
Abstract
Though the ischemic penumbra has been classically described on the basis of blood flow and physiologic parameters, a variety of ischemic penumbras can be described in molecular terms. Apoptosis-related genes induced after focal ischemia may contribute to cell death in the core and the selective cell death adjacent to an infarct. The HSP70 heat shock protein is induced in glia at the edges of an infarct and in neurons often at some distance from the infarct. HSP70 proteins are induced in cells in response to denatured proteins that occur as a result of temporary energy failure. Hypoxia-inducible factor (HIF) is also induced after focal ischemia in regions that can extend beyond the HSP70 induction. The region of HIF induction is proposed to represent the areas of decreased cerebral blood flow and decreased oxygen delivery. Immediate early genes are induced in cortex, hippocampus, thalamus, and other brain regions. These distant changes in gene expression occur because of ischemia-induced spreading depression or depolarization and could contribute to plastic changes in brain after stroke.
On the eve of knowing the sequence of the mouse and human genomes, the prospects for this information helping to diagnose and treat stroke and other polygenic neurological disorders has enormous potential. This is not meant to be a review of gene regulation following ischemia. Rather, it is an attempt to show how specific changes of gene expression may be used to infer mechanisms of injury or recovery after stroke that might lead to novel therapy.
Gene induction in brain, particularly stroke, cannot be studied in isolation. That is, the spatial, temporal, and cellular basis for the changes of expression must be known before speculations regarding therapeutic potential can be addressed. For example, genes induced after temporary ischemia in brain might reflect the prominent role of free radicals and oxidative stress (Chan, 1994), whereas the same genes might play a less important role in permanent arterial occlusion (Chan et al., 1993). Genes induced in inflammatory cells in the core of an infarct have different implications for mechanisms of injury and stroke therapy than do genes induced in neurons outside an infarct during the same time periods (del Zoppo, 1997; Dirnagl et al., 1999). Genes induced days after ischemia may be related to plasticity and recovery rather than to damage.
This paper could have been titled multiple molecular, spatial, temporal, and cellular penumbras after focal ischemia. Every gene in every cell can vary spatially and temporally with varying degrees of ischemia, making any representation a tremendous oversimplification (Fig. 1). Figure 1 attempts to condense large amounts of data. It is important to devise strategies to look at the most important genes because the changes of the majority of genes after stroke probably do not mediate either injury or protection. At this point it is difficult to know what the most important genes are. We refer to genes that are either the most familiar, or that have been the most studied, with the realization that much still needs to be learned and that many important genes for understanding the pathogenesis of stroke have yet to be identified.
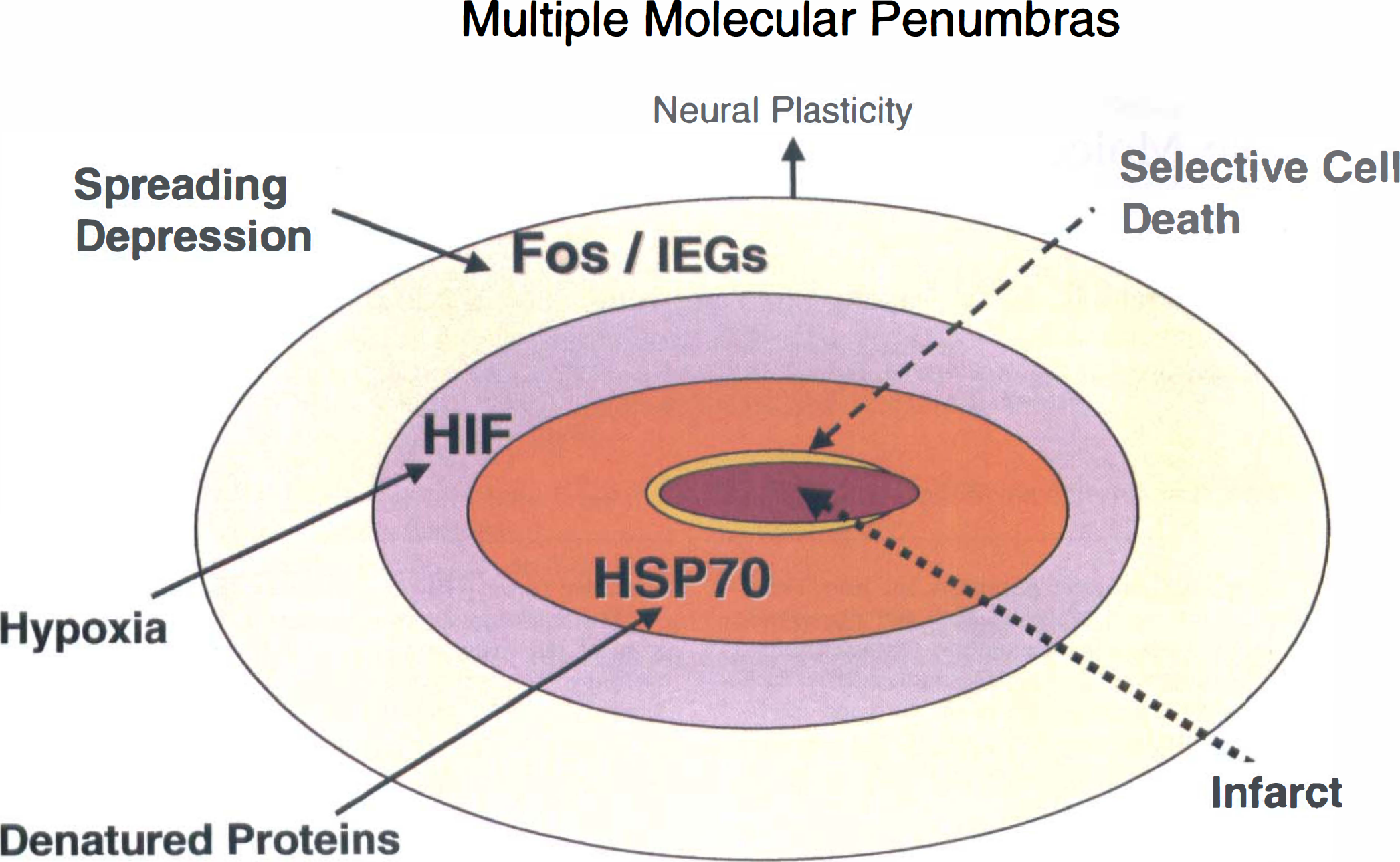
Schematic of multiple molecular penumbras after a stroke. A zone of selective neuronal cell death borders the infarct. The zone of protein denaturation extends outside of this and is demarcated by HSP70 protein expression in injured neurons. Hypoxia inducible factor is induced in areas where blood flow is persistently decreased and oxygen delivery is impaired. This may be co-incident with HSP70 or extend over more widespread regions depending on collaterals. Ischemia-induced spreading depression induces c-fos and many other immediate early genes at some distances from the infarct, including the ipsilateral rat occipital and frontal lobes, contralateral cortex and many subcortical structures.
It was often difficult to determine where most genes were induced after a stroke. The imprecise descriptions in most studies as to which cells in which brain regions express a gene hampers the interpretation of the factors that might induce these genes. We have made some assumptions about where each set of genes is induced after ischemia in order to compare this presumptive data with more precise information on gene induction in different brain regions. Genes that have well described mechanisms of induction through oxygen, free radicals, denatured proteins, pH, and so on can provide molecular and biochemical insights into the injury. Though there are assumptions about how the genes may be induced in the intact brain, the inferences may be useful.
Though mRNA is frequently studied, in terms of effectors that mediate injury, it is essential that protein expression, or protein function (enzymatic activity and so on), be examined. Hence, if a gene is believed to mediate injury after stroke, its protein must be expressed. Because most proteins are not expressed in the core of a stroke, it is important to examine protein expression in the core, at the margins, and at some distance from the strokes.
Relatively few genes are discussed because of the enormity of the subject. Global ischemia was purposely excluded in order to focus on focal ischemia penumbras. However, there are broad reviews available on gene regulation after cerebral ischemia (Chan, 1994; Dirnagl et al., 1999; Koistinaho and Hokfelt, 1997; Kogure and Kato, 1993; Massa et al., 1996; del Zoppo, 1997; Chen and Simon, 1997; Feuerstein et al., 1997; Nowak and Jacewicz, 1994; Nowak, 1999). The speed and widespread availability of information today will cause this review to be out of date even before publication; however, we hope the general ideas will prove useful.
INFARCT CORE - PROTEIN SYNTHESIS
A decrease or block of protein synthesis is one of the first biochemical changes to occur after focal cerebral ischemia (Fig. 2). This occurs when blood flow decreases approximately 50% (Mies et al., 1991). A decrease in ATP is not the signal for a block in protein synthesis because ATP does not decrease until flow falls to 20% of the normal level (Mies et al., 1991; Jacewicz et al., 1986). Ribosomal protein synthesis appears to be the sensitive step that responds to this reduced blood flow, occurring because of in inactivation of initiation factor 2 (eIF2), guanine nucleotide exchange factor (eIF-2-GTP complex factor), and eukaryotic elongation factor (eEF-2) (Marin et al., 1997; Massa et al., 1996). Glutamate-dependent phosphorylation of eEF-2 (Marin et al., 1997) provides a direct link between ischemia-induced increases of extracellular glutamate and ischemia-induced inhibition of protein synthesis (Fig. 2). Phosphorylation of eIF-2 by Protein Kinase R also provides a control point in protein synthesis that is sensitive to oxygen and/or adenosine monophosphate (Srivastava et al., 1998).
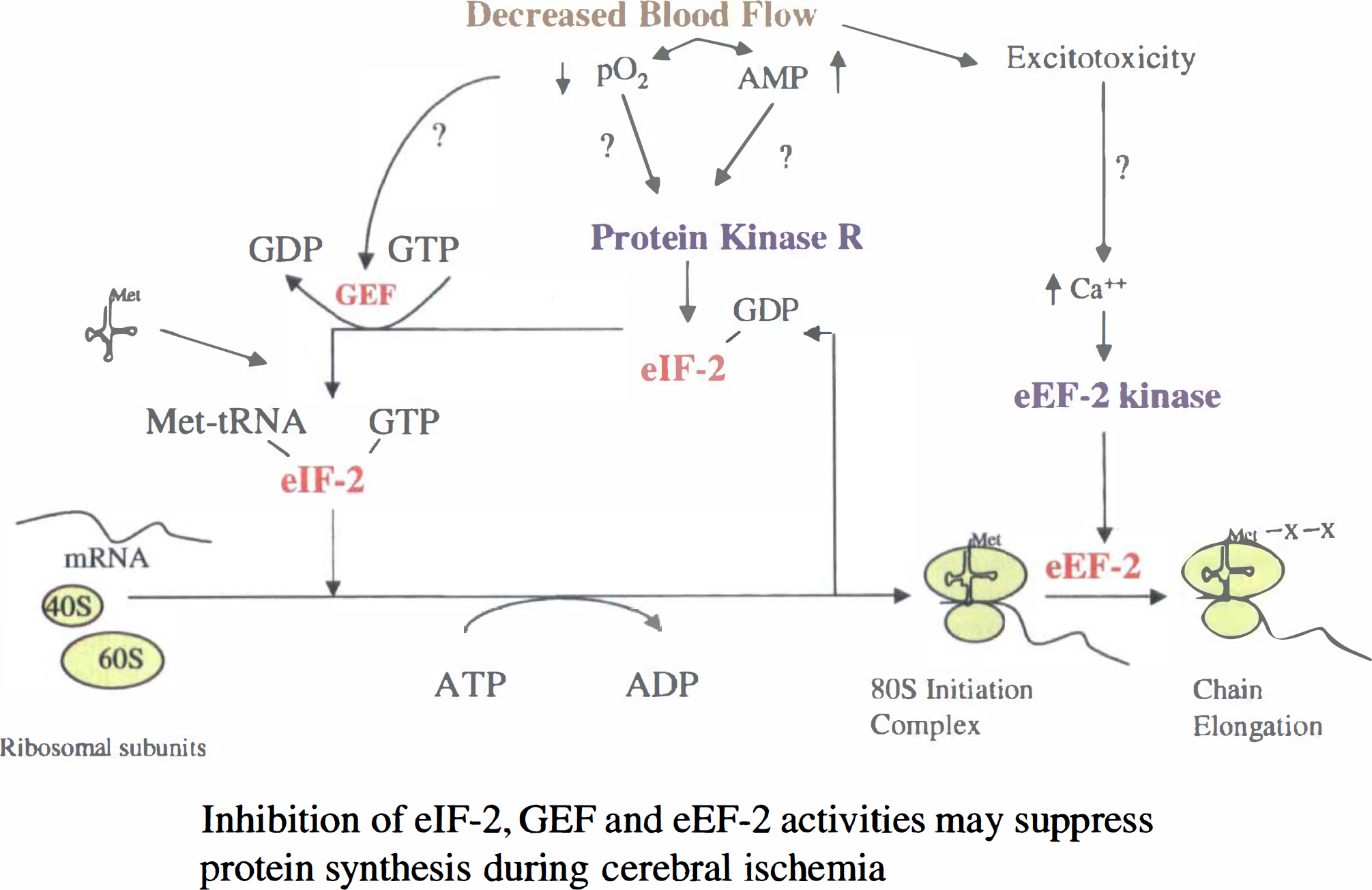
Protein synthesis on the ribosomes can be completely suppressed during focal ischemia through inhibition of several elongation factors, including eIF-2, GEF, and eEF. EIF-2 phosphorylation by PKR is affected by increases of adenosine monophosphate and decreased oxygen that occur during ischemia. Increased concentrations of glutamate released during ischemia can directly down-regulate eEF-2 kinase that regulates eEF-2 function during ischemia.
There is little or no synthesis of new proteins in neurons or astrocytes in the “core” of a cerebral infarct (Kleihues and Hossmann, 1971; Cooper et al., 1977; Mies et al., 1990, 1991; Hossmann, 1993, 1994). Most cerebral ischemia scientists use a change of histologic staining—hematoxylin and eosin, Nissl, or mitochondrial staining—to define the edges and volumes of infarction. Staining proteins by immunocytochemistry and Western blots within the core of an infarction is a function of the half-life of a protein. Proteins with short half-lives will disappear rapidly, without synthesis or rapid degradation. Because of their slow degradation proteins with long half-lives will continue to be detected long after histologic evidence of tissue infarction.
Protein synthesis continues in cells that survive an infarct. In some infarcts some or all of the blood vessels survive. When this occurs, protein synthesis can continue within blood vessels. For example, heat shock protein 70 (HSP70) continues to be expressed in blood vessels in an infarct (Gonzalez et al., 1989, 1991; Kinouchi et al., 1993a, b ), as well as iNOS, eNOS (Iadecola et al., 1996) and many other genes. Importantly, there may be expression of cell adhesion molecules, cytokines, and chemokines by vascular cells within infarcts and at the margins of infarcts. Finally, inflammatory cells inside of infarcts, including neutrophils and macrophages, mount a specific genomic response to the dying and dead neurons and glia.
CORE - ADHESION MOLECULES, CYTOKINES, CHEMOKINES
Intercellular adhesion molecule-1 (ICAM-1) is expressed by vessels in the core of the infarction and at the edges of an infarction (Yang et al., 1999c). ICAM-1 mRNA and endothelial leukocyte adhesion molecule-1 (ELAM-1) and selectin are induced by 3 hours and 6 hours, respectively, after ischemia and peak at 6 to 12 hours (Zhang et al., 1995b; del Zoppo, 1997; Feuerstein et al., 1997; Wang et al., 1994a; Wang and Feuerstein, 1995; Amberger et al., 1997). ICAM-1 protein is expressed mainly within the core of the infarct on endothelial cells (Kim, 1996) and plays a role in neutrophil invasion of ischemic tissue. Cytokine-induced neutrophil chemo-attractant protein (CINC) is also induced mainly within an infarct and at its margins (Liu et al., 1993; Yamasaki et al., 1995). CD11 positive neutrophils appear within a day at the infarct site and are numerous by 3 days (Kato et al., 1996). Many studies that show that a reduction in inflammatory cells or inhibition of adhesion molecules lessens injury in experimental models of stroke suggest that adhesion molecules and inflammatory cells play a role in mediating focal ischemic brain injury (Chopp et al., 1996; del Zoppo, 1997; Feuerstein et al., 1997; Kitagawa et al., 1998; Soriano et al., 1999). This suggestion is balanced by a recently completed study of an anti-ICAM antibody in humans that failed to show benefit. This might relate in part because maximal treatment of infarction could be dependent upon reperfusion of the core so that antibodies reach all areas of ischemia, particularly because anti-ICAM antibodies work best after temporary ischemia (Zhang et al., 1995a, 1999).
Integrin alpha beta 3 is expressed primarily in the core of an infarct and is likely related to vascular responses (Abumiya et al., 1999). Monocyte-chemoattractant protein-1 and macrophage inflammatory protein-1 alpha (MIP-1) are induced primarily in the core and adjacent areas of ischemia (Kim et al., 1995). MIP-1 is induced first in the core, where the greatest damage occurs, and then in the regions adjacent to the infarction, (Takami et al., 1997) where damage is less severe and possibly where macrophages and microglia engulf single cells or small groups of cells that might die more slowly. CD18 positive macrophages, which immunostain for heme oxygenase-1 (Bergeron et al., 1997), begin to appear at 2 to 3 days in the core of an infarct and are quite numerous by 7 days (Kato et al., 1996).
MATRIX METALLOPROTEINASES
The matrix metalloproteinases (MMPs) include MMP-7 (matrilysin), MMP-3,-10,-11,-13 (stromelysins), MMP-14 (membrane MMP), and MMP-2 and MMP-9 (gelatinase A and B, respectively) (Rosenberg et al., 1996; Mun-Bryce and Rosenberg, 1998). MMP-2 and MMP-4 have been the subject of recent studies because they attack type IV collagen, laminin, and fibronectin, the major components of the basal lamina around cerebral blood vessels. MMP-2 is expressed constitutively in brain and may play a role in ischemia (Clark et al., 1997; Gasche et al., 1999; Rosenberg et al., 1996). MMP-9, the 92kD type IV collagenase, is not expressed in normal brain. After ischemia, ProMMP-9 is induced in the core within 2 hours with enzymatic activity and mRNA induction being detected by 4 hours (Fujimura et al., 1999a; Gasche et al., 1999). Induction of MMP-9 mRNA could be mediated by a NF-kB site in the MMP-9 promoter (Mun-Bryce & Rosenberg, 1998) (Fig. 3). Activation of MMP-9 correlates with blood-brain barrier break down (Gasche et al., 1999) and in at least one study correlated with areas of hemorrhagic conversion after focal ischemia (Heo et al., 1999). MMPs may be important for producing increases of blood brain barrier permeability and brain edema after stroke (Gasche et al., 1999). MMPs may also promote tissue invasion of neutrophils and macrophages, and contribute to hemorrhages that result after reperfusion of ischemic tissue (Mun-Bryce & Rosenberg, 1998; Heo et al., 1999).
