
Abstract
The goal of this study was to test the hypothesis that administration of L-arginine, a substrate for the synthesis of nitric oxide, restores endothelium-dependent dilatation of the basilar artery during diabetes mellitus. We measured the diameter of the basilar artery in vivo in nondiabetic and diabetic (streptozotocin; 50–60 mg/kg i.p.) rats in response to endothelium-dependent agonists (acetylcholine and bradykinin) and an endothelium-independent agonist (nitroglycerin) before and during application of L-arginine. Topical application of acetylcholine (1.0 and 10 μM) and bradykinin (1.0 and 10 μM) produced dilatation in nondiabetic rats of the basilar artery which was impaired in diabetic rats. Topical application of nitroglycerin (0.1 and 1.0 μM) produced similar dilatation of the basilar artery in nondiabetic and diabetic rats. Topical application of L-arginine (0.1 and 3 mM) did not enhance dilatation of the basilar artery in response to acetylcholine and bradykinin in diabetic rats. Thus, impairment of dilatation of the basilar artery in diabetic rats in response to acetylcholine and bradykinin appears to be related to a mechanism unrelated to the availability of L-arginine for nitric oxide synthase.
Diabetes mellitus impairs endothelium-dependent relaxation of large and small cerebral blood vessels (Fujii et al., 1992; Pelligrino and Albrecht, 1991; Pelligrino et al., 1992; Dandona et al., 1978; Mayhan, 1989, 1992, 1993; Mayhan et al., 1991). The mechanism(s) by which diabetes impairs endothelium-dependent dilatation of cerebral blood vessels appear to differ. We have suggested that impaired endothelium-dependent dilatation of rat pial (cortex) arterioles is related to the production of a cyclooxygenase constrictor substance that, presumably, activates the prostaglandin H2/thromboxane A2 receptor (Mayhan et al., 1991). Other investigators have suggested that impaired endothelium-dependent dilatation of rat pial arterioles was related to the activation of protein kinase-C (Pelligrino et al., 1994). Thus, alterations in several cellular pathways may account for impaired endothelium-dependent reactivity of cerebral arterioles.
We were the first to examine endothelium-dependent responses of the basilar artery in vivo during diabetes mellitus (Mayhan, 1992). Similar to that which we have reported for pial arterioles (Mayhan et al., 1991; Mayhan, 1989; Pelligrino et al., 1994), endothelium-dependent responses of the basilar artery to acetylcholine and bradykinin were profoundly impaired in diabetic rats (Mayhan, 1992). However, in contrast to that reported for pial arterioles (Mayhan et al., 1991), impaired dilatation of the basilar artery during diabetes mellitus could not be explained by the production of a cyclooxygenase constrictor substance and/or activation of the prostaglandin H2/thromboxane A2 receptor (Mayhan, 1992). Thus, other cellular mechanisms must account for impaired endothelium-dependent dilatation of the basilar artery during diabetes mellitus.
A recent study (Pieper and Peltier, 1995) suggests that impaired relaxation of the thoracic aorta in rats during diabetes mellitus could be enhanced by treatment with L-arginine (3 mM), a substrate for nitric oxide synthase. Thus, it appears that impaired endothelium-dependent responses during diabetes mellitus may be related to a decreased availability of L-arginine to nitric oxide synthase, leading to a decreased agonist-induced synthesis/release of nitric oxide. The goal of the present study was to examine whether impaired endothelium-dependent dilatation of the basilar artery during diabetes mellitus could be enhanced by L-arginine.
METHODS
Induction of diabetes
Male Sprague–Dawley rats (200–220 g body wt) were divided randomly into nondiabetic and diabetic groups. All rats were housed in hanging cages and had access to food and water ad libitum. One group of rats was injected with streptozotocin (50–60 mg/kg i.p.) to induce diabetes. The second group of rats (nondiabetic) was injected with vehicle. Blood samples, for measurement of blood glucose concentration, were obtained at 2 weeks after injection of streptozotocin or vehicle, and on the day of the experiment. Other than a failure to gain as much weight as the nondiabetic rats, there were no differences in the general condition of nondiabetic and diabetic rats.
Preparation of animals
Rats were prepared for studies at 2.5–3.5 months after injection of streptozotocin or vehicle. Rats were anesthetized (pentobarbital sodium, 50 mg/kg body wt i.p.), and a tracheotomy performed. Animals were ventilated mechanically with room air and supplemental oxygen. A catheter was inserted into a femoral vein for injection of drugs, and a femoral artery was cannulated for measurement of arterial pressure. Skeletal muscle paralysis was induced with gallamine triethiodide (5–10 mg/kg i.v.). Supplemental anesthesia was administered at a dose of 10–20 mg/kg/h i.v. No differences were observed between nondiabetic and diabetic rats with regard to the amount of pentobarbital and gallamine triethiodide (mg/kg body wt) required to induce/maintain anesthesia and paralysis, respectively.
After placement of all catheters, the animal was placed in a head holder in a supine position. The larynx and esophagus were retracted rostrally and laterally and the musculature covering the basioccipital bone removed. Then, a craniotomy was made in the bone at the base of the skull. The dura was incised to expose the basilar artery. We (Mayhan, 1991; Mayhan, 1990b) and others (Faraci, 1990) have used this method to expose the basilar artery previously.
The cranial window was suffused with artificial cerebral spinal fluid (CSF) (38°C) and bubbled continuously to maintain gases within normal limits. Blood gases were monitored and maintained within normal limits throughout the experiment.
Diameter of the basilar artery was measured on-line using a video image shearing device (Model 908, Instrumentation for Physiology and Medicine, San Diego, CA, U.S.A.).
Experimental protocol
The basilar artery preparation was allowed to equilibrate for 30 min after the craniotomy. Then, we examined responses of the basilar artery in nondiabetic and diabetic rats to agonists [acetylcholine (1.0 and 10 μM) and bradykinin (1.0 and 10 μM)] that, presumably, produce dilatation via the synthesis/release of nitric oxide or a nitric oxide-containing compound. We also examined dilatation of the basilar artery in nondiabetic and diabetic rats in response to an endothelium-independent agonist [nitroglycerin (0.1 and 1.0 μM)]. Drugs were mixed in artificial cerebral spinal fluid and superfused over the basilar artery preparation. The application of vehicle did not affect vessel diameter and the application of agonists was randomized. The diameter of the basilar artery was measured immediately before the application of agonists and every min for 5 min during application of the agonists. Steady-state responses to the agonists were reached within 2–3 min after application, and the diameter of the basilar artery returned to control within 2–4 min after application of the agonists was stopped.
After examining responses to the agonists under control conditions, we then examined the possibility that impaired responses of the basilar artery to acetylcholine and bradykinin may be related to an alteration in the availability of L-arginine to nitric oxide synthase. To test this possibility, we again examined responses of the basilar artery in nondiabetic and diabetic rats to the agonists 30 min after starting a continuous topical application of L-arginine (0.1 or 3 mM). A previous study (Faraci, 1990) has shown that a concentration of L-arginine similar to that used in the present study is efficacious in the basilar artery of rats. Vasoconstriction produced with an enzymatic inhibitor of nitric oxide was completely reversed by 0.1 mM L-arginine (Faraci, 1990). In additional studies, we examined the effect of D-arginine (3 mM) on responses of the basilar artery.
Statistical analysis
An unpaired r-test was used to compare values between different groups of animals. A paired r-test was used to compare responses of the basilar artery before and after application of L-arginine. A p-value of 0.05 was considered to be significant.
RESULTS
Control conditions
Baseline diameter of the basilar artery was 281 ± 35 μm in nondiabetic rats and 318 ± 49 μm in diabetic rats (mean ± SD; p > 0.05). Blood pressure was 114 ± 18 mm Hg in nondiabetic rats and 123 ± 19 mm Hg in diabetic rats (mean ± SD; p > 0.05). On the day of the experiment, blood glucose concentration was 106 ± 55 mg/dl in nondiabetic rats and 371 ± 48 mg/dl in diabetic rats (mean ± SD; p < 0.05 versus nondiabetic rats). Body weight was 412 ± 53 g in nondiabetic rats and 294 ± 55 g in diabetic rats (mean ± SD; p < 0.05 versus nondiabetic rats). These values are similar to those reported previously by us (Mayhan, 1989; Mayhan et al., 1991; Mayhan, 1990a).
Responses in nondiabetic and diabetic rats
Dilatation of the basilar artery in response to acetylcholine and bradykinin was significantly impaired during diabetes mellitus. Acetylcholine (1.0 and 10 μM) dilated the basilar artery by 21 ± 12 and 32 ± 17% (mean ± SD), respectively, in nondiabetic rats but by only 5 ± 5 and 8 ± 6%, respectively, in diabetic rats (p < 0.05) (Fig. 1). Bradykinin (1.0 and 10 μM) dilated the basilar artery by 21 ± 15 and 29 ± 17%, respectively, in nondiabetic rats, but by only 3 ± 4 and 5 ± 8%, respectively, in diabetic rats (p < 0.05) (Fig. 1).
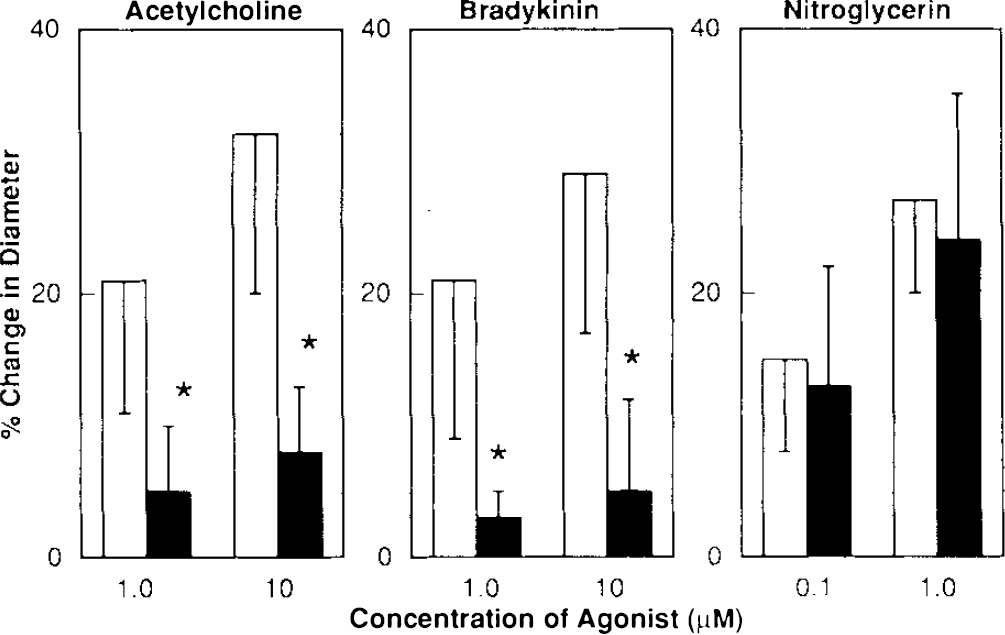
Responses of the basilar artery to acetylcholine, bradykinin and nitroglycerin in nondiabetic (open bars) and diabetic (closