
Abstract
A low dose (0.5 mg/kg) of lipopolysaccharide (LPS), administered 72 hours before 60-minute middle cerebral artery occlusion, induced a delayed neuroprotection proven by the significant decrease (–35%) of brain infarct volume in comparison with control, whereas infarct volumes remained unchanged in rats treated 12, 24, or 168 hours before ischemia. This delayed neuroprotective effect of LPS was induced only with low doses (0.25 to 1 mg/kg), whereas this effect disappeared with a higher dose (2 mg/kg). The delayed neuroprotection of LPS was induced in the cortical part of the infarcted zone, not in the subcortical part. The beneficial effect of LPS on consequences of middle cerebral artery occlusion was suppressed by dexamethasone (3 mg/kg) and indomethacin (3 mg/kg) administered 1 hour before LPS, whereas both drugs had no direct effect on infarct volume by themselves, suggesting that activation of inflammatory pathway is involved in the development of LPS-induced brain ischemic tolerance. Preadministration of cycloheximide, an inhibitor of protein synthesis, also blocked LPS-induced brain ischemic tolerance suggesting that a protein synthesis is also necessary as a mediating mechanism. Superoxide dismutase (SOD) could be one of the synthesized proteins because lipopolysaccharide increased SOD brain activity 72 hours, but not 12 hours, after its administration, which paralleled the development of brain ischemic tolerance. In contrast, catalase brain activity remained unchanged after LPS administration. The LPS-induced delayed increase in SOD brain content was suppressed by a previous administration of indomethacin. These data suggest that the delayed neuroprotective effect of low doses of LPS is mediated by an increased synthesis of brain SOD that could be triggered by activation of inflammatory pathway.
There has been recent interest in the possibility that the brain can be protected against the consequences of ischemia by a number of procedures applied a few days previously. Such delayed neuroprotection could be the result of ischemic preconditioning, heat stress, and cortical spreading depression (Chen and Simon, 1997). Recently, it has been demonstrated that the administration of a low dose of bacterial endotoxin (lipopolysaccharide (LPS) derived from Escherichia coli) can reduce the infarct size induced by a permanent middle cerebral artery occlusion performed 2 to 4 days later (Tasaki et al., 1997; Dawson et al., 1999; Ahmed et al., 2000).
Several mechanisms have been evoked in the initiation of ischemic tolerance, such as adenosine A1 receptors, muscarinic M2 receptors, opioid receptors, and K+-ATP dependent channels (Chen and Simon, 1997; Losano et al., 1996). In LPS-induced ischemic tolerance, beneficial effect has been nullified by inhibition of tumor necrosis factor-alpha (TNFα) suggesting that the release of this cytokine could be a trigger for delayed neuroprotection (Tasaki et al., 1997). The finding that cytokines can activate inflammatory processes through prostanoid pathway (Cirino, 1998) suggests that inflammation could also play a role in triggering the cascade of induction of ischemic tolerance induced by LPS and that antiinflammatory drugs might abolish LPS-induced protective effect in brain as demonstrated with dexamethasone in heart preconditioning (Vegh et al., 1992).
The time required for occurrence of ischemic tolerance also implies synthesis of new proteins or enzymes that could be generated by trigger mechanisms of preconditioning. In myocardial and cerebral preconditioning the role of induction of heat shock protein (HSP), antioxidant enzymes, or antiapoptotic proteins have been evoked (Chen and Simon, 1997; Losano et al., 1996). In a previous study the authors failed to find any induction of HSP after LPS administration suggesting that this pathway could not be involved in LPS-induced ischemic tolerance contrasting with preconditioning (Puisieux et al., 2000). Because cytokines and inflammation have been previously demonstrated to induce antioxidant enzymes, this pathway could be involved as a mediating mechanism in the delayed neuroprotective effect of LPS.
In the current study, the authors have investigated the delayed neuroprotective effect of low doses of LPS in an experimental model of cerebral ischemia—reperfusion. The authors studied the role of inflammatory pathway as a triggering mechanism and the role of protein synthesis, specifically antioxidant enzymes, as a mediating mechanism and their relationships.
