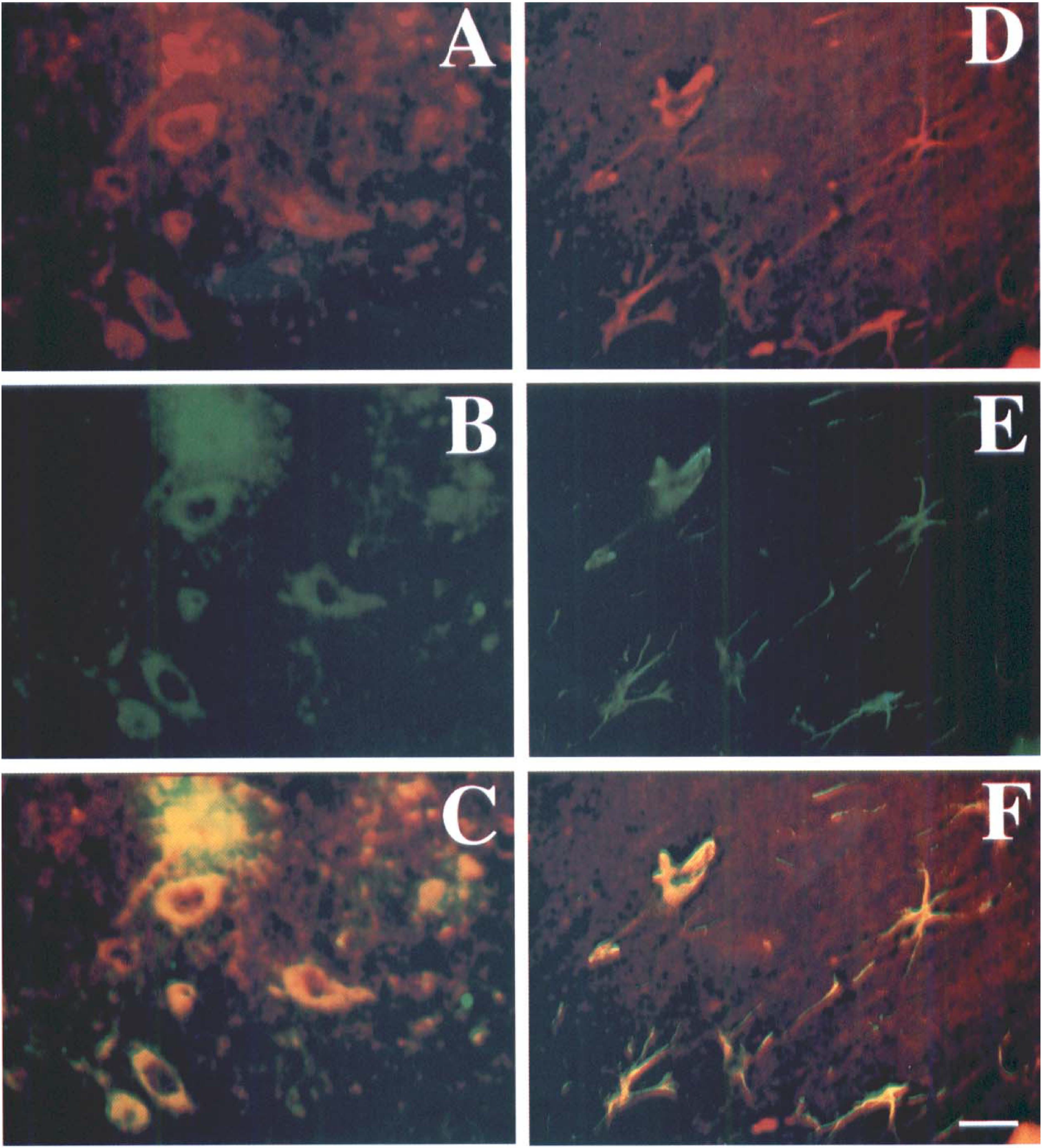
Photomicrographs showing double-labeled immunofluorescence staining in the mouse brain after 30 minutes of middle cerebral artery occlusion (MCAO). Phosphorylated SAPK/JNK positive stained cells (
DISCUSSION
In the current study, the authors observed time courses for ERK1, ERK2, p38, and SAPK/JNK phosphorylation in the mouse brain during MCAO. The duration and magnitude of ERK1, ERK2, p38, and SAPK/JNK phosphorylation were different. SAPK/JNK phosphorylation occurred earlier than that of p38 and ERK1 and ERK2 (5 minutes vs. 10 and 30 minutes). Interestingly, p38 phosphorylation peaked the earliest, whereas SAPK/JNK phosphorylation was the most marked among the three MAPKs. Phosphorylated p38 expression returned to normal levels by 1 hour, whereas ERK1 and ERK2 and SAPK/JNK expression returned to normal by 6 hours. These results suggest that focal cerebral ischemia can induce early short-lasting increases in phosphorylation of all three MAPKs. However, the timing and magnitude of ERK1, ERK2, p38, and SAPK/JNK phosphorylation are distinct.