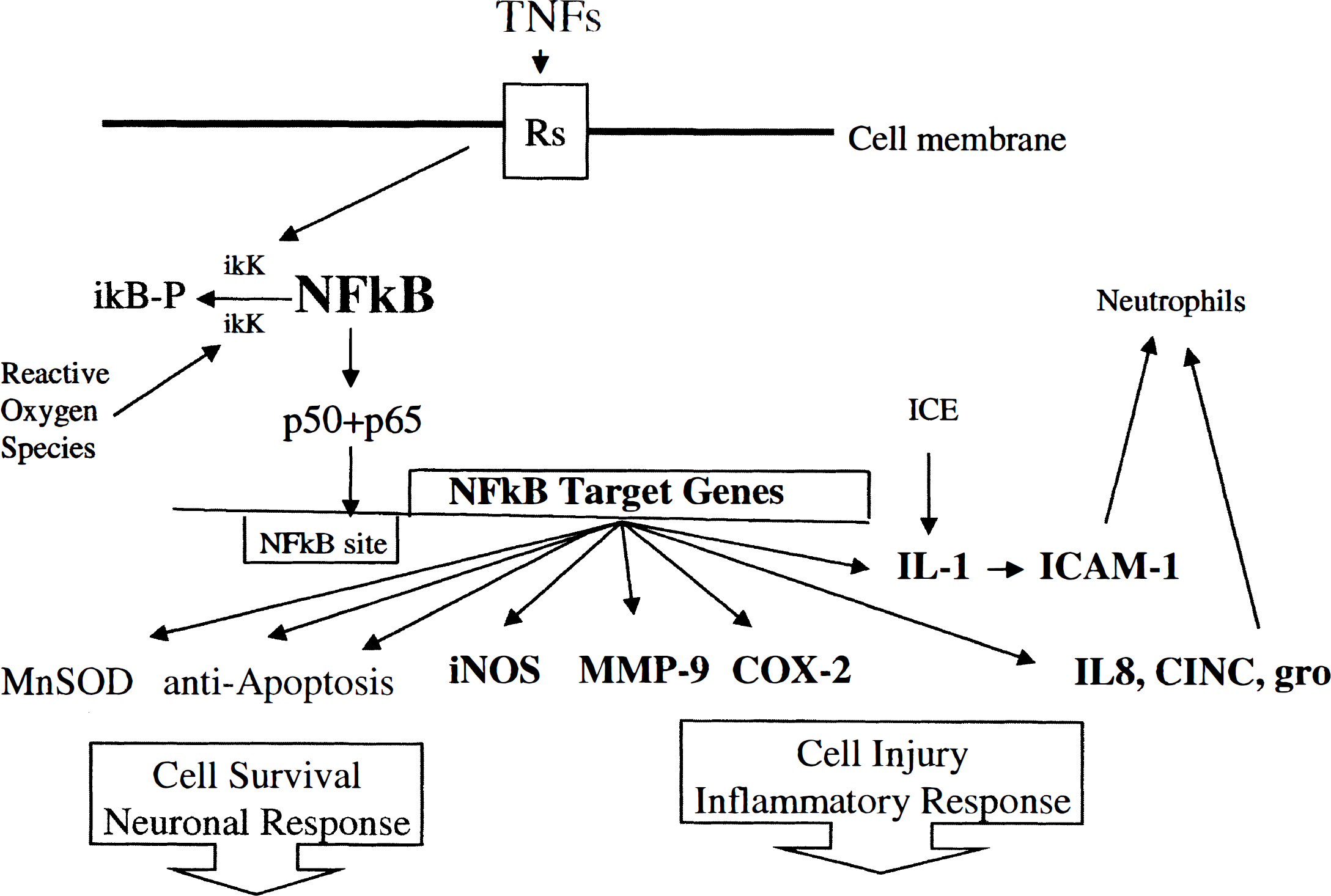
Postulated mechanisms by which TNF and NFkB might mediate either cell survival or cell injury. This would depend upon which cells these genes are induced in and which target genes are induced. Note that NFkB is activated by phosphorylation of ikB and its release resulting in NFkB activation and binding of the p50-p65 complex to NFkB sites in target genes.
Therapeutic significance of the core
Many of the above genes are expressed mainly in the core of an infarct. When there is no reperfusion of the core, there would seem little hope of rescuing the core with current technology and little reason to think that manipulating the above genes might affect the outcome. However, if the core were reperfused either spontaneously or with tPA, the induction of these inflammatory molecules (Fig. 3) might prove to be important therapeutic targets (Zhang et al., 1999).
ZONE OF SELECTIVE NEURONAL DEATH ADJACENT TO INFARCTS
The edges of infarcts appear to be fairly well demarcated on Nissl and hematoxylin and eosin-stained tissue sections at the gross level. However, on hematoxylin and eosin-stained sections there are eosinophilic cells, most of which are neurons, that are outside an infarct. This appears to represent a rim of selective neuronal cell death (Nedergaard, 1987; Nedergaard et al, 1987) (see Fig. 1). These histologic findings have been supported by TUNEL staining. TUNEL-stained neurons with fragmented DNA are found immediately outside the areas of infarction, at most a centimeter from the infarct, and vary in number depending upon the severity of ischemia and the brain region examined (Li et al., 1995c; Li et al., 1995b; States et al., 1996). Consequently, there is a zone of selective neuronal death that occurs adjacent to, and just outside of histologically defined infarct margins.
APOPTOSIS, DNA DAMAGING INDUCIBLE AND DNA REPAIR GENES
DNA damage-inducible and DNA repair genes tend to be expressed either within the core or within the regions adjacent to the infarction. It is possible that these genes contribute to selective neuronal cell death, or contribute to tissue infarction itself, or both. Bax, the pro-apoptotic gene, is induced in the core of infarcts (Gillardon et al., 1996) and in cells just outside the infarct that have evidence of DNA fragmentation by TUNEL staining (Matsushita et al., 1998; Isenmann et al., 1998). Bcl-2 tends to decrease in cells that appear to be lethally injured. Bcl-2 and Bcl-x1, the anti-apoptotic genes, tend to be induced in cells that are immediately adjacent to an infarct (Asahi et al., 1997) and probably survive ischemia (Chen et al., 1995) (Isenmann et al., 1998). Bcl-2 can be induced in the entire middle cerebral artery (MCA) territory with less severe degrees of ischemia (Chen et al., 1995). The cleaved portion of caspase 3, the protease associated with programmed cell death, is found in the MCA core and in the region adjacent to the core (Asahi et al., 1997; Namura et al., 1998).
Genes associated with DNA damage are expressed in and around the core after focal ischemia. Gadd45, a protein induced in response to DNA damage, was expressed at the edge of infarcts in cells that had little evidence of DNA fragmentation (Hou et al., 1997). This contrasted with another study that suggested that p53, Bax, MDM2, and Gadd45 were induced in cells that were dead or expected to die (Li et al., 1997). DNA repair proteins decrease in cells expected to die in the core (Fujimura et al., 1999b, c ) and are induced in cells immediately adjacent to the core that appear to survive the ischemia (Li et al., 1997). Cell cycle genes and proteins can be induced in cells that survive ischemia and possibly in proliferating cells like microglia and astrocytes (Wiessner et al., 1996).
There is conflicting data about some of these genes. One study shows that p21 mRNA and protein and cyclin G1 increase; whereas p53 and Bax messenger RNA and protein levels, and protein levels of p27, cyclin-dependent kinase 5, p35, and cyclin E decrease in the infarct core and border areas after middle cerebral artery occlusion (MCAO) (van Lookeren Campagne and Gill, 1998). Some of the disparity between these studies could be attributed to differences in how much of the core or adjacent surviving brain was sampled, because protein levels in the core for most genes would decrease. Also of interest is that at least one study shows induction of GADD45, growth arrest and DNA damage-inducible in neurons throughout an ischemic hemisphere, in areas inside and outside any region of ischemia (Jin et al., 1996). This pattern of gene induction is similar to that of immediate early genes (IEGs) described below; hence, it is likely induced in response to spreading depression rather than damaged DNA (though once induced it could respond to DNA damage after focal ischemia).
CD95 and tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) have also been studied after reversible MCAO in adult rats (Martin-Villalba et al., 1999). Both CD95 ligand and TRAIL were expressed in the rim around the infarct. Recombinant CD95 ligand and TRAIL proteins induced apoptosis in primary neurons and neuron-like cells in vitro. FK506 prevented postischemic expression of these death-inducing ligands both in vivo and in vitro and abolished phosphorylation, but not expression, of the c-jun transcription factor involved in the transcriptional control of CD95 ligand. In addition, in Ipr mice expressing dysfunctional CD95 reversible MCAO resulted in infarct volumes significantly smaller than those found in wild-type animals (Martin-Villalba et al., 1999).
Therefore, the evidence suggests that this narrow zone of selective neuronal injury, where various apoptosis-related and DNA damage and repair genes are expressed, could be important for stroke outcome. This is supported by recent studies showing that inhibiting caspases or introducing bcl-2 into brain improve outcome from stroke (Cheng et al., 1998; Lawrence et al., 1996a; Endres et al., 1998; Martinou et al., 1994). Itispossible that the region of selective cell death might convert to an area of infarction with continuing or worsening ischemia (Du et al., 1996). Adjacent, previously unaffected regions could then develop selective cell death and so on. It remains to be seen whether manipulating apoptotic-related proteins will improve stroke outcomes.
