
Abstract
Using an open cranial window technique, the authors investigated the mechanisms associated with the suppressed CO2 reactivity after mild controlled cortical impact (CCI) injury in rats. The dilation of arterioles (n = 7) to hypercapnia before injury was 38 ± 12%, which was significantly reduced both at 1 hour (23 ± 15% dilation) and at 2 hours after injury (11 ± 19% dilation). In the presence of L-arginine (10 mmol/L topical suffusion, 300 mg/kg intravenous infusion), the dilation of pial arterioles (n = 6) to hypercapnia was partially restored to 30 ± 6% at 2 hours after injury. In the presence of the nitric oxide (NO) donor, S-nitroso-N-acetylpenicillamine (SNAP) (10−8 mol/L topical suffusion), the dilation of pial arterioles (n = 5) to hypercapnia remained diminished (5 ± 7%) at 2 hours after injury. The dilation of pial arterioles (n = 4) to hypercapnia also remained suppressed (5 ± 6%) with topical suffusion of the free radical scavengers, polyethylene glycol-superoxide dismutase (60 units/mL) and polyethylene glycol-catalase (40 units/mL). The authors have shown that L-arginine at least partially restores the diminished response to hypercapnia after mild CCI injury. Furthermore, these data suggest that the beneficial effects of L-arginine are mediated by a combination of providing substrate for NO synthase and scavenging free radicals.
The cerebrovascular response to changes in PCO2 (or pH) appears to be a fundamental mechanism involved with maintaining adequate CBF (for review see Brian, 1998). Although the mechanism of action of CO2 remains the focus of current investigation, the mechanism may be partially involved with the coupling of CBF to energy metabolism (Kjallquist et al., 1969).
After traumatic brain injury (TBI), impaired CO2 reactivity has been a common finding both in the clinic and the laboratory. Clinically, CO2 reactivity has been shown to be slightly depressed in the acute stages after TBI, with restoration to normal after 24 hours (Cold et al., 1977; Enevoldsen and Jensen, 1978). Complete absence of CO2 reactivity carries a poor prognosis and usually is a terminal event. In animal studies, mild TBI impairs and severe TBI abolishes the CBF response to increased CO2 in the acute stages after injury (Saunders et al., 1979; Wei et al., 1980; Golding et al., 1999). Previously, cerebrovascular responsiveness to hypocapnia has been shown to be restored by oxygen radical scavengers and cyclooxygenase inhibitors, suggesting that the impaired cerebrovascular function after TBI is mediated by the generation of free radicals associated with increased prostaglandin production (Wei et al., 1981; Ellis et al., 1991).
Aside from impaired cerebrovascular function, nitric oxide (NO) production is reduced after TBI in patients (Jacob et al., 1993) and in experimental animals (Cherian et al., 1999). Moreover, treatment with L-arginine, the precursor for NO, has been shown to prevent the post-traumatic hypoperfusion after TBI (DeWitt et al., 1997). Because NO plays a critical role in the hypercapnic response (Iadecola, 1992; Wang et al., 1992), we tested the hypothesis that L-arginine restores the suppressed CO2 reactivity after mild controlled cortical impact (CCI) injury in the rat.
MATERIALS AND METHODS
Experiments were carried out in accordance with NIH guidelines for the care and use of laboratory animals and were approved by the Animal Protocol Review Committee at Baylor College of Medicine.
Surgery
Adult male Long-Evans rats (300 to 380 g, n = 53) were maintained on rodent chow and water before surgery. Anesthesia was induced with 4% isoflurane. Animals were tracheotomized (14-gauge catheter) and mechanically ventilated with 2% isoflurane in room air supplemented with 100% oxygen (to maintain PO2 at approximately 100 mm Hg). The tail artery was cannulated (22-gauge catheter) for monitoring mean arterial blood pressure, and the left femoral artery was cannulated with polyethylene tubing (PE-50) for sampling blood to measure gases. In some animals, the right femoral vein also was cannulated for drug infusion. Rectal temperature was maintained at 37°C using a heating pad and a temperature controller (Digi-Sense; Cole-Palmer, Vernon Hills, IL, U.S.A.). The head of each rat was secured in a stereotaxic frame. The skull was exposed by a midline incision, and the scalp (including the periosteum) and temporal muscle were reflected. Bleeding from the bone was controlled with bone wax.
Open cranial window preparation
An open skull technique with constant suffusion of artificial CSF (aCSF) was used to view the parietal cortex of rats after sham injury or mild CCI injury (for detailed description see Harper and Bohlen, 1984). Briefly, a dam of dental acrylic was constructed surrounding the right parietal cortex. Two 20-gauge needles were positioned on opposite sides of the dam and embedded in dental acrylic to act as inlet and outlet ports of aCSF. A 10-mm diameter craniotomy then was performed in the center of the dam with a dental drill using the bregma, lambda, and sagittal sutures as boundaries. To prevent thermal injury to the cortex, a stream of cool air was directed at the drilling site. Under an operative microscope, the dura mater was incised with a 27-gauge needle (slightly curved at the tip). With the aid of fine forceps and microscissors, the dura-arachnoid complex was reflected around the pial arteriole that was to be visualized. Bleeding from the dural vessels was controlled by constriction with the forceps. The exposed pial surface was continuously suffused with aCSF at a rate of 2 mL/min.
Suffusion of artificial CSF
The aCSF had the following composition (Baumbach et al., 1989): 131.9 mmol/L NaCl, 3.0 mmol/L KCl, 24.6 mmol/L NaHCO3, 6.7 mmol/L urea, 3.7 mmol/L glucose, and 1.5 mmol/L CaCl2. The aCSF was continuously bubbled with 10% CO2 balanced in N2 and directly connected to a pump and custom-built heat exchanger. After equilibration, the CSF sampled from the dam had a pH of 7.3 ± 0.02, PCO2 of 37.9 ± 2.0 mm Hg, PO2 of 117.9 ± 3.6 mm Hg, and a temperature of 37.2 ± 0.3°C (mean ± SD, n = 10).
Measurement of pial arteriole diameter
Pial arterioles with resting diameters of 16 to 83 μm were tested. Vessel diameter was monitored through an Olympus microscope (64× objective) connected to a video monitor, a time-date generator, a videotape recorder, and an image-shearing monitor (Instrumentation for Physiology and Medicine, San Diego, CA, U.S.A.).
Traumatic brain injury
Traumatic brain injury was induced using a CCI device as described in detail by Dixon et al. (1991). Briefly, a cylinder (30° from the vertical) having a rounded impactor tip of 9.5 mm in diameter was pneumatically driven into the brain at the site of the craniotomy. To induce a mild level of injury, the CCI device was adjusted so that the cylinder velocity was 3 m/sec with a 50-millisecond duration and a 2.5-mm deformation (Cherian et al., 1994). Sham controls underwent identical surgical manipulations as the CCI-injured rats with the exception of injury.
CO2 reactivity
Reactivity of CO2 was tested by measuring pial arteriole diameter under control conditions and after 10 minutes of hypercapnia (by adding 10% CO2 to the inspired air), at which time a steady state had been obtained. Two blood samples were obtained to measure blood gases, one before and one during each hypercapnic test. During hypercapnia, mean PCO2 = 75.3 ± 3.8 mm Hg (mean ± SD, n = 15). At the end of the study, animals were killed by intravenous injection of concentrated potassium chloride solution.
Protocols
Animals were randomly assigned into one of five protocols (see later). After completion of surgery and open cranial window preparation, animals were allowed 30 minutes for stabilization. Changes in pial arteriole diameter then were assessed in response to hypercapnia (hypercapnia test 1). After return to normocapnia, sham injury or mild CCI injury was induced. Changes in pial arteriole diameter were again assessed 1 hour later in response to hypercapnia (hypercapnia test 2). Drugs or vehicle then were suffused topically or infused intravenously. For the final time, changes in pial arteriole diameter were assessed in response to hypercapnia at 2 hours after sham or mild CCI injury (hypercapnia test 3). The specific protocols are described later.
Protocol 1. Effect of hypercapnia
Changes in pial arteriole diameter in response to hypercapnia (10 minutes) were made before and 1 and 2 hours after sham (n = 8) or mild CCI injury (n = 7). The sham-injured animals served as a time control to establish the reproducibility of responses to hypercapnia.
Protocol 2. Effect of L-arginine
Changes in pial arteriole diameter in response to hypercapnia (10 minutes) were made before and 1 and 2 hours after mild CCI injury. At 1 hour after mild CCI injury, L-arginine (n = 6) was topically suffused (10 mmol/L for 1 hour) and infused intravenously (300 mg/kg over 20 minutes). The efficacy of L-arginine as a NO substrate was verified in naive animals (n = 6) by assessing changes in arteriole diameter in response to hypercapnia in the presence of Nω-nitro-L-arginine (L-NAME) (10−1 mmol/L) before and after topical suffusion of L-arginine (10 mmol/L for 1 hour) and intravenous infusion (300 mg/kg for 20 minutes).
Protocol 3. Effect of glibenclamide
In contrast to others, one laboratory (Kontos and Wei, 1996) reports a role of the ATP-sensitive potassium (KATP) channels in hypercapnia-induced vasodilation. To ascertain whether the KATP channels are involved in the hypercapnic response, and therefore involved in the compromised response after CCI injury, we assessed changes in pial arteriole diameter (n = 6) in response to hypercapnia before and after topical suffusion of glibenclamide (10−5 mol/L for 15 minutes). The efficacy of glibenclamide as an inhibitor of KATP channels was established by cosuffusion of the KATP agonist, pinacidil (10−8 mol/L, n = 4).
Protocol 4. Effect of SNAP
Changes in pial arteriole diameter (n = 5) in response to hypercapnia were made before and 1 and 2 hours after mild CCI injury. At 2 hours after mild CCI injury, the NO donor SNAP was topically suffused for 5 minutes (10−8 mmol/L). The efficacy of SNAP as a NO donor was verified in naive animals (n = 3) by assessing the changes in arteriole diameter in response to hypercapnia in the presence of L-NAME (10−1 mmol/L) before and after topical suffusion of SNAP (10−8 mol/L for 5 minutes).
Protocol 5. Effect of polyethylene glycol-superoxide dismutase and polyethylene glycol-catalase
Changes in pial arteriole diameter (n = 4) in response to hypercapnia were made before and 1 and 2 hours after mild CCI injury. At 1 hour after mild CCI injury, polyethylene glycol-superoxide dismutase (PEG-SOD) (60 units/mL) and polyethylene glycol-catalase (PEG-catalase) (40 units/mL) were topically suffused concomitantly for 1 hour. The efficacy of PEG-SOD and PEG-catalase as free radical scavengers was verified in naive animals (n = 4) after topical suffusion of bradykinin (10-6 mmol/L) (Sobey et al., 1997).
Drugs
All drugs were purchased from Sigma Chemicals (St. Louis, MO, U.S.A.). Stock solutions of L-arginine (1 mmol/L), SNAP (10−4 mmol/L), L-NAME (10−1 mmol/L), PEG-SOD (4000 units/mL), and PEG-catalase (40,000 units/mL) were dissolved in distilled water. Stock solutions of bradykinin (10−2 mmol/L) were dissolved in saline (Sobey et al., 1997). Stock solutions of pinacidil (10−3 mmol/L) and glibenclamide (10−2 mmol/L) were made first by making 2×10−3 mmol/L and 2×10−2 mmol/L solutions, respectively, in dimethyl sulfoxide and then dissolving the solutions 1:1 with saline so that the final concentrations were 10−3 mmol/L and 10−2 mmol/L, respectively (Sobey et al., 1997). The vehicle for pinacidil and glibenclamide (0.05% dimethyl sulfoxide) had no effect on pial arteriole diameter (data not shown).