The relation between the MAPKs pathway and ischemia and reperfusion injury has been examined in studies beginning in the middle 1990s. Pombo et al. (1994) showed that ERK1 and ERK2 (1.5-fold) and SAPK/JNK (5-to 8-fold) were significantly increased in the rat kidney undergoing 40 minutes of ischemia followed by 5, 20, or 90 minutes of reperfusion. Bendinelli et al. (1996) reported that ERK1 and ERK2 and SAPK/JNK were activated in the rat liver while the animal was undergoing 60 minutes of ischemia followed by 30, 60, or 120 minutes of reperfusion. Several other groups then demonstrated that ERK1 and ERK2, SAPK/JNK, or p38 are activated in heart ischemia and reperfusion in vitro and in vivo (Aikawa et al., 1997; Bogoyevitch et al., 1996; Fukunaga and Miyamoto, 1998; Shimizu et al., 1998). However, it is unclear whether or which MAPKs were activated by focal cerebral ischemia.
Walton et al. (1998) first reported the effect of cerebral ischemia on MAPKs. These authors found that p38 and MAP kinase-activated protein 2 (MAPKAP2) activity increased four days after global forebrain ischemia. Immunohistochemistry also demonstrated that p38 and MAPKAP2 are expressed in microglia. Recently, Barone et al. (1999) demonstrated that SB239063, a potent p38 inhibitor, can significantly reduce infarct volume and neurologic deficit in rat after MCAO. Alessandrini et al. (1999) confirmed that PD98059, a specific ERK1 and ERK2 inhibitor, dramatically reduced ischemia and reperfusion brain injury, suggesting a role for this pathway in the initial stages of stroke pathophysiology.
Recently, several upstream kinases responsible for MAPK phosphorylation have been characterized. Mitogen-activated protein kinase family may play three separate but interconnected roles during ischemia. These kinases show specificity for the three subfamilies of MAPK; for example, MKK1 and MKK2 activate ERK1 and ERK2 (Hunter, 1995), MKK3 and MKK6 activate p38, and MKK4 and MKK7 activate SAPK/JNK (Mielke et al., 1999). Upstream regulators of MAPK vary with the extracellular stimulus and the cell type. Transient activation is universal in the SAPK/JNK response to stress (Raingeaud et al., 1995) and is not mediated through a Ras-dependent pathway (Guan et al., 1996; Raingeaud et al., 1995). In contrast, transient ERK activation is associated with a proliferative response (Marshall, 1995) and is activated by growth factors through a Ras-dependent signal-transduction pathway (Egan and Weinberg, 1993; Guan et al., 1996). Interestingly, although the signal pathway of JNK is very similar to p38 in in vitro study (Davis, 1994), the timing and magnitude of JNK activation are distinct from p38 activation in cerebral ischemic mice. These results suggest that all three MAPKs are involved in cerebral ischemia, but through different pathways.
Immunohistochemical studies provided evidence that phospho-specific ERK1, ERK2, p38, and SAPK/JNK expression correlated with the intensity and duration of the results from the Western blot analysis. Double-labeled immunostaining demonstrated that these phospho-specific ERK1, ERK2, p38, and SAPK/JNK positive cells were neurons and astrocytes, and expression occurred predominantly in neuronal cell bodies and extensive dendritic arborizations. In addition, clearly immunopositive staining was seen in the axon hillocks and proximal axons of pyramidal neurons. The presence of ERK1, ERK2, p38, and SAPK/JNK immunoreactivity in neurons and neurofibrils after MCAO suggests that MAPK expressed in ischemic neurons may be delivered through axonal transport.
Knowledge of MAPK phosphorylation presents an attractive pharmacologic opportunity considering its rapid initiation and progression after cerebral ischemia. Proper intervention of the MAPK pathway may be critical for cell survival and may attenuate ischemic infarction. Further studies using specific inhibitory peptides may be needed to clarify the mechanisms of ERK1, ERK2, p38, and SAPK/JNK after ischemia and reperfusion.
Footnotes
Abbreviations used
Acknowledgements
The authors thank Dr. Richard F. Keep, Director of the Crosby Neurosurgical Laboratory, for valuable discussion, and Ms. Kathleen Donahoe for editorial assistance.