METHODS
Animals and drugs administration
Lipopolysaccharide derived from Escherichia coli (serotype 055:B5) was administered intraperitoneally (IP) to male Wistar rats (IFFA Credo France), weighing 280 to 320 g, in a dose of 0.5 mg/kg, 12 hours, 24 hours, 72 hours, or 168 hours before cerebral ischemia. Control animals received an equivalent volume (0.5 mL) of 0.9% w/v normal saline 12 hours (n = 2), 24 hours (n = 2), 72 hours (n = 2), or 168 hours (n = 2) before ischemia. A second series of experiments was designed to explore the dose-related protective effect of LPS on infarct size. Lipopolysaccharide (doses of 0.25, 0.5, 1, or 2 mg/kg) or an equivalent volume of saline were administered to animals 72 hours before focal cerebral ischemia. In a third series of experiments, saline (0.5 mL) or LPS (0.5 mg/kg) was administered either alone or 1 hour after injection IP of dexamethasone (3 mg/kg), or indomethacin (3 mg/kg), or vehicle 72 hours before middle cerebral artery occlusion. In a fourth series of experiments, saline (0.5 mL) or LPS (0.5 mg/kg) was administered either alone or 1 hour after injection of cycloheximide (1 mg/kg, IP) or vehicle. Superoxide dismutase (SOD) and catalase in brain were measured 12 hours or 72 hours after administration of LPS 0.5 mg/kg or an equivalent volume of saline. In additional groups, SOD in the brain was measured 72 hours after LPS (0.5 mg/kg) or saline administration injected 1 hour after an administration of indomethacin (3 mg/kg). In all experiments, 1 mg LPS represented 1.45.106 UI of endotoxin. All experiments were performed in strict accordance with the guidelines of the National Institutes of Health and French Department of Agriculture.
Middle cerebral artery occlusion method
Anesthesia was induced with chloral hydrate administered IP at a dose of 300 mg/kg. A rectal probe was inserted and core temperature maintained by the use of a heating pad and a heating lamp at 37 ± 0.5°C. The caudal artery was exposed and cannulated with a 24G polyethylene catheter and connected to a blood pressure monitor. Mean arterial blood pressure (mm Hg) was monitored throughout the experiment and blood samples were taken before, during, and after ischemia to measure blood pH, arterial Pa
The ostium of the right middle cerebral artery (MCA) was occluded intraluminally with a method previously described (Bastide et al., 1999). The right carotid arteries were exposed through a midline cervical incision and the common carotid and external carotid arteries were ligated with a silk suture. The pterygopalatine artery was exposed by developing a plane alongside the internal carotid artery, and was ligated at its origin with a fine silk. Aneurysm clip was placed across internal carotid artery and an arteriotomy was made in the common carotid artery stump allowing the introduction of a 4/0 monofilament nylon suture with its tip rounded by flame heating. This was secured in place and the aneurysm clip on the internal carotid artery was removed. The suture was gently advanced into the internal carotid artery and passed into the intracranial circulation to lodge in the narrower lumen of the proximal anterior cerebral artery (20 to 22 mm distal to the carotid bifurcation) thereby occluding the origin of the MCA. Mild resistance to this advancement indicated that the intraluminal occluder had entered the anterior cerebral artery. After 60 minutes the suture was carefully removed until its tip was blocked by ligature placed on common carotid artery to permit reperfusion. The caudal arterial catheter was removed and the artery was ligated to prevent bleeding. The animals were placed in cages to recover from anesthesia at room temperature and were allowed to eat and drink freely.
Histology and measurement of infarct size
Twenty-four hours after reperfusion, the rats were killed by an overdose of pentobarbital injected IP, and brains were rapidly removed, frozen, and coronally sectioned into 50-μm-thick slices on a cryostat at 12 levels separated by 1-mm intervals, according to stereotactic sections maps (Paxinos and Watson, 1986). Sections were stained with Cresyl Fast Violet. The unstained area of the brain sections was defined as infarcted. Cortical and subcortical uncorrected infarcted areas and total hemispheric areas were calculated separately for each coronal slice by an image analysis software (Color Image 1.32) after digitization by scanner process. Total, cortical, and subcortical infarct volumes and hemispheric volumes (mm3) were calculated by the use of numerical integration of the respective areas for all the sections per animals and the distance between them. A corrected total infarct volume was calculated to compensate for the effect of brain edema (Bastide et al., 1999). The corrected volume was calculated using the following equation: corrected infarct volume = infarct volume − (right hemisphere volume − left hemisphere volume).
Measurement of cerebral antioxidant enzymes
Brain (30 g) was rapidly removed, pounded, crushed, and homogenized in a saline solution (7 mL).
Measurement of SOD
The inhibition of the autooxidation of pyrogallol by SOD was measured at 420 nm, comparatively to a calibration curve, made with known amounts of the enzyme. Results were expressed in mg/mL of homogenate (Marklund and Marklund, 1974).