Data analysis
Data are expressed as mean ± SD, and statistical comparisons were performed using either a one-way analysis of variance with repeated measures (RM ANOVA) or two-way RM ANOVA. Post hoc comparisons were made using a Student-Newman-Keuls test. Significance was taken to be P < 0.05.
RESULTS
Arterial blood variables
The mean arterial blood pressure, arterial pH, and PCO2 levels were measured in all groups before and during each hypercapnic challenge. Table 1 presents these values for the sham-injured and mild CCI-injured animals. Similar values were observed for the remaining treatment groups (data not shown). In all animals, arterial PO2 levels were maintained at 122.7 ± 16.0 mm Hg (n = 53). During hypercapnia, both pH and PCO2 levels were significantly different from normocapnia (P < 0.05; two-way repeated measures ANOVA followed by Student-Newman-Keuls test); however, there were no differences between sham- and CCI-injured animals in the response to hypercapnia.
The mean arterial blood pressure and blood gases measured in each group
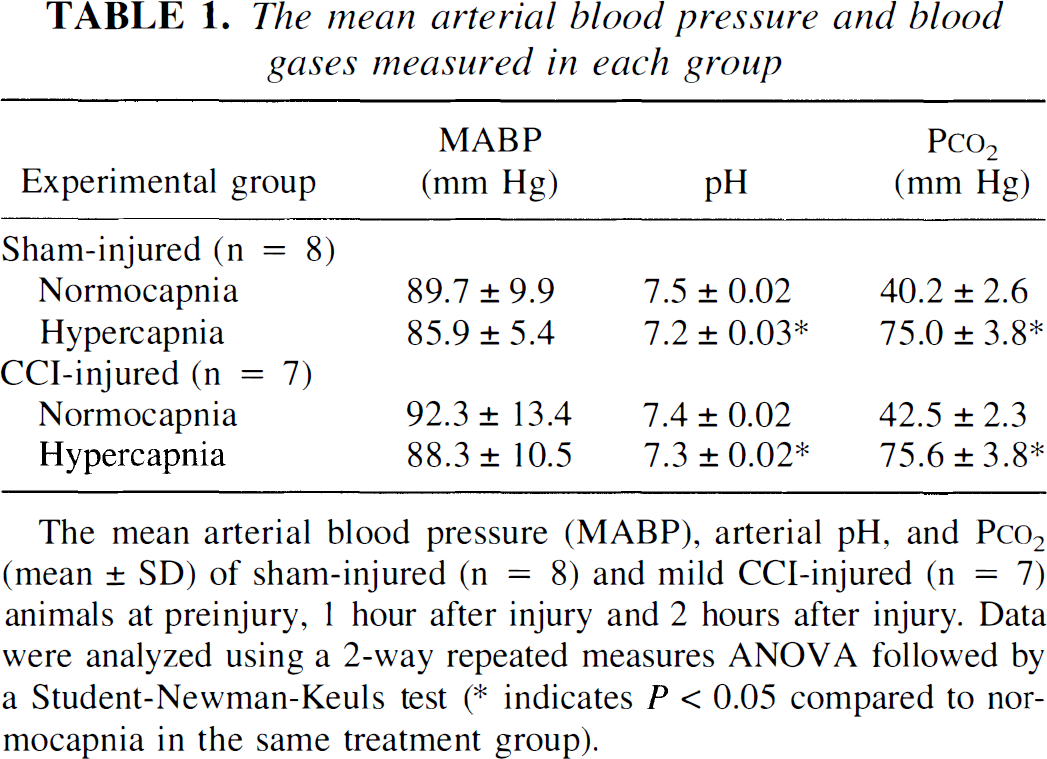
The mean arterial blood pressure (MABP), arterial pH, and P
Protocol 1. Effect of hypercapnia
Figure 1A demonstrates that there was no significant difference in responses of pial arterioles to hypercapnia over the 3-hour monitoring period (P = ns; one-way RM ANOVA followed by Student-Newman-Keuls test). This established the reproducibility of the hypercapnic response over time. In injured animals, the dilation of pial arterioles to hypercapnia before injury was 38 ± 12%, which was significantly reduced both at 1 hour (23 ± 15% dilation) and at 2 hours (11 ± 19% dilation) after mild CCI injury (P < 0.05 compared with before injury; one-way RM ANOVA followed by Student-Newman-Keuls test; Fig. 1B). Before injury, pial arterioles (n = 4) dilated from 59 ± 20 μm to 85 ± 34 μm after 10 minutes of hypercapnia. At 2 hours after mild CCI injury, pial arterioles (n = 4) dilated from 49 ± 17 to 55 ± 17 μm in response to hypercapnia.
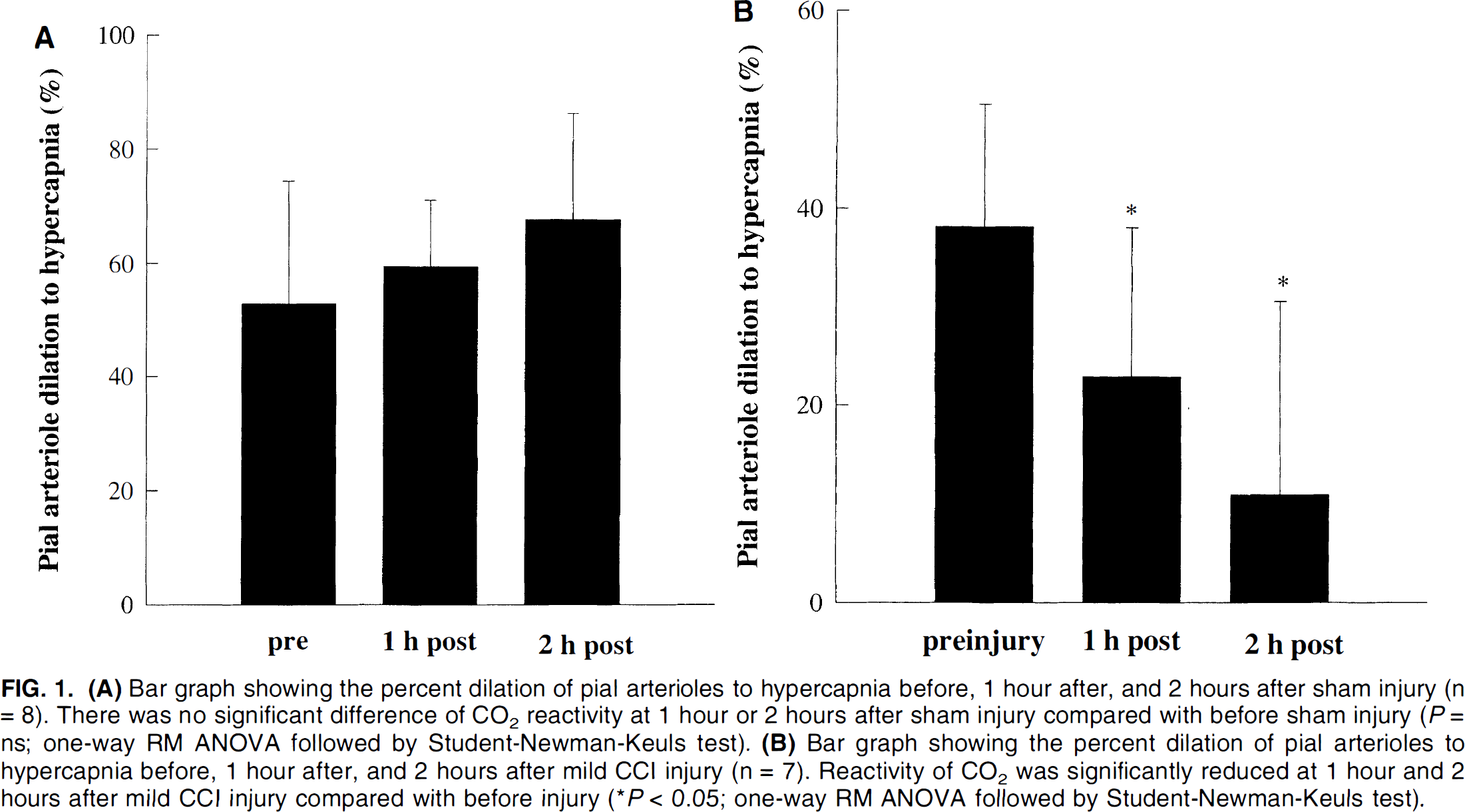
Protocol 2. Effect of L-arginine
When L-arginine (10 mmol/L topical suffusion, 300 mg/kg intravenously) was given at 1 hour after mild CCI injury, the dilation to hypercapnia was partially restored to 30 ± 6% at 2 hours after injury (P = ns compared with before injury; one-way RM ANOVA followed by Student-Newman-Keuls test; Fig. 2A). No significant changes in pial arteriole diameter were noted after 1 hour of suffusion with L-arginine. L-Arginine suffusion and infusion did not augment hypercapnic vasodilation in a control group (data not shown). Similar to untreated animals, at 1 hour after mild CCI injury (before L-arginine administration), the dilation to hypercapnia was significantly reduced at 5.5 ± 10.5% (P < 0.05; one-way RM ANOVA followed by Student-Newman-Keuls test). The efficacy of L-arginine as a NO substrate was confirmed in naive animals by restoring the L-NAME-induced reduction of the hypercapnic response (Fig. 2B). No significant changes in pial arteriole diameter were observed after 1 hour of suffusion with L-NAME. This agrees with previous reports showing that pial arteriole diameter (Pelligrino et al., 1995) or CBF (Fabricius and Lauritzen, 1994) remains unchanged after topical suffusion of nitric oxide synthase (NOS) inhibitors. In the presence of L-NAME, vasodilation to hypercapnia was significantly inhibited by 53 ± 20% (P < 0.05 compared with before L-NAME; one-way RM ANOVA followed by Student-Newman-Keuls test), which was restored in the presence of L-arginine (Fig. 2B).