NOS AND NO
Immediately after focal infarction, NO (nitric oxide) is derived mainly from neuronal nNOS (NOS-1) and endothelial eNOS (NOS-3) in the core and margin of the infarct (Iadecola, 1997; Ashwal et al., 1998). Inhibiting nNOS at this point appears to improve stroke outcome (Iadecola et al., 1994, 1997; Dirnagl et al., 1999), with NO injury occurring through actions on the N-methyl-
Inducible NOS (iNOS, NOS-2) is induced many hours after ischemia (Nagafuji et al., 1994; Iadecola, 1997). It is induced either in neutrophils or macrophages, in the core of the infarct, or in microglia, blood vessels, or astrocytes at the margins of the infarct (Iadecola et al., 1996; Coeroli et al., 1998; Forster et al., 1999; Loihl et al., 1999). Inhibition of iNOS (NOS-2) appears to improve stroke (Iadecola et al., 1995, 1996, 1997; Dirnagl et al., 1999).
TNF AND NFkB
Some cytokines are expressed only within the infarct, whereas others are expressed at a considerable distance, possibly even in the opposite hemisphere. In addition, there are many differences in the temporal induction of the various genes (Hill et al., 1999). Tumor necrosis factor-α (TNF-α) is induced early, whereas interleukin-1 (IL-1) and transforming growth factor (TGF) are delayed (Hill et al., 1999). TGF expression can persist for weeks after a stroke (Wang et al., 1995b), as can interferon-inducible protein-10 (Wang et al., 1998b).
TNF-α
TNF-α is induced in the core and in the region adjacent to the infarction in neurons, astrocytes, and endothelial cells (Feuerstein et al., 1994; Gong et al., 1998; Liu et al., 1994; Feuerstein et al., 1997) within 1 to 6 hours the ischemia (Yang et al., 1999a). TNF-α expression can be observed in neurons and macrophages (Liu et al., 1994). Although TNF-α is mainly induced in the ischemic hemisphere, it can also be induced in the ipsilateral hippocampus (Gong et al., 1998) and the contralateral nonischemic hemisphere (Zhai et al., 1997).
TNF-α has often been thought to mediate injury and apoptotic cell death (Dawson et al., 1996; Mizuno and Yoshida, 1996; Yang et al., 1998), although more recent studies suggest that it can also be protective. Some investigators have shown that administration of TNF worsens infarcts, and binding TNFs with ligands or antibodies decreases stroke size (Barone et al., 1997; Dawson et al., 1996; Nawashiro et al., 1997). However, other studies have suggested that mice with knockouts of their TNF receptors have larger infarcts and greater injury because of excitotoxins (Bruce et al., 1996; Sullivan et al., 1999). These differences in the actions of TNF could depend upon the following: (1) which TNF is induced; (2) which receptor it acts on; and (3) which cells the TNF is induced in (Fig. 3). TNF induction in neutrophils and endothelial cells could mediate injury, whereas TNF induction in neurons could be protective (Bruce et al., 1996; Mattson, 1997) (Fig. 3).
NFkB
Stimulation of TNF-α receptors leads to activation of NFkB, with phosphorylation and release of ikB from the NFkB complex (May and Ghosh, 1999; Li and Karin, 1999). The p50 and p65 dimer of NFkB then acts on NFkB target genes (Ghosh et al., 1998) (see Figure 3). The precise role of NFkB in ischemia remains unclear because some studies show that NFkB might mediate injury, whereas others suggest that it could protect brain.
For example, one study shows that NFkB/p65 decreases after focal ischemia (Botchkina et al., 1999a). However, other studies show that, although absolute amounts of p50 and p65 may not be predictive, NFkB DNA binding activity is increased after focal and global ischemia (Schneider et al., 1999; Carroll et al., 1998; Gabriel et al., 1999; Howard et al., 1998; Salminen et al., 1995). In one study stroke volumes were decreased in p50 knockout mice (Schneider et al., 1999) suggesting a harmful role for NFkB. However, blocking NFkB activity can exacerbate excitotoxic injury (Botchkina et al., 1999b), and stroke and excitotoxic injury in p50 knockout mice has been reported to be worse (Yu et al., 1999).
In addition to being regulated by TNF and reactive oxygen species (Schreck et al., 1991; Meyer et al., 1993), NFkB is also regulated by ikB kinases (ikKs). ikKs phosphorylate ikB, which activates the p50-p65 complex of NFkB. TNF activates NFkB by activating ikKs (Schottelius et al., 1999). Aspirin and NSAIDs down-regulate NFkB and TNF (Shi et al., 1999) in part by inhibiting ikKs (Pierce et al., 1996; Stevenson et al., 1999). IL-10 decreases NFkB activity in part by inhibiting ikKs, and also by blocking NFkB binding to target promoter elements (Schottelius et al., 1999).
Although NFkB is a major sensor and effector of oxidative stress in cells, it is not entirely clear how this occurs (Li and Karin, 1999). Thioredoxin (TRX) is a small disulfide protein induced in response to oxidative stress. TRX interacts with and activates NFkB (Weichsel et al., 1996). Although TRX expression decreases in an infarct, it is markedly induced adjacent to an infarct (Takagi et al., 1998). TRX overexpression in transgenic mice protects them against focal ischemia (Takagi et al., 1999).
NFkB could mediate cell protection and cell damage because of its many downstream target genes. Genes with NFkB binding sites in their promoters that would protect cells include MnSOD (Darville et al., 2000; Xu et al., 1999); calbindin (Bruce-Keller, 1999); bcl family genes, including bcl-2 and bcl-x1 (Wang et al., 1998a; Chen et al., 1999a; Zong et al., 1999; Tsukahara et al., 1999; Tamatani et al., 1999); and TNFR-associated factors (TRAFs) and inhibitor of apoptosis proteins (IAPs) (Wang et al., 1998a).
A number of other genes can be regulated at least in part by NFkB can mediate cellular injury. These include IL-1, ICAM-1, CINC, IL-8, and gro, that promote neutrophil adhesion (Roebuck, 1999); COX-2, that metabolizes arachidonic acid (Lee and Burckart et al., 1998; Kotake et al., 1998; Plummer et al., 1999); MMP-9, that cleaves type 4 collagen at the blood-brain barrier (Mun-Bryce & Rosenberg, 1998; Lee and Burckart, 1998); heme oxygenase-1, that metabolizes heme to release iron; and iNOS, that releases NO and contributes to oxidative stress (Kotake et al., 1998). Lastly, the pro-apoptotic gene bcl-xs has a NFkB element in its promoter that is activated during brain ischemia (Dixon et al., 1997).
NFkB and TNF might promote cell injury or cell protection depending upon the cells and the circumstances of their induction (Li & Karin, 1999). For example, COX-2 is induced in smooth muscle cells but not through NFkB (Chen et al., 1999b). As a working hypothesis, it is conceivable that TNF and NFkB expression in neutrophils and endothelial cells might induce neural injury related genes after stroke (Schneider et al., 1999). However, TNF and NFkB expression in neurons may induce neuroprotective genes and prevent injury caused by stroke and excitotoxins (Bruce-Keller, 1999) (Bruce et al., 1996) (Mattson, 1997) (Yu et al., 1999a). This dual action of TNF and NFkB is shown in Fig. 3.
COX-2
Phospholipids are metabolized to arachidonic acid (AA) by phospholipase A2. AA is metabolized to prostaglandins by COX-2 and metabolized to leukotrienes by 5-lipoxygenase. COX-2 is induced by focal ischemia (Kinouchi et al., 1999b; Planas et al., 1995). COX-2 inhibitors decrease stroke volumes in some, but not all, studies (Nagayama et al., 1999; Hara et al., 1998). Cell protection produced by COX-2 inhibitors appears to be linked to iNOS mediated injury (Nagayama et al., 1999). It should be noted that spreading depression induces COX-2 throughout a hemisphere (Miettinen et al., 1997; Koistinaho et al., 1999) so that variable degrees of focal ischemia may induce COX-2 throughout one-half of the brain. This may explain why COX-2 is induced at great distances from the region of ischemia in rodents (Koistinaho et al., 1999) and in human stroke patients (Sairanen et al., 1998). This also suggests that spreading depression, not AA itself, is the likely stimulus for COX-2 induction after focal ischemia.
INTERLEUKINS
IL-1
IL-1 is markedly induced after focal ischemia (Szaflarski et al., 1995; Betz et al., 1996; Rothwell and Relton, 1993; Yabuuchi et al., 1994; Rothwell et al., 1997). IL-1 is induced in the ischemic ipsilateral cortex and in the contralateral, nonischemic cortex (Zhai et al., 1997). It peaks at 6 hours after ischemia and persists for several days (Wang et al., 1994b). The bilateral IL-1 induction appears to occur in cerebral endothelial cells and microglia (Giulian et al., 1986; Zhang et al., 1998b). This suggests that although ischemia may only occur in one hemisphere, adverse cytokine responses may appear in the opposite hemisphere (Zhai et al., 1997). This could occur through ischemia induced spreading depression as described below to explain bilateral induction of fos and other IEGs after stroke. IL-1 appears to worsen ischemic injury (Betz et al., 1996; Stroemer and Rothwell, 1998), and to produce selective neuronal cell death and edema (Holmin and Mathiesen, 2000). Blocking IL-1 decreases ischemic injury (Loddick et al., 1997; Rothwell et al., 1997; Betz et al., 1996; Yang et al., 1999b). This could occur in part because IL-1 induces ICAM-1 and other proinflammatory molecules (Rothwell et al., 1997; Yang et al., 1999c) (Fig. 3).
IL-6 is also induced diffusely in brain after ischemia (Loddick et al., 1998). It is induced in neurons and microglia and is found in the ischemic hemisphere, the ipsilateral hippocampus, and contralateral cortex (Suzuki et al., 1999). Mice deficient in IL-6 have markedly decreased astrocytic and microglial responses to injury (Klein et al., 1997) and administration of IL-6 protected against stroke (Loddick et al., 1998).
IL-10 is also induced by ischemia, although it is induced only in the ischemic hemisphere (Zhai et al., 1997). This monocyte chemo-attractant is induced mainly in regions of injury at early times after stroke and continues expression for days after stroke (Wang et al., 1998b).
TGF AND PLASMINOGEN ACTIVATOR INHIBITOR
TGF-b1 mRNA is induced in the ischemic MCA territory, including cortex and striatum, and in the ischemic cingulate cortex (Lehrmann et al., 1998). Microglia and macrophages are the major source of TGF-β1 after ischemia (Lehrmann et al., 1998). Expression of mRNA was detected by 6 to 12 hours after ischemia (Wang et al., 1995b), was highly expressed at 1 week, and continued expression in a rim around the infarct at 3 weeks after infarction (Lehrmann et al., 1998). TGFβ appears to protect against focal ischemia (Ruocco et al., 1999).
The plasminogen activator inhibitor-1 (PAI-1) is a TGF target gene. Focal ischemia induces PAI-1 without any effect on protease nexin-a, neuroserpin, or tissue plasminogen activator. PAI-1 is expressed in astrocytes (Docagne et al., 1999). PAI-1 was modulated by TGF-β1 treatment through a TGF-β-inducible element contained in the PAI-1 promoter (CAGA box) (Docagne et al., 1999). TGF-beta and activin induced the overexpression of PAI-1 in astrocytes; whereas bone morphogenetic proteins, glial cell line-derived neurotrophic factor, and neurturin did not. Protective effects of TGFβ may be mediated in part through binding of TGFb to PAI-1 and its downstream effects. Others have confirmed that focal ischemia induces PAI-1 mRNA without effects on tPA or u-PA mRNAs (Ahn et al., 1999), although u-PA enzymatic activity increases and tPA enzymatic activity decreases after stroke (Rosenberg et al., 1996a). TGF could induce PAI-1 mRNA through the combined actions of two sets of transcriptional activators, Smad3 and Smad4 in cooperation with AP-1 (Fig. 6) (Zhang et al., 1998a).
GROWTH FACTORS
bFGF
bFGF is induced mainly in astrocytes in the MCA territory and ipsilateral hippocampus after ischemia (Lin et al., 1997). Others have noted global up-regulation of bFGF in the ischemic hemisphere (Lippoldt et al., 1993), including nonischemic regions of cingulate cortex, temporal cortex, and some nonischemic subcortical structures (Speliotes et al., 1996). This suggests that bFGF is probably induced by ischemia induced spreading depression (see below). Although bFGF protected against stroke in rodent models, it failed to protect in a recent human trial. However, it is still possible that bFGF and other growth factors might promote more rapid stroke rehabilitation and perhaps improve long-term recovery (Fisher and Finklestein, 1999; Ay et al., 1999).
BDNF
BDNF is also induced throughout an ischemic hemisphere (Hsu et al., 1993; Kokaia et al., 1998; Guegan et al., 1998; Kokaia et al., 1993). It is likely that BDNF is induced through spreading depression induction of Fos (An et al., 1993a) that then induces BDNF through an AP-1 site in its promoter (Cui et al., 1999). bFGF may be induced through a similar mechanism of spreading depression induction of fos that then acts on AP-1 sites in the promoter of the bFGF gene. Inflammatory cytokines like IL-1 also induce bFGF through an AP-1 site in its promoter (Faris et al., 1998). Once induced, BDNF, bFGF, and other growth factors have a large number of target genes that they also regulate (Aho et al., 1997; Semkova and Krieglstein, 1999; Shieh and Ghosh, 1999; Black, 1999).
Glial derived nerve growth factor is also induced after stroke (Abe et al., 1997), as is EGFr (Planas et al., 1998). Insulin growth factors and binding proteins (Gluckman et al., 1992; Lee et al., 1996), platelet derived growth factor (Iihara et al., 1994), ciliary neurotrophic factor (Lin et al., 1998) and growth inhibitory factor (metallothionein III) (Yuguchi et al., 1997) are also induced after focal ischemia. Insulin growth factor like receptor-II (IGF-II) is induced in pyramidal neurons primarily in the core (Stephenson et al., 1995) Interestingly, growth inhibitory factor is induced throughout the hemisphere after a focal stroke, again suggesting possible induction of this gene through spreading depression. Fibroblast growth factor and ciliary neurotrophic factor have been reported to attenuate the thalamic atrophy that occurs after MCAOs in animals (Yamada et al., 1991; Kumon et al., 1996). When tested, the administration of most of the neurotrophic factors protects against focal ischemia (Semkova and Krieglstein, 1999; Wang et al., 1997).
HEAT SHOCK PROTEINS
HSP70 - zone of protein denaturation and renaturation
The induction of heat shock proteins (HSPs) after focal and global ischemia continues to be of interest because they are unique among most of the genes studied because they are specifically induced in cells responding to injury (Nowak and Jacewicz, 1994; Massa et al., 1996; Nowak, 1999; Welch and Brown, 1996), and these genes protect against a wide variety of injuries (Massa et al., 1996; Rajdev et al., 1997, 2000; Chen and Simon, 1997).
HSP70 is the major inducible heat shock protein, being expressed at low levels in all cells (Welch and Gambetti, 1998; Craig et al., 1993). Any injury that contributes to protein denaturation appears to produce transcriptional activation of hsp70, including ischemia, heat shock, heavy metals, hypoglycemia, low pH, and disease states (Lindquist, 1992; Brown, 1995; Morimoto et al., 1997; Welch and Gambetti, 1998). The presence of the denatured proteins appears to be the major stimulus for hsp70 induction (Ananthan et al., 1986). The transcriptional activation of hsp70 occurs through heat shock factors (HSFs). HSFs are bound to HSP90 in normal cells in an inactivated state (Zou et al., 1998; Gass et al., 1994; Schumacher et al., 1996). With the appearance of denatured proteins, HSP90 binds to the denatured proteins releasing HSFs (Zhou et al., 1996; Zou et al., 1998). HSFs are activated—that is, phosphorylated and form a trimer—and bind to heat shock elements on hsp70 and other heat shock genes to stimulate the heat shock response (Zou et al., 1998).
Thus, the zone of HSP70 induction after focal ischemia can be viewed as the zone of protein denaturation associated with the injury (see Figures 1 and 4). Since HSP70, in concert with HSP90 and other heat shock protein chaperones, acts to renature the denatured proteins, hsp70 induction represents the zone of protein denaturation and attempted protein renaturation. After permanent ischemia or severe temporary focal ischemia hsp70 mRNA may not be expressed in the core of an infarct if ATP is limiting (Nowak, 1999; Welsh et al., 1992; Kobayashi and Welsh, 1995). However, even with vessel occlusions that lead to MCA infarction, hsp70 mRNA can be expressed inside and outside the region of infarction, with most of the hsp70 mRNA within the infarct being expressed in vessels (Kinouchi et al., 1993a, b , 1994b).
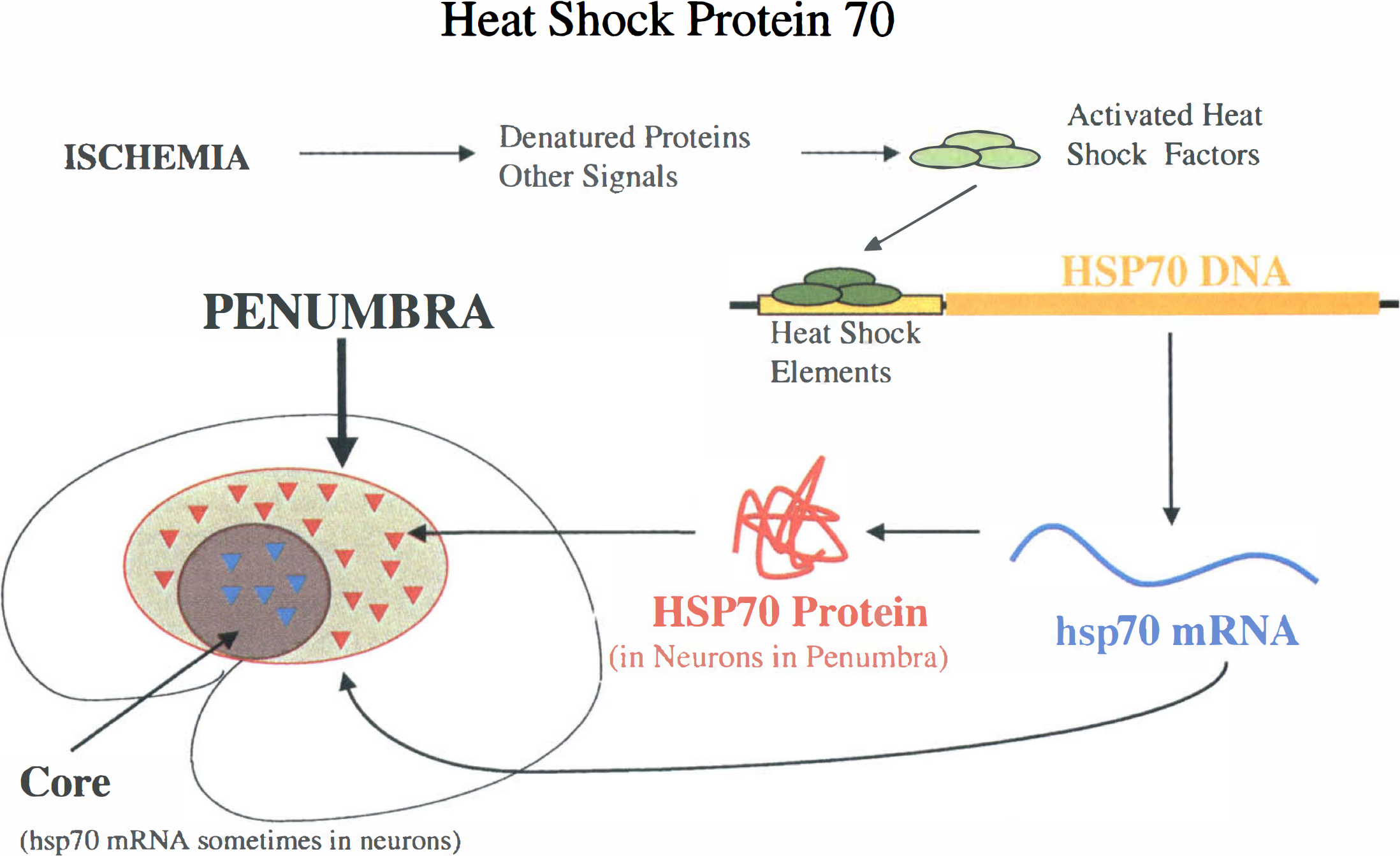
The denatured protein penumbra. Induction of HSP70 after stroke delineates a region that includes the core of the infarct and regions adjacent to the infarct where the presence of denatured proteins within the cells signals the induction of hsp70. hsp70 mRNA can be detected in the core of infarct with moderate focal ischemia, but may not be synthesized after permanent vessel occlusions when ATP is rapidly depleted. hsp70 mRNA and protein are expressed in some glia and in many neurons at some distance from the infarct in a penumbra defined entirely upon the expression of a molecule.
HSP70 protein is expressed mainly in blood vessels and sometimes in scattered microglia and astrocytes in areas inside an infarction (Kinouchi et al., 1993a; Soriano et al., 1994; Planas et al., 1997). HSP70 protein is expressed in glia at the margins of infarcts, and HSP70 protein is expressed in glia and neurons outside areas of infarction (Kinouchi et al., 1993a, b ; Li et al., 1992, 1993; Soriano et al., 1994). As noted above, the neuronal expression of HSP70 protein can be interpreted as a molecularly defined penumbra of protein denaturation (Sharp et al., 1999) (Figs. 1 and 4). The zone of protein denaturation (HSP70) extends beyond the zone of selective neuronal cell death. This is based upon the finding that after a stroke, TUNEL-stained cells occur immediately adjacent to the infarct, whereas HSP70-stained neurons are found at much greater distances from the infarct in the same brains (States et al., 1996; Planas et al., 1997; Li et al., 1993, 1995a, c ).
HSP70 expression appears to protect cells against various types of injury (Xu and Giffard, 1997; Chen et al., 1996; Mailhos et al., 1994; Wagstaff et al., 1998; Yenari et al., 1998). However, it may not protect against apoptosis or relatively severe injury (Wagstaff et al., 1998). Most important, HSP70 overexpression can protect heart (Marber et al., 1995; Trost et al., 1998) and hippocampus (Plumier et al., 1997d) against ischemia. We have found that transgenic mice that overexpress HSP70 protein in brain are protected against strokes produced by permanent MCAO (Rajdev et al., 1998, 2000).
HO-1 plays an important role in metabolizing heme released from hemoglobin after subarachnoid hemorrhage and intracerebral hemorrhage in brain (Matz et al., 1996; Matz et al., 1997; Turner et al., 1998). Heme-iron acts on the HO-1 promoter and is a potent HO-1 inducer (Alam et al., 1994). The major source of heme in ischemic brain however, probably comes from the heme found in mitochondrial electron transport heme proteins released after cell injury.
HO-1 is also induced after cerebral ischemia (Paschen et al., 1994; Geddes et al., 1996). After focal ischemia HO-1 is induced in vessels in the core of an infarct, and in microglia, scattered neurons, and astrocytes at the margin of an infarct (Nimura et al., 1996). At 1 day after a stroke, HO-1 protein is induced in microglia well outside regions of ischemia, including cingulate cortex—a pattern postulated because of spreading depression (Nimura et al., 1996). In fact, spreading depression can induce HO-1 (Koistinaho et al., 1999), c-fos, and COX-2 as described below (Koistinaho et al., 1999). This HO-1 induction could occur through spreading depression-fos-activated AP-1 sites in its promoter (Alam and Den, 1992). Although hypoxia can induce HO-1 through a hypoxia-inducible factor site (Lee et al., 1997) we found little evidence for hypoxia induction of HO-1 in neonatal brain (Bergeron et al., 1997).
HO-1 continues to be expressed in microglia and macrophages at very long time periods after stroke (Koistinaho et al., 1996; Bergeron et al., 1997). This could occur through NFkB sites in the HO-1 promoter (Lavrovsky et al., 1994), suggesting that HO-1 may play a role in inflammation as well (Ewing and Maines, 1993; Willis et al., 1996).
HYPOXIA INDUCIBLE FACTOR - ZONE OF HYPOXIA
HIF-1 is a recently recognized transcription factor that is induced by changes in molecular oxygen levels in tissue (Semenza, 1999; Ratcliffe et al., 1998; Wang et al., 1995a). Mutations in the HIF-1α gene result in the inability to induce erythropoietin and increase red blood cells after hypoxia (Semenza, 2000). HIF-1α mRNA is induced by hypoxia and not by inhibitors of mitochondrial respiration. This suggests that HIF-1 is activated by molecular oxygen sensor, possibly a heme-protein (Bunn and Poyton, 1996; Huang et al., 1999).
Once induced, HIF-1α protein binds to HIF-1β, which is constitutively expressed in most cells (Wood et al., 1996). The HIF-1 dimerization that stabilizes both proteins leads to binding to hypoxia response sequences in various target genes. Hypoxia-inducible genes that have HIF-1 sites in their genes and may be regulated at least in part by hypoxia include erythropoietin (Huang et al., 1997), tyrosine hydroxylase (Millhorn et al., 1997), inducible NOS (iNOS, NOS2) (Melillo et al., 1997; Keinanen et al., 1999), vascular endothelial growth factor (VEGF) (Forsythe et al., 1996), glucose transporter-1 (GLUT-1), HO-1 (HSP32) (Lee et al., 1997), transferrin (Rolfs et al., 1997; Lok and Ponka, 1999) and all of the glycolytic enzymes including phosphofructokinase and lactate dehydrogenase (Firth et al., 1994, 1995; Semenza et al., 1996) (Fig. 5).
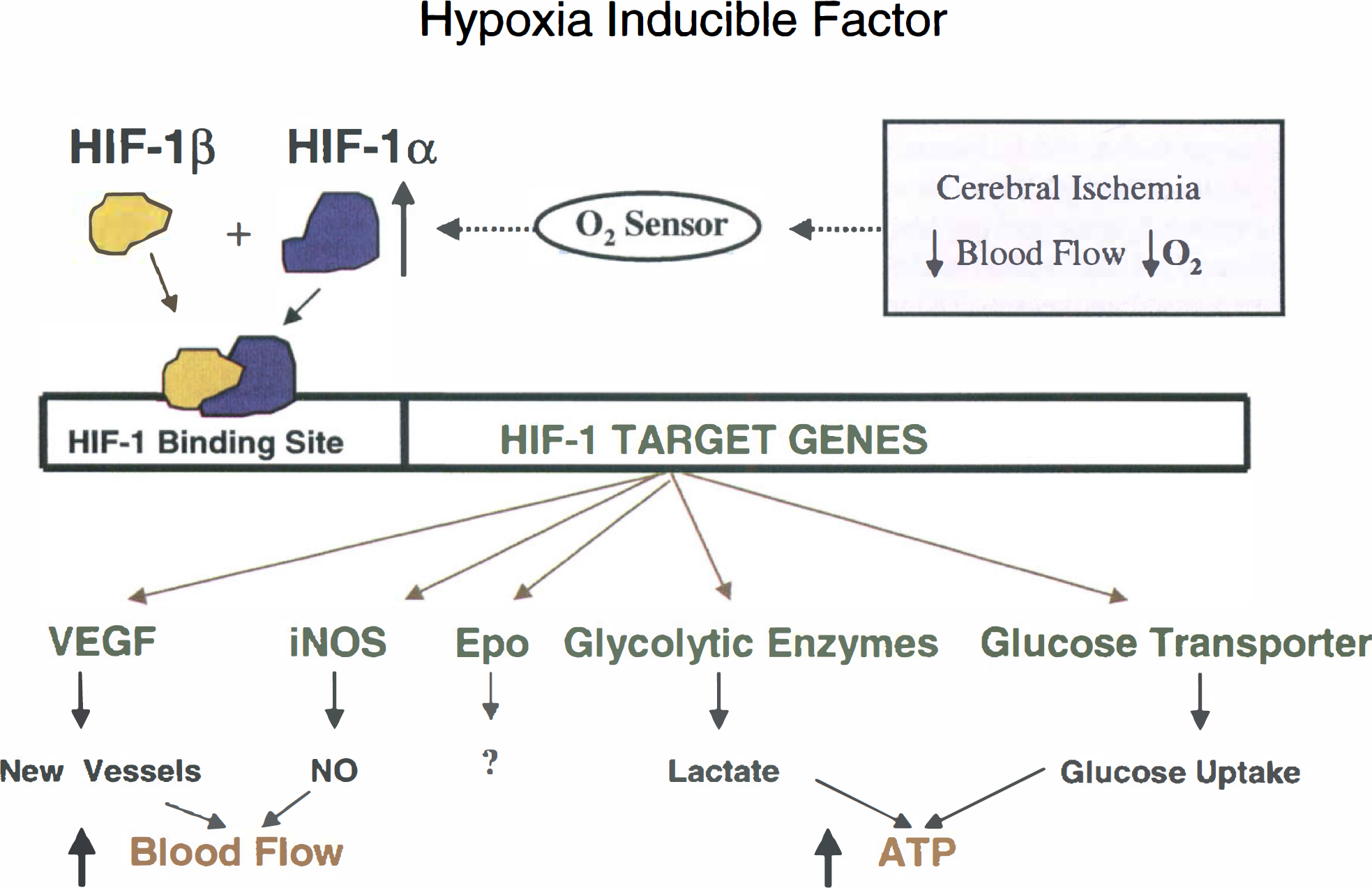
Proposed mechanism by which stroke induces the hypoxia inducible-factor (HIF) in brain. Decreased blood flow decreases oxygen delivery that is presumed to be detected by a sensor of molecular oxygen. This induces HIF-1α, but not HIF-1β mRNA. The HIF-1α and HIF-1β proteins form a dimer that then bind to response elements in target genes. This interaction then induces the target genes, including the glucose transporter and glycolytic enzymes.