Measurement of catalase
Homogenates were extracted by mixing in 10 vol of 0.25% sodium cholate (Sigma-Aldrich, Saint Quentin Fallavier, France). After centrifugation at 4000 × g for 10 minutes at 4°C the supernatants were frozen at −80°C until use. The following dilutions of extracts were made with phosphate buffer, pH 7.0 (1:20 to 1:40) immediately before assay. Catalase activity was assessed in diluted extracts by measuring the rate of reduction of H2O2 substrate, followed spectrophotometrically at 240 nm on a UV/visible spectrophotometer, Lambda 20 (Perkin-Elmer, Courtaboeuf, France). One unit of catalase equals 1 μmol H2O2 decomposed by 1 minute at 25°C. The protein was determined in the extracts using a Pierce kit (Rockford, IL, U.S.A). Standard bovine serum albumin was made up in TBS (pH 7.6) giving a concentration range 0.2 to 2.0 mg/mL (Aebi, 1984).
Statistical analysis
All data are expressed as mean ± SD. Continuous variables (mean arterial blood pressure, blood gases, infarct volumes, SOD, and catalase dosages) were compared with a one-way analysis of variance, then followed by a post hoc protected least significant difference Fisher test if analysis of variance was significant. A value of P < 0.05 was considered significant.
RESULTS
Time course of ischemic tolerance induced by bacterial endotoxin
After LPS (0.5 mg/kg), administered 12, 24, 72, or 168 hours before MCA occlusion, there were no statistically significant differences before and during ischemia in physiologic parameters compared with the saline-treated group. Pretreatment with LPS (0.5 mg/kg, administered IP 72 hours before MCA occlusion) resulted in a significant decrease of 35% in the infarct volume (Fig. 1), in comparison with the infarct volume of the saline-treated group. The infarct volumes resulting from MCA occlusion performed 12, 24, or 168 hours after pretreatment with LPS (0.5 mg/kg) were not significantly different in comparison with the saline-treated group.
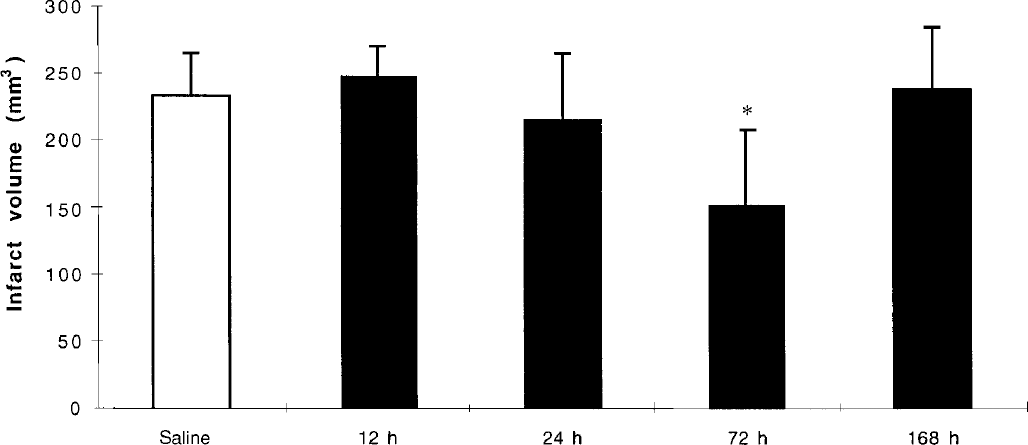
Time-dependence of the effect of lipopolysaccharide (LPS) on total infarct volumes (corrected for edema) induced by 60-minute focal cerebral ischemia. Injections of LPS 0.5 mg/kg were given intraperitoneally 12 hours (n = 5), 24 hours (n = 6), 72 hours (n = 6), or 168 hours (n = 6) before cerebral ischemia. Comparison versus saline-treated group (n = 8; 4 hours, n = 2; 24 hours, n = 2; 72 hours, n = 2; 168 hours, n = 2). Values are mean ± SD. *P < 0.01 as compared with saline.
Dose-related ischemic tolerance induced by bacterial endotoxin
There were no statistically significant differences, before and during ischemia, in physiologic parameters in rats receiving different doses of LPS in comparison with the saline-treated group. The decrease in total infarct volume by 48% was significantly maximal in rats receiving 0.25 mg/kg LPS (Fig. 2). In doses of 0.5 and 1 mg/kg, the infarct volumes were also significantly (P < 0.05) decreased with LPS, whereas in a dose of 2 mg/kg the infarct was not significantly different from the volume in saline-treated rats (Fig. 2). Delayed neuroprotective effect induced by the different doses of LPS was observed in the cortical part of the infarcted zone, whereas the subcortical part of infarct remained unchanged under the different conditions (Fig. 2).
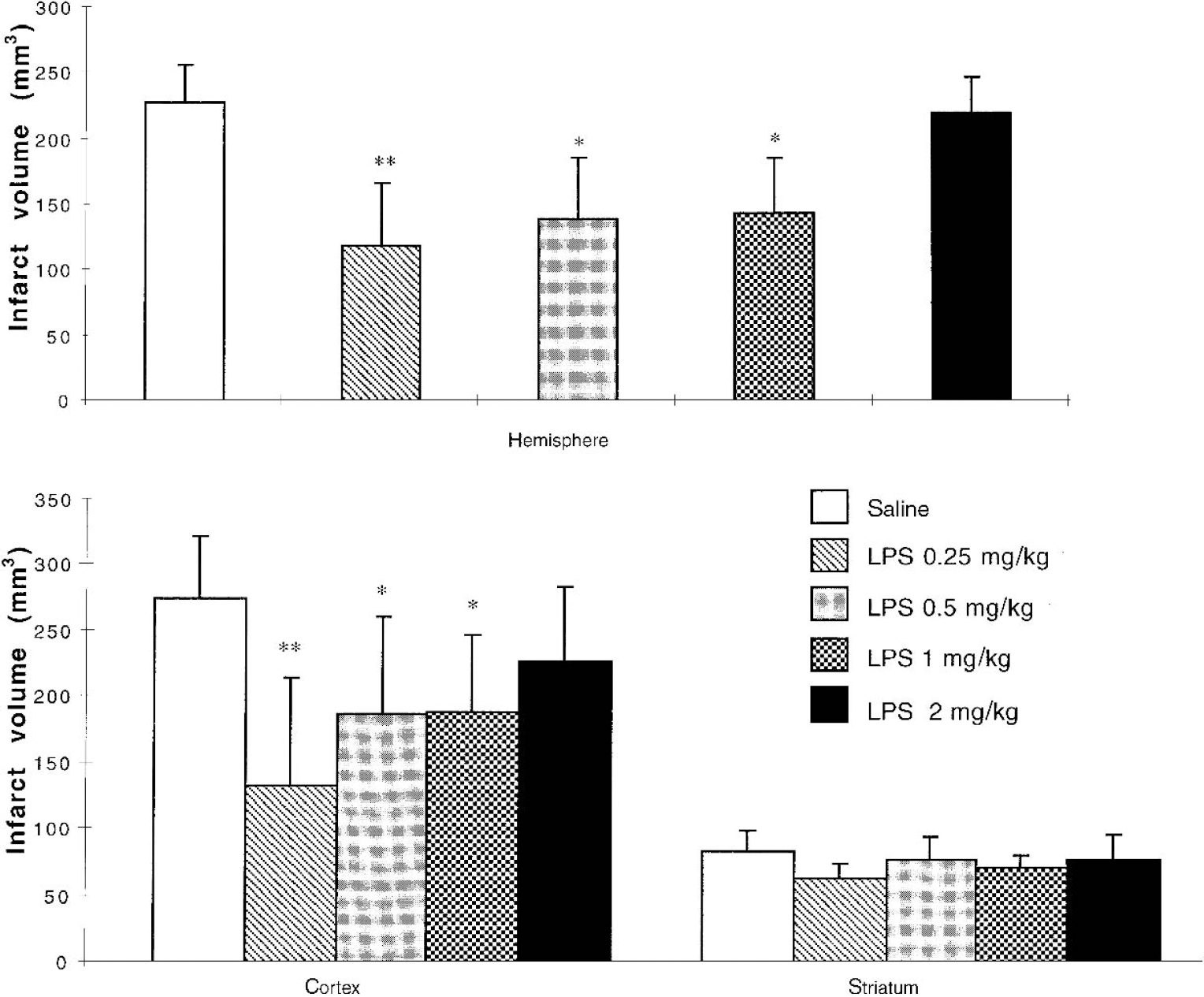
Dose-dependence of the effects of lipopolysaccharide (LPS) on infarct volumes (total hemispheric, cortical, and striatal infarcts) induced by 60-minute focal cerebral ischemia. Injections were given intraperitoneally 72 hours before cerebral ischemia: saline (n = 6), LPS 0.25 mg/kg (n = 7), LPS 0.5 mg/kg (n = 6), LPS 1 mg/kg (n = 5), LPS 2 mg/kg (n = 5). Total hemispheric infarct volumes were corrected for edema. Cortical and striatal parts of infarct volumes were not corrected for edema. Values are mean ± SD. *P < 0.05; **P < 0.01 as compared with saline.
Effects of dexamethasone and indomethacin on ischemic tolerance induced by bacterial endotoxin
Dexamethasone (3 mg/kg) or indomethacin (3 mg/kg) administered alone, 72 hours previously to MCA occlusion, had no significant effect on infarct volume or on physiologic parameters (Fig. 3). The weak decrease in infarct volume after dexamethasone administration remained insignificant. In LPS-treated animals, before administration of dexamethasone or indomethacin had no effect on physiologic parameters. Infarct volumes in rats receiving LPS associated with a previous administration of dexamethasone or indomethacin were significantly (P < 0.05) greater than in rats challenged with LPS alone. Infarct volumes in these two groups were not statistically different in comparison with infarct volume in the saline-treated group (Fig. 3).
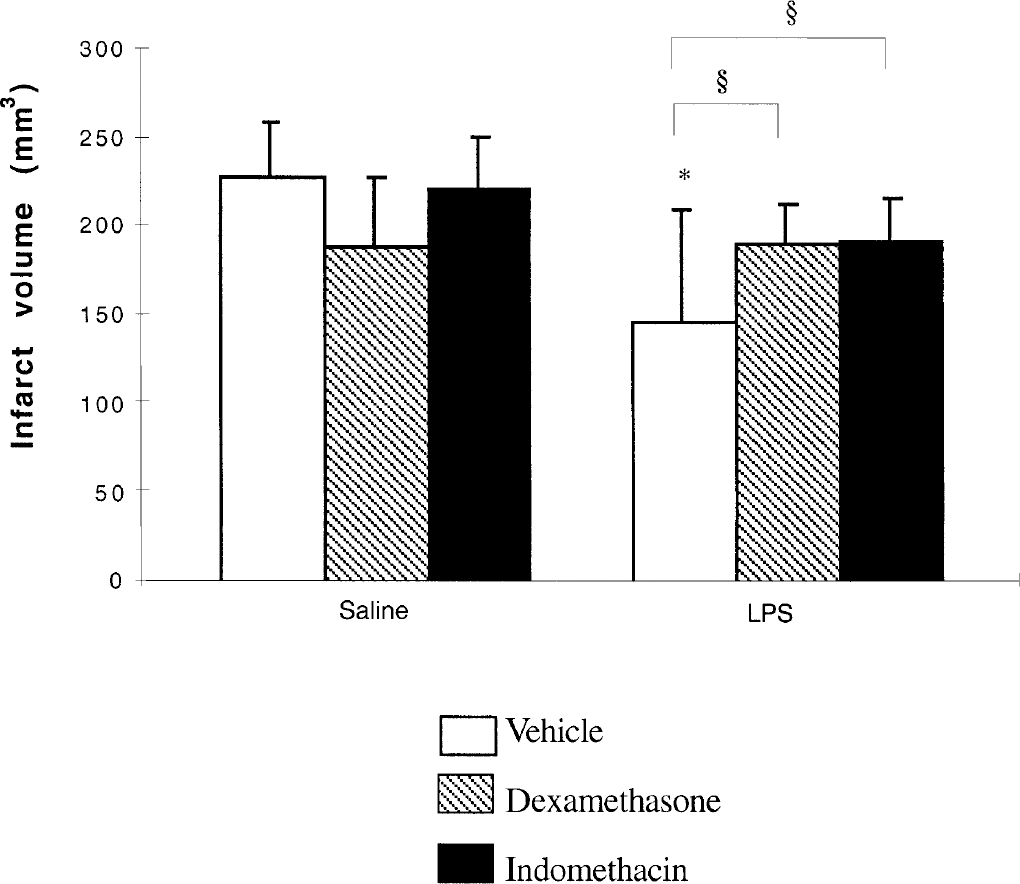
Effect of dexamethasone or indomethacin on delayed neuroprotection induced by lipopolysaccharide (LPS). Vehicle, dexamethasone (3 mg/kg), or indomethacin (3 mg/kg) were intraperitoneally injected 1 hour before administration of saline (0.5 mL) or LPS (0.5 mg/kg), 72 hours before cerebral ischemia. Values are mean ± SD (n = 5 to 7 per group). *P < 0.05 as compared with vehicle + saline; §P < 0.05 as compared with vehicle + LPS.
Effects of cycloheximide on ischemic tolerance induced by LPS
In all conditions, physiologic parameters remained unchanged before and during ischemia. Cycloheximide administered alone, 72 hours previously to MCA occlusion, had no significant effect on infarct volume (Fig. 4). Infarct volumes in rats receiving LPS associated with a previous administration of cycloheximide were significantly greater than in rats challenged with LPS alone, but were not different from infarct volumes in saline-treated rats.
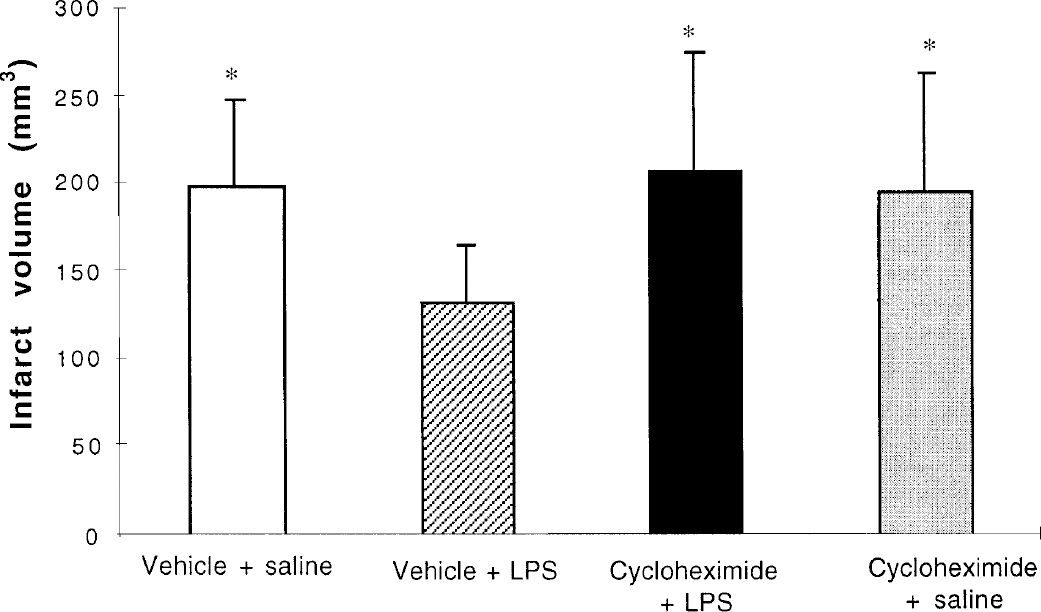
Effect of cycloheximide on delayed neuroprotection induced by lipopolysaccharide (LPS). Vehicle or cycloheximide (1 mg/kg; IP) were intraperitoneally injected 1 hour before administration of saline or LPS (0.5 mg/kg), 72 hours before cerebral ischemia. Values are mean ± SD (n = 6 to 7 per group). *P <0.01 as compared with vehicle + LPS.
Antioxidant enzymes measurement
Superoxide dismutase brain activity was significantly increased 72 hours after LPS administration, whereas activity remained unchanged 12 hours after LPS administration compared with activity in control rats (Fig. 5). The increase in SOD brain activity after LPS administration was significantly abolished by a previous administration of indomethacin, whereas administration of indomethacin alone had no effect on SOD brain activity. In contrast, catalase activity remained unchanged in brain after lipopolysaccharide administration as compared with control (Fig. 5).
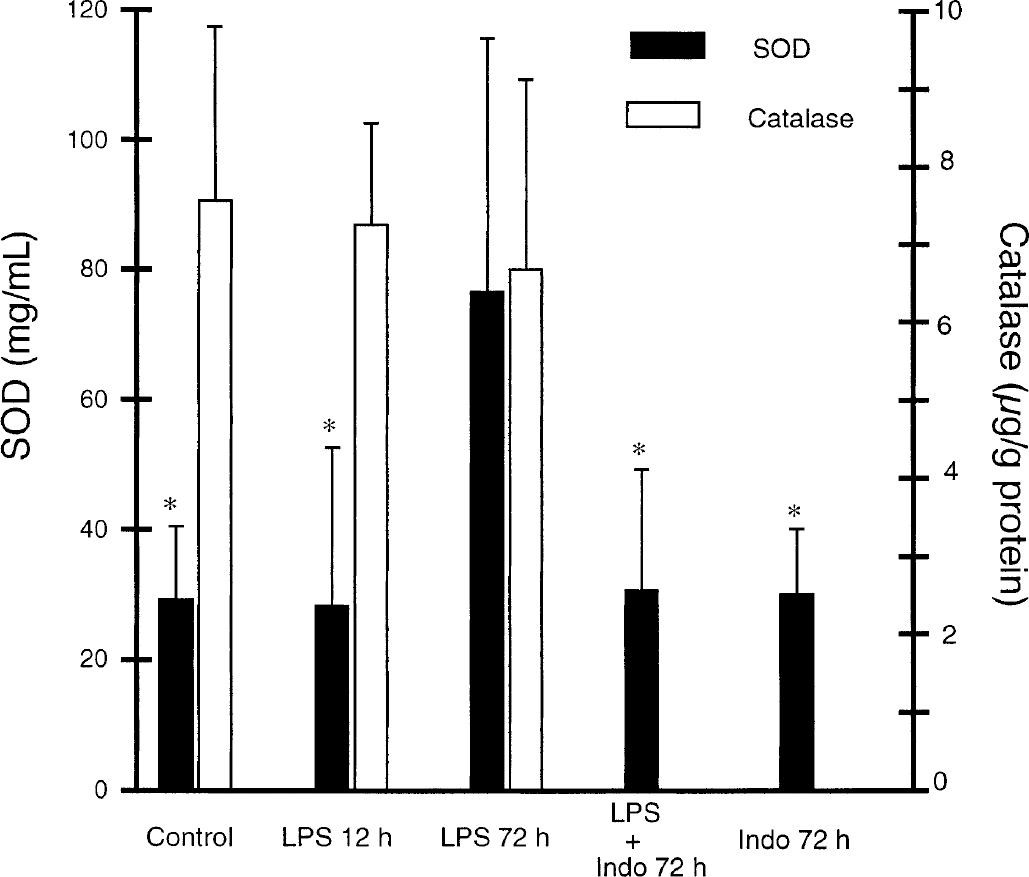
Effect of lipopolysaccharide (LPS) on superoxide dismutase (SOD) and catalase brain content. Vehicle or LPS (0.5 mg/kg) were administered, with or without simultaneous administration of indomethacin (Indo) (3 mg/kg), 12 or 72 hours before brain SOD or catalase activity measurement. All animals receiving vehicle alone (12 or 72 hours before measurement) were pooled to constitute a control group. Values are mean ± SD (n = 6 to 8 per group). *P < 0.01 as compared with LPS at 72 hours.
DISCUSSION
These results demonstrate that the previous administration of a low dose of LPS is effective in reducing the consequences of MCA occlusion, in terms of infarct size. Several studies have demonstrated that LPS protects the heart against various consequences of ischemia—reperfusion injury, whereas such an effect has been reported more recently in the brain with a model of permanent focal ischemia (Tasaki et al., 1997; Dawson et al., 1999). Using another focal model with reperfusion, we confirm here that a delay of three days is necessary to observe such a protection. There is no protective effect when LPS is administered one day or seven days before ischemia. Moreover, our results also support the absence of deleterious effects of LPS at the used doses as shown, particularly the absence of infarct size increase 12 hours after LPS administration, despite its inflammatory effect in brain (Ahmed et al., 2000). The time course of ischemic tolerance appears to be different in the brain and in the heart for LPS and preconditioning (Chen and Simon, 1997; Parratt and Szekeres, 1995). In the heart there is an early window and a delayed second window of protection, whereas in brain there is only a delayed window of protection induced by LPS or preconditioning, suggesting that the mechanisms could not be totally similar.
Delayed neuroprotective effect induced by LPS has been obtained with doses 5- to 20-fold lower than those required to induce septic shock (Liu et al., 1996). The dose used in the first report of LPS-induced brain ischemic tolerance was 0.9 mg/kg, a dose considered as optimal after a series of preliminary dose-finding studies (Tasaki et al., 1997). Delayed neuroprotection has been reported with a dose 5- to 10-fold smaller than the dose we used (Ahmed et al., 2000), suggesting that a very weak activation of biochemial targets of LPS is necessary to induce ischemic tolerance. In contrast, our data prove that brain ischemic tolerance disappears when the doses are increased suggesting that the mechanisms of neuroprotection may become deleterious when their activation levels are greater or other deleterious pathways are activated. The neuroprotection afforded by LPS has been demonstrated to be mediated by cytokine release, in particular by TNFα (Tasaki et al., 1997). These protective effects of TNFα are in contrast to the detrimental effects of TNFα synthesized during ischemia (Barone et al., 1997). The level of activation of TNFα may be important to determine deleterious or beneficial effect contributing to the dose-related protective effect of LPS in brain.
Because maximal induction of TNFα protein is observed 7 hours after administration of LPS, when there is no ischemic tolerance (Buttini et al., 1997), TNFα may be only a trigger for ischemic tolerance inducing a cascade of events leading to protection and not the effector of the protection (Liu et al., 2000). The inhibition of LPS-induced ischemic tolerance by prior injection of dexamethasone could be explained by the inhibition of TNFα released in brain because glucocorticoïds, such as methylprednisolone, are recognized to inhibit LPS-induced TNFα in the rat brain (Buttini et al., 1997). Another triggering mechanism could be the LPS-induced activation of the prostanoid pathway, which is also inhibited by dexamethasone (Masferrer et al., 1992). In the current study, the effect of indomethacin, a cyclooxygenase inhibitor, supports the hypothesis of involvement of prostanoids. There is also good evidence that prostanoïds, such as prostacyclin, induce a delayed cardioprotective effect, explaining why prostacyclin has been considered as an endogenous myocardial protective substance (Parratt and Szekeres, 1995). In brain, it has been also found that prostacyclin analogs may have a neuroprotective effect in the acute phase of ischemia, whereas a potential delayed effect is not demonstrated (Dogan et al., 1996).
The time frame required for induction of ischemic tolerance in brain and its persistence over days also suggests new protein synthesis. Inhibition of LPS-induced brain ischemic tolerance by cycloheximide, a protein synthesis inhibitor, favors this hypothesis. Such an inhibition has not been yet demonstrated in LPS-induced brain ischemic tolerance although it has been demonstrated for cerebral ischemic preconditioning (Barone et al., 1998) or in 3-nitroproprionic acid-induced brain ischemic tolerance (Wiegand et al., 1999). Although in both preconditioning and LPS-induced brain ischemic tolerance protein synthesis is involved, the proteins in cause are not obligatory the same in the two models. In a previous study, we failed to demonstrate a delayed heat shock protein synthesis after LPS administration suggesting that, in contrast with preconditioning, this pathway was not involved in mediating mechanisms of LPS-induced ischemic tolerance (Puisieux et al., 2000). Our data here favor involvement of enhanced endogenous antioxidant enzymes as proven by the increase in SOD brain activity parallel to the neuroprotective effect of LPS. Such an involvement of SOD has been previously demonstrated in brain preconditioning (Ohtsuki et al., 1992; Kato et al., 1995). As in brain preconditioning, LPS could increase the activity of manganese SOD, the inducible form of SOD (Kato et al., 1995). In another model of brain ischemic tolerance induced by administration of 3-nitroproprionic acid, the authors were unsuccessful in showing an increased activity of SOD (Wiegand et al., 1999). Lipopolysaccharide has been recently found to induce delayed cardioprotection and an increase in SOD mRNA expression in the same dose that was used in the current study (Meng et al., 1998). The monophosphoryl lipid A, the nontoxic fraction of LPS, is also responsible for parallel delayed cardioprotection and an increase in SOD activity (Yamashita et al., 1999). Lipopolysaccharide is also responsible for induction of SOD in cultured neuronal and glial cells (Kifle et al., 1996; Yu et al., 1999). The overexpression of SOD could contribute, by a scavenging process, to decrease the deleterious effect of the free radicals on neurons during ischemia—reperfusion. In addition, of its neuronal role in brain tolerance induction, SOD could also exert a vascular role. It has been recently demonstrated that the monophosphoryl lipid A induced a protective effect against vasospasm after subarachnoid hemorrhage parallel to an increase of SOD activity, which could contribute to preventing the endothelium-dependent dysfunction likely responsible for vasospasm occurrence (Toyoda et al., 1998). The ability of LPS to increase SOD expression could in part explain the delayed endothelium-dependent relaxation improvement that we have recently demonstrated and that could contribute to LPS antiischemic properties (Pu et al., 1999). The dissociate effect of LPS on SOD and catalase brain content remains unclear but has been yet reported in other conditions (Steeves et al., 1994).
Our data also suggest that the inflammatory processes (cytokines and prostanoids release) initiated by LPS are responsible for the delayed increase of antioxidant enzyme activity. To our knowledge, such an interaction between the two pathways has not yet been reported in brain. Nevertheless, it has been previously demonstrated that cytokines, such as TNFα or IL-1, induced, in a delayed manner (72 hours), SOD expression in lung, which explains the protective effects of these cytokines against pulmonary oxygen toxicity (White et al., 1989). Moreover, the cytokine-induced overexpression of SOD was suppressed by cyclooxygenase inhibitors in lung. Such an increase in expression of SOD was also reproduced by endotoxin administration, which explains its pulmonary protective effect (Lewis-Molock, 1994). Inflammatory cytokines are also able to induce SOD in corpus luteum that may play an important role in protecting luteal cells from inflammation-mediated oxidative damage (Sugino et al., 1998). Nitric oxide, which also has been involved as a triggering mechanism of LPS-induced brain ischemic tolerance (Puisieux et al., 2000) could also induce the increase in SOD brain content because nitric oxide donors have been previously reported to up-regulate SOD expression (Sugaya et al., 1997). A transient burst of oxygen free radicals, which has been involved as a trigger in 3-nitroproprionic acid-induced ischemic tolerance (Wiegand et al., 1999) could also be responsible for an up-regulation of antioxidant enzymes while this hypothesis remains to be demonstrated.
In conclusion, our data demonstrate that low doses of LPS induce ischemic tolerance in brain, with a maximum three days after its administration. This delayed neuroprotective effect is abolished by previous administration of substances with properties that inhibit cytokine release and the prostanoid pathway suggesting that inflammatory processes could be involved as triggering mechanisms in brain ischemic tolerance. This delayed neuroprotective effect also parallels an LPS-induced increase in synthesis of brain SOD suggesting that enhanced endogenous antioxidant activity could be one of the mechanisms mediating the neuroprotection. The suppression of LPS-induced increase in antioxidant enzymes by blockade of the prostanoid pathway suggests a possible direct relationship between triggering and mediating mechanisms. All of these pathways are potential pharmacologic targets in the prevention of deleterious effects of brain ischemia.