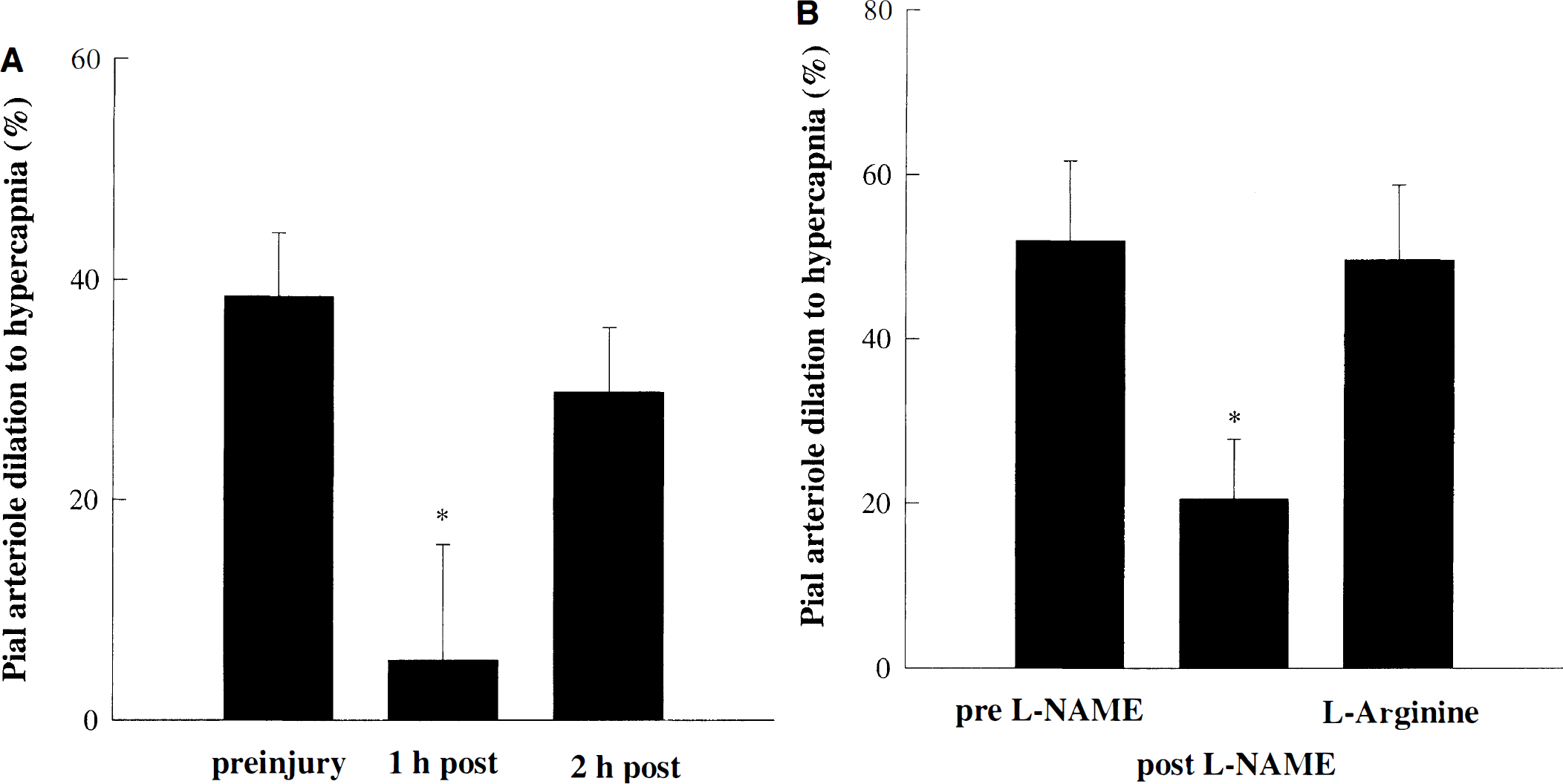
Protocol 3. Effect of glibenclamide
Figure 3A demonstrates that topical suffusion of glibenclamide (10−5 mmol/L) had no effect on pial arteriole dilation to hypercapnia. Pial arteriole dilation to hypercapnia in the absence of glibenclamide was 29 ± 9% compared with 35 ± 16% in the presence of glibenclamide (P = ns; one-way RM ANOVA followed by Student-Newman-Keuls test). Glibenclamide had no effect on the diameter of cerebral arterioles under control conditions. We confirmed the efficacy of glibenclamide as an inhibitor of KATP channels by suppressing the dilation of the KATP agonist, 10−8 mmol/L pinacidil (Fig. 3B). Pial arteriole dilation to pinacidil under control conditions was 37 ± 14% compared with 9 ± 3% in the presence of glibenclamide (P < 0.05; one-way RM ANOVA followed by Student-Newman-Keuls test).
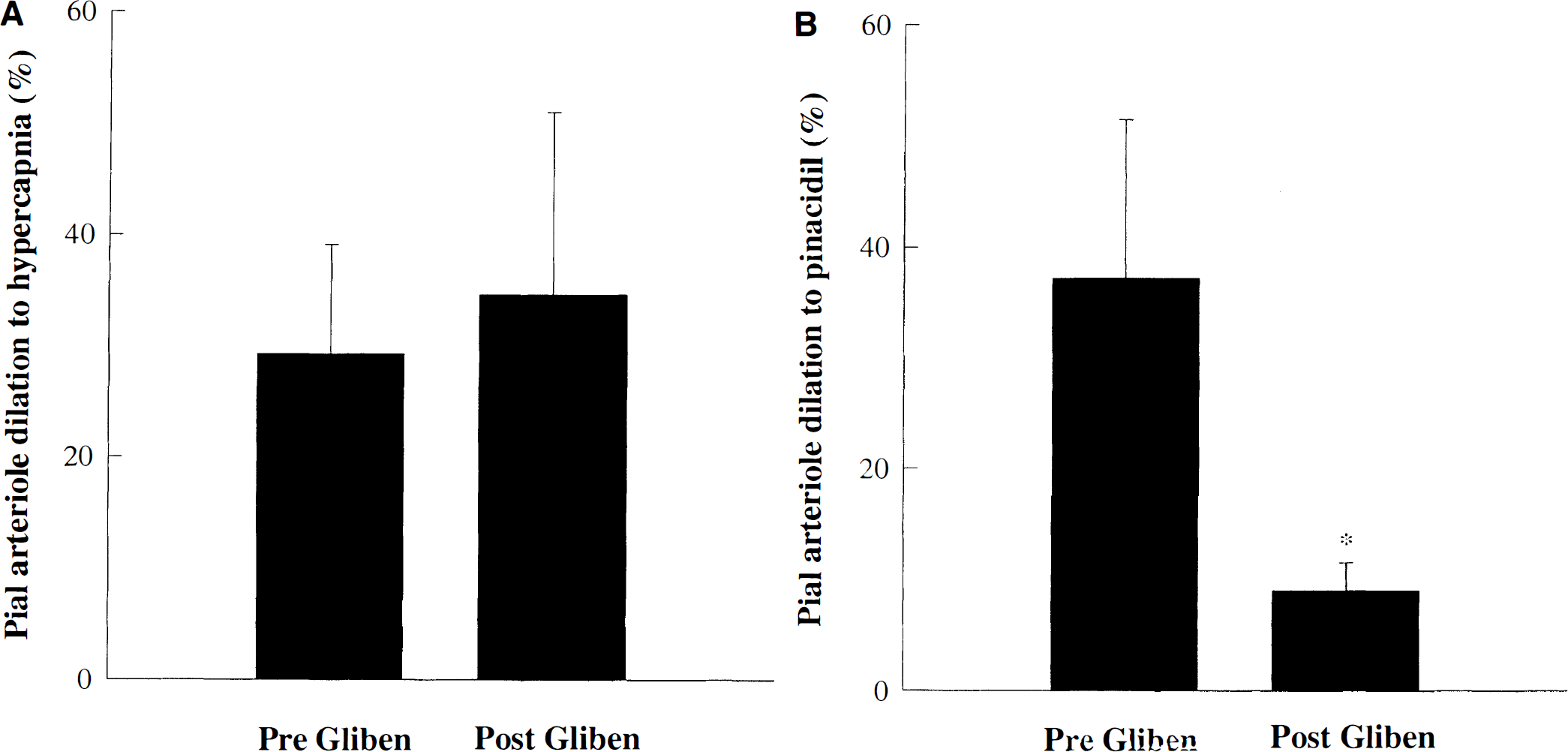
Protocol 4. Effect of SNAP
At 2 hours after mild CCI injury, the NO donor SNAP (10−8 mmol/L) was topically suffused for 5 minutes (Fig. 4A). However, the dilation to hypercapnia still was significantly suppressed (5 ± 7%). The efficacy of SNAP as a NO donor was confirmed in naive animals by restoring the L-NAME-induced reduction of the hypercapnic response (Fig. 4B). Notice that this is the highest concentration of SNAP that does not produce consistent vasodilation (Kontos and Wei, 1996). In other words, this concentration of SNAP establishes a subthreshold level of NO.
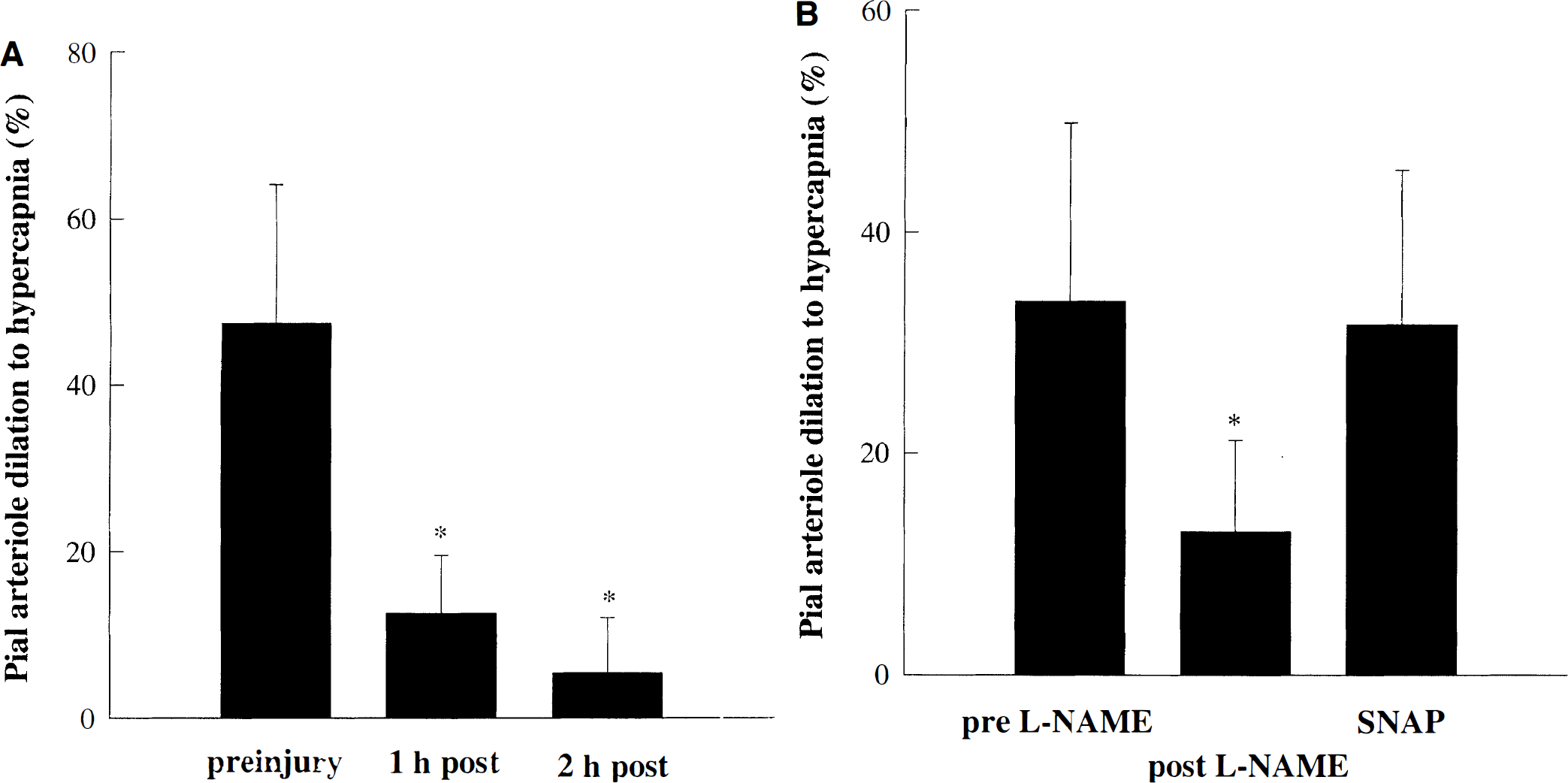
Protocol 5. Effect of PEG-SOD and PEG-catalase
At 1 hour after mild CCI injury, the free radical scavengers, PEG-SOD and PEG-catalase, were topically suffused for 1 hour (Fig. 5A). However, the dilation to hypercapnia still was significantly suppressed (5 ± 6%) at 2 hours after injury (P < 0.05 compared with before injury; one-way RM ANOVA followed by Student-Newman-Keuls test). The efficacy of PEG-SOD and PEG-catalase as free radical scavengers was confirmed in naive animals by inhibiting the dilation to 10−6 mmol/L bradykinin topical suffusion (Sobey et al., 1997) (Fig. 5B). Under control conditions, bradykinin dilated pial arterioles by 25 ± 2%; however, in the presence of PEG-SOD and PEG-catalase, dilations were significantly inhibited to 5 ± 2% (P < 0.05; one-way RM ANOVA followed by Student-Newman-Keuls test).
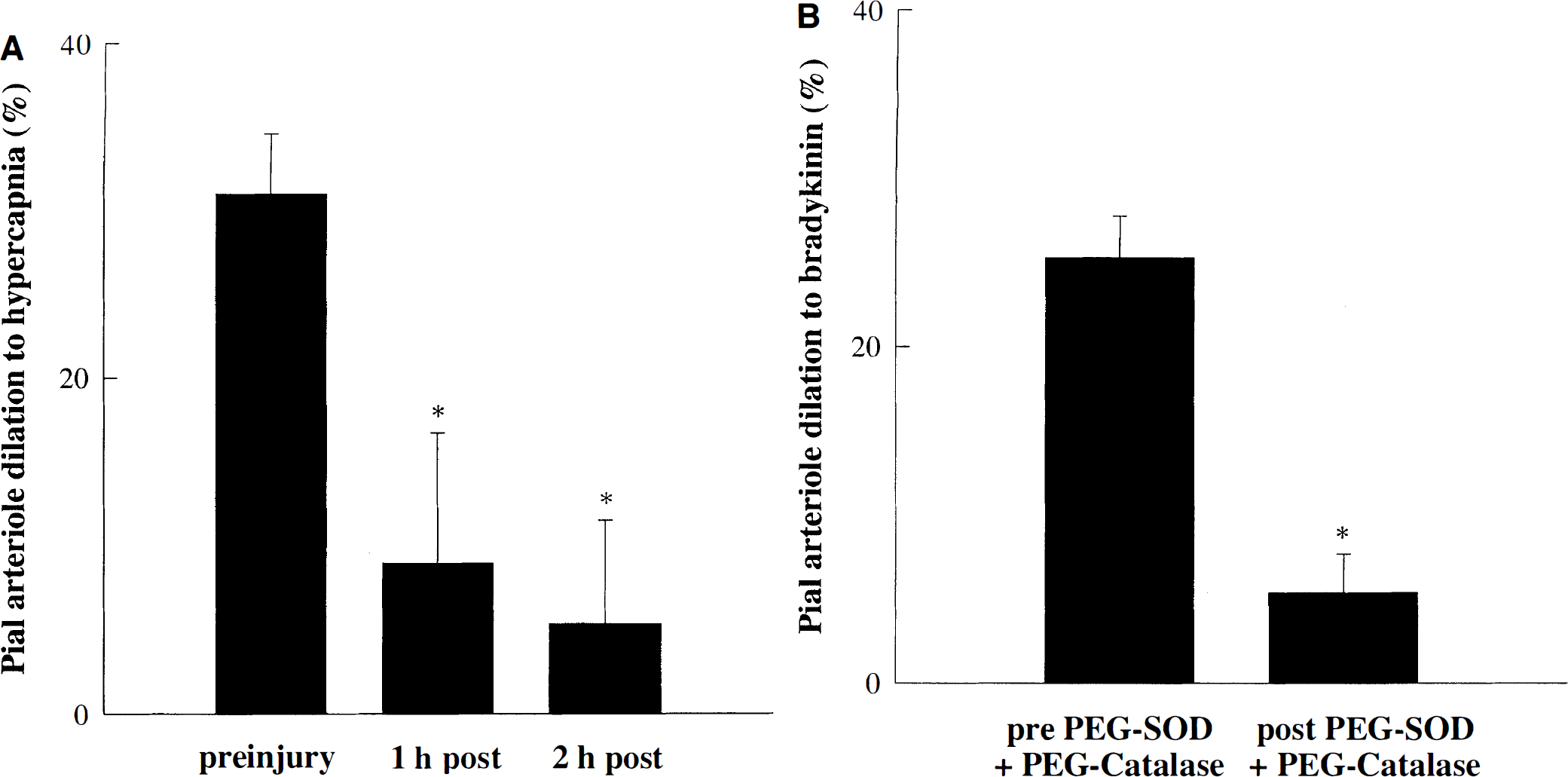
DISCUSSION
There are two major findings in the current study. First, L-arginine at least partially restored the suppressed response to hypercapnia when administered at 1 hour after mild CCI injury in the rat. This was not found with the NO donor, SNAP, or the free radical scavengers, PEG-SOD and PEG-catalase. Second, our data suggest that the beneficial effects of L-arginine after TBI are mediated by a combination of providing substrate for NOS and scavenging free radicals.
Pial arteriole dilation to hypercapnia is diminished after CCI injury
We have shown, using an open cranial window technique, that the response to hypercapnia is suppressed for at least 2 hours after mild CCI injury. This confirmed our previous findings showing that the cortical perfusion response (using laser Doppler flowmetry) to hypercapnia was severely attenuated (Golding et al., 1999) and extends previous reports demonstrating impairment of CO2 reactivity after more severe levels of TBI (Wei et al., 1980; Lewelt et al., 1982). Wei et al. have shown that the suppressed reactivity to CO2 may be preserved by preventing the rise in blood pressure after severe head injury (Wei et al., 1980). However, blood pressure did not rise after mild CCI injury (data not shown) and, therefore, could not be an attributing factor in our study.
L-Arginine restores the diminished hypercapnic response after CCI injury
We have shown that L-arginine was able to at least partially restore the pial arteriole response to hypercapnia after mild CCI injury. These data supplement previous reports suggesting a beneficial role of L-arginine after TBI (DeWitt et al., 1997). Whereas DeWitt et al. have demonstrated complete prevention of cerebral hypoperfusion when L-arginine was administered 5 minutes after TBI, the current study shows that L-arginine may still elicit beneficial effects when administered at 1 hour after injury. This is of particular clinical interest because of the larger temporal window of efficacy that it offers.
KATP channels do not play an integral role in the pial arteriole dilation to hypercapnia
One laboratory reports that (1) KATP channels are involved with the dilation to hypercapnia, and (2) L-arginine modulates KATP channels (Kontos and Wei, 1996). If KATP channels were involved with the hypercapnic vasodilation in our studies, it could be speculated that the restoration of the response to hypercapnia produced by L-arginine could be mediated by modulation of the KATP channels. However, our current findings show a lack of effect of KATP channel inhibition on hypercapnia-induced cerebrovasodilation. This agrees with many other reports in the literature demonstrating that at this level of hypercapnia, cerebral vasodilation during hypercapnia is dependent largely on production of NO (Faraci et al., 1994; Reid et al., 1995; Wang et al., 1998).
SNAP does not restore the diminished hypercapnic response after CCI injury
In contrast to L-arginine, we have shown that the NO donor SNAP did not restore the diminished hypercapnic response after mild CCI injury. It could be speculated that the inhibition of NO production by TBI may be different than simply NOS inhibition in naive rats. It has been established that NOS inhibition diminishes the response to hypercapnia (Wang et al., 1992; Reid et al., 1995; current results). In addition, subthreshold NO, or cyclic GMP, restores the L-NAME-induced inhibition of the hypercapnic response (Wang et al., 1998). It therefore could be predicted that if TBI interfered with NO production in a manner similar to that of NOS inhibition, then the NO donor, SNAP, would restore the reduced response to hypercapnia after TBI. However, we have shown that SNAP did not restore the diminished hypercapnic response after mild CCI injury. It is possible that the diminished CO2 reactivity after TBI is affected by both a limitation of NO and excessive free radical production. Because NO donors are known to produce peroxynitrite (Holm et al., 1998), this would rationalize the finding that SNAP did not restore CO2 reactivity. Our results confirm those of Wada et al., who report that L-arginine, but not the NO donor SIN-1, provides histologic protection after TBI (Wada et al., 1997).
PEG-SOD and PEG-catalase do not restore the diminished hypercapnic response after CCI injury
Because of their high reactivity and capacity to produce cellular damage, oxygen radicals have been implicated as mediators of tissue damage after TBI (Wei et al., 1981). Previous studies by Wei et al. (Wei et al., 1981; Kontos and Wei, 1992) have shown that the reduced CO2 reactivity observed after brain injury can be restored by topical application of the free radical scavengers PEG-SOD and PEG-catalase. Moreover, SOD has been shown to improve the posttraumatic cerebral hypoperfusion after TBI (Muir et al., 1995; DeWitt et al., 1997). However, in these previous studies, the free radical scavengers were administered either before injury or immediately after injury. It is possible that the temporal window of efficacy of SOD, in contrast to L-arginine, is limited, which would explain why we did not see a beneficial effect in the current study.
CONCLUSION
The beneficial effects of L-arginine after mild CCI injury could be multifactorial. First, L-arginine may simply provide substrate for NO production. Data from our laboratory demonstrate that both brain tissue NO and interstitial nitrates and nitrites are significantly decreased after CCI injury (Cherian et al., 1999). Moreover, systemic administration of L-arginine after injury (300 mg/kg intravenously) was shown to increase brain tissue NO production (Cherian et al., unpublished observations). Taken together along with the results of the current study, these data suggest that L-arginine may be limiting after TBI. Alternatively, L-arginine may be dissociated from NOS after TBI. Whereas L-arginine is concentrated primarily in astrocytes in the normal adult rat brain (Aoki et al., 1991), neuronal NOS (the isoform involved in the hypercapnic response) is located in neurons. It is possible that CCI injury may simply prevent astrocytic L-arginine from gaining access to neuronal NOS. Aside from the direct involvement of NO with L-arginine supplementation, metabolites of L-arginine may serve as neuroprotective agents. To illustrate, L-arginine can be decarboxylated to form agmatine, which has properties of an endogenous neurotransmitter (Reis and Regunathan, 1999). Agmatine also is a precursor of brain putrescine, which blocks the ligand-gated N-methyl-D-aspartate (NMDA) receptor channel at sites distinct from polyamine sites. Because activation of NMDA receptor channels has been shown to play a major role in the excitotoxicity after TBI (Faden et al., 1989; Palmer et al., 1993), agmatine may provide beneficial effects by acting as a unique NMDA receptor channel antagonist.
In addition to diminishing NO production, the absence (or limitation) of L-arginine stimulates brain NOS to generate superoxide and peroxynitrite, which are known to be neurotoxic (Heinzel et al., 1992; Pou et al., 1992; Klatt et al., 1993). Supplementation of L-arginine would, therefore, not only provide substrate for NO production but also prevent production of toxic free radicals.
Second, L-arginine itself may be providing protective free radical scavenging effects (Rehman et al., 1997). Because L-arginine has been shown to scavenge the hydroxyl radical (Rehman et al., 1997), it is possible that the beneficial effects of L-arginine are mediated by its capacity to scavenge free radicals. We did not observe any beneficial effect of free radical scavengers alone, which suggests that the suppressed CO2 reactivity after mild CCI injury is mediated by factors related to both NO generation and free radicals.
In conclusion, the results of the current study suggest that L-arginine at least partially restores the suppressed response to hypercapnia after mild CCI injury in the rat and that this is mediated by a combination of providing substrate for NOS and scavenging free radicals.
Footnotes
Acknowledgments
The authors wish to express their sincere thanks to Dr. Christopher Sobey (University of Melbourne, Australia) and Dr. Frank Faraci (University of Iowa, Iowa City, Iowa, U.S.A.) for their guidance with the cranial window preparation.