During reoxygenation, the HIF-1 protein complex has a very short half-life, a matter of minutes (Semenza, 2000). The rapid proteolysis of HIF occurs through the proteasome (Salceda and Caro, 1997; Srinivas et al., 1999; Kallio et al., 1999) that is regulated by the Van Hippel Landau tumor suppressor protein (Maxwell et al., 1999).
HIF is induced in brain after focal ischemia (Bergeron et al., 1999). Although HIF-1α and HIF-1β mRNA are both present in normal brain, only HIF-1α mRNA is induced in the cingulate cortex adjacent to an infarct after suture-induced MCAOs (Bergeron et al., 1999). HIF-1α and HIF-1β protein increase, possibly because of hypoxia-induced stabilization of the dimer. HIF-1 target genes also were induced in the cingulate cortex after MCA inclusion, including GLUT-1, phosphofructokinase, lactate dehydrogenase and others (Bergeron et al., 1999). Because blood flow decreased in this region of HIF-1 expression outside the infarction, we proposed that this region of HIF-1 gene expression represented a region of chronically decreased blood flow that was also chronically hypoxic and that resulted in HIF-1α gene induction. Hence, the region of HIF-1α gene expression after a stroke could be interpreted to be the region of chronic hypoxia around the region of infarction (Figs. 1 and 5).
The region of HIF-1α expression could be co-incident with HSP70 expression. However, the region of HIF-1 expression may be larger than that for HSP70 (Fig. 1), because after suture-induced MCAOs, HIF-1α was expressed in the cingulate cortex (Bergeron et al., 1999) where HSP70 is rarely induced (Kinouchi et al., 1994a). The HIF induction in cingulate that occurs with the suture occlusion MCA model is likely caused by anterior cerebral artery ischemia without infarction because of a patent anterior communicating artery (Longa et al., 1989).
It is possible that many experimental models of ischemia may not induce HIF. Global ischemia models produce transient ischemia with only temporary hypoxia that may not induce HIF. Transient focal ischemia models in which blood flow and oxygen delivery are rapidly restored may not induce HIF.
Other hypoxia inducible factors have also been recognized, including HIF-2/HRF and EPAS-1 (Tian et al., 1997; Erna et al., 1997; Flamme et al., 1997). Because EPAS1 is expressed in capillary endothelial cells (Tian et al., 1997), it might play a primary role in inducing vascular target genes that might be somewhat different or only partially overlap the target genes for HIF-1 (Conrad et al., 1999; Kobayashi et al., 1999). For example, EPAS1, HIF-related genes, or both might regulate VEGF expression in vessels (Ema et al., 1997; Flamme et al., 1997; Badr et al., 1999); whereas HIF-1 itself might regulate glycolytic enzymes to a greater degree in neurons (Bergeron et al., 1999; Badr et al., 1999). EPAS-1 is crucial for normal survival and oxygen sensing during development (Tian et al., 1998). Metal transcription factor-1 is another transcription factor that appears to mediate metal response element responses to hypoxia in metallothionein genes (Murphy et al., 1999).
Genes with HIF-EPAS sites often have other sites in the gene that mediate gene activation through other mechanisms. These include a glucose-regulated element in the glucose transporter GLUT-1 gene (Ebert et al., 1995) and NFkB sites in iNOS and HO-1. The HIF site in p53 does not appear to mediate hypoxic induction of p53 (Wenger et al., 1998). Ischemic induction of HO-1 in brain does not appear to occur through hypoxia, hence, the HIF site in the HO-1 gene (Bergeron et al., 1997). These results are important for showing that the presence of a particular promoter or enhancer element in a gene does not mean that that element is used, or that a particular stimulus uses that promoter element in a particular cell (Chen et al., 1999b).
Although the genes induced by HIF-1 and other hypoxia responsive transcription factors generally tend to increase blood flow, glucose delivery, and maintenance of energy after chronic hypoxia (Fig. 5), the role of HIF-1 in acute focal cerebral ischemia is unclear. For example, increased NO from iNOS (Palmer et al., 1998), dopamine from tyrosine hydroxylase (Millhorn et al., 1997) and lactate from lactate dehydrogenase (Semenza et al., 1996) may worsen ischemia. However, increased glucose transporter expression (Lawrence et al., 1996b; Vannucci et al., 1998) or erythropoietin (Bernaudin et al., 1999; Semenza, 1994) expression might protect brain. The role of HIF-1 remains to be determined. HIF-1 may have harmful roles in some cell types and beneficial roles in other cell types, with the ultimate harm or protection depending on the model, timing, and mode of the HIF induction in brain (Halterman et al., 1999; Zaman et al., 1999). HIF-1 could play a role in mediating hypoxia-induced tolerance to cerebral ischemia (Gidday et al., 1994,1999; Bergeron et al., 1999).
VEGF
VEGF is a potential HIF-1 target gene (Levy et al., 1995, 1997) that is induced by focal ischemia. VEGF is induced in the core and ischemic border zone after focal ischemia Kovacs et al., 1996; Cobbs et al., 1998), although some have reported bilateral cortical induction of VEGF after focal ischemia (Lennmyr et al., 1998). In primate ischemic models, noncapillary vessels in the ischemic core and the periphery of an infarct express VEGF mRNA (Abumiya et al., 1999). VEGF mRNA is located in both microglia-macrophages and in endothelial cells in regions adjacent to rodent infarcts (Plate et al., 1999). The VEGF receptors Flt-1 and Flk-1 were induced after ischemia as well, Flt-1 on neurons, glia, and endothelial cells; and Flk-1 mainly on glial and endothelial cells (Lennmyr et al., 1998). Induction of the VEGF receptors and other VEGF target genes could be mediated by Ets-1, a vascular-related transcription factor (Valter et al., 1999). The regulation of VEGF may involve multiple sites on the gene and may involve several transcription factors (Dibbens et al., 1999). HIF-like or EPAS-1 induction of VEGF could mediate the formation of new vessels after stroke (LaManna et al., 1998; Ment et al., 1997; Shweiki et al., 1992). However, because the formation of new vessels is considerably delayed, it seems unlikely that this would influence the outcome of an acute infarct. Expression of VEGF could influence the permeability of existing vessels and contribute to ischemia-induced edema, however (Ment et al., 1997; LaManna et al., 1998; van Bruggen et al., 1999).
IMMEDIATE EARLY GENE INDUCTION
Hemispheric spreading depression
After focal ischemia, a number of IEGs, including the c-fos gene, are induced throughout the entire hemisphere of the rat brain, and in the frontal, parietal, occipital, and limbic cortex including cingulate cortex (An et al., 1993; Hsu et al., 1993; Welsh et al., 1992; Kinouchi et al., 1994a; Lindsberg et al., 1996). Because most rodent MCAO models only produce infarction in the MCA distribution, it has been suggested that spreading depression accounts for induction of c-fos and other genes in the nonischemic portions of the hemisphere (Gass et al., 1992; Kinouchi et al., 1994d; Mancuso et al., 1999). This is supported by evidence showing that preventing ischemia induced spreading depression with NMDA antagonists, like MK-801, prevents c-fos induction in frontal and occipital poles after MCAOs (Gass et al., 1992; Kinouchi et al., 1994d; Collaco-Moraes et al., 1994). In addition, spreading depression produced by applying potassium chloride to the cortex, or by producing small cortical lesions induces these genes throughout the entire hemisphere (Herdegen et al., 1993; Sharp et al., 1989, 1990; Herrera and Robertson, 1989, 1990; Kobayashi et al., 1995; Koistinaho et al., 1999).
A large number of genes in addition to c-fos are induced throughout an ischemic hemisphere; hence, these genes are likely to be induced by spreading depression or repeated ischemic depolarizations (Koistinaho and Hokfelt, 1997). These genes include junB (Comelli et al., 1993; Hsu et al., 1993; Kamii et al., 1994a; Kinouchi et al., 1994a), Zac 1 and PACAP (Gillardon et al., 1998), NGFIA,B,C (Lin et al., 1996; Honkaniemi et al., 1997), egr (Honkaniemi et al., 1997), Rheb (Kinouchi et al., 1999a), Arc (Kunizuka et al., 1999) and probably other IEGs. Hsp27, COX2, and PKC are induced by spreading depression (Plumier et al., 1997c; Miettinen et al., 1997; Koponen et al., 1999).
Some of the genes induced by ischemia induced spreading depression are likely to be fos-jun target genes (Fig. 6). BDNF, bFGF and GFAP are induced throughout a hemisphere after spreading depression (Kraig et al., 1991; Kokaia et al., 1993) and may play a role in protecting brain against stroke (Matsushima et al., 1998). Since BDNF, bFGF and GFAP have AP-1 sites in their promoters, members of the fos and jun families could induce these genes. However, the induction of any of these genes could be complex. Although GFAP could be induced through AP-1 sites, there are also NFkB-like sites in the GFAP gene that renders it responsive to both TGF-1 and IL-1 (Krohn et al., 1999). Other genes with AP-1 sites in their promoters that could be induced by fos-jun family members could include dynorphin, enkephalin, NPY, iNOS, HO-1, APP, tyrosine hydroxylase, GAP43, NGF, and many others (Nowak, 1999; Morgan and Curran, 1995). Hypoxia induction of tyrosine hydroxylase occurs specifically through junB/c-fos dimers binding to the AP-1 site in the tyrosine hydroxylase promoter (Norris and Millhorn, 1995; Millhorn et al., 1997).
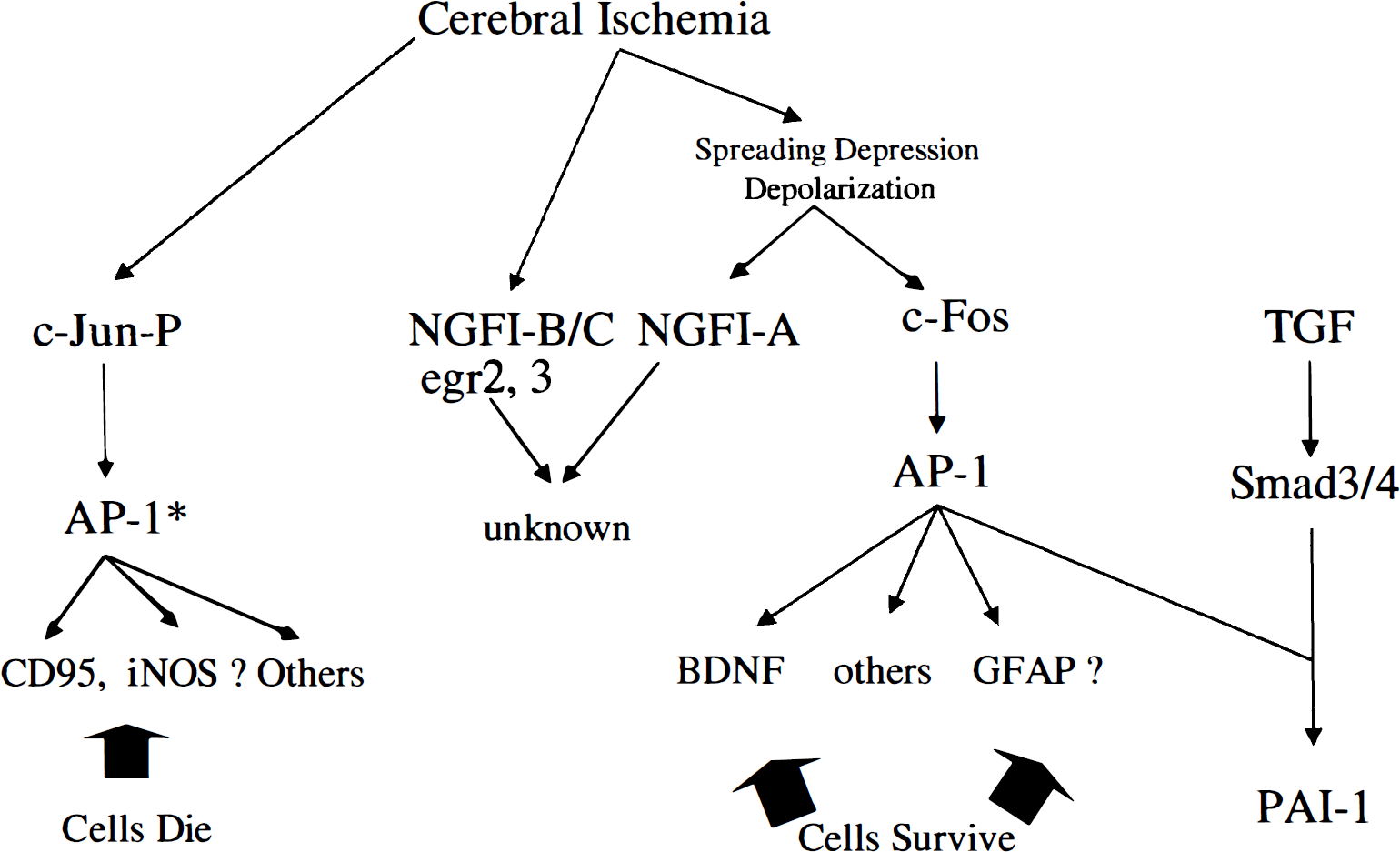
Ischemia induces a large number of transcription factors in brain. Spreading depression appears to be the stimulus for the induction of c-fos, NGFI-A and a large number of other immediate early genes. Though c-jun may be widely induced after focal ischemia, phosphorylated c-jun (c-jun-P) is associated with cell death in many paradigms and probably has different AP-1* mediated target genes than c-fos mediated AP-1 target genes in areas of spreading depression where there is no cell death. Notably, AP-1 can interact with many other transcription factors including Ets, Smad3/4, and others to presumably regulate different sets of target genes.
c-jun
c-jun mRNA also appears to be induced throughout an ischemic hemisphere (Gass et al., 1992; An et al., 1993; Kinouchi et al., 1994a; Munell et al., 1994). The role of c-jun and its other family members is complex because different family members likely have different target genes in different cells. In addition, c-jun can form a homodimer as well as complex with c-fos family members. Therefore, it is not too surprising that c-jun expression has been associated with cell survival as well as cell death. C-jun is expressed in axotomized motor neurons that will survive axotomy, and c-jun is expressed in ischemia-induced tolerance models in which cells survive ischemia (Herdegen and Leah, 1998; Sommer et al, 1995; Kato et al., 1995a). However, phosphorylated c-jun (c-jun-P) appears to be expressed in cells that undergo apoptosis and may be expressed in ischemic cells that are dying or dead (Gillardon et al., 1999; Matsuoka et al., 1999; Domanska-Janik et al., 1999; Walton et al., 1999). Phosphorylated c-jun is coexpressed with possible jun/AP-1* target genes, APP, and CPP32 (caspase 3) in ischemic neurons (Walton et al., 1999).
Hippocampus
A number of genes are induced in hippocampus after MCAOs in rodents, particularly using the suture model. This includes c-fos and c-jun family members, the zinc finger immediate early genes, and a variety of other genes including COX-2 (Kinouchi et al., 1994a, b , c , 1999a, b ; Honkaniemi et al., 1997; Kamii et al., 1994b; Koistinaho et al., 1999). There are several possible explanations for such gene induction. First, there may be some hippocampal ischemia using the suture model. This is supported by TUNEL positive CA1 neurons in hippocampus using this model that is also associated with bilateral induction of HSP70 in CA1 pyramidal neurons (States et al., 1996). In addition, models that produce infarctions restricted to cortex do not generally induce the IEGs in hippocampus (Gass et al., 1992; Lindsberg et al., 1996).
It is also possible that IEGs are induced in hippocampus through excitatory mechanisms. Middle cerebral artery occlusions can produce repeated cortical spreading depressions that depolarize entorhinal cortex (Busch et al., 1995). Activation of entorhinal cortical inputs to hippocampus could account for the induction of many genes in hippocampus after MCAOs. This is supported by the suppression of hippocampal gene induction by MK-801 that prevents cortical spreading depression (Kinouchi et al., 1994c; Gass et al., 1992).
Gene induction in contralateral cortex and subcortical structures
Some studies also demonstrate that ipsilateral thalamus, ipsilateral substantia nigra, and contralateral cortex show induction of c-fos and other immediate early genes after an ipsilateral MCAO (Kinouchi et al., 1994a, b , c ). NGFI-A is induced bilaterally after MCAOs (Lin et al., 1996). IL-1 and TNF-α are not only induced in the ischemic hemisphere, but they are also induced in the contralateral hemisphere at lower levels (Buttini et al., 1996; Zhai et al., 1997). HSP27 can be induced in both hemispheres after a unilateral MCA stroke (Kato et al., 1995b). Arc is induced bilaterally in hippocampus and amygdala after MCAOs (Kunizuka et al., 1999). GLUT-1 and GLUT-3 can be induced in both hemispheres after unilateral stroke (Lee and Bondy, 1993; Urabe et al., 1996). Changes of gene expression in cerebellum are of interest because of the well-described phenomenon of cerebellar diaschisis (Ginsberg, 1990). However, there is little information on gene regulation in cerebellum after stroke. cGMP changes in cerebellum after MCAOs (Kader et al., 1993) and biliverdin reductase is induced in cerebellum after permanent MCAOs (Panahian et al., 1999).
The gene induction in the contralateral hemisphere, cerebellum, and many other subcortical regions is not caused by ischemia. Gene induction in these remote regions may be caused in part by acute ischemia-induced depolarization in the period immediately after a focal stroke. At longer times, changes in gene expression likely represent plastic changes in neurons and glia that must occur in the contralateral cortex, pons, cerebellum, spinal cord, and other brain regions that are connected directly or indirectly to the cortex and basal ganglia that were infarcted by the MCAOs. These changes of gene expression offer fruitful possibilities for possibly enhancing brain plasticity and behavioral recovery after stroke. Changes in GABA receptor subunits many days after stroke may be related to plastic responses of GABAergic neurons that could mediate recovery mechanisms (Neumann-Haefelin et al., 1999). There are bilateral changes of NMDA receptors in cortex after focal ischemia (Que et al., 1999) that may play a role in plastic changes in cortex and cortical motor and sensory maps (Nudo and Friel, 1999; Johansson, 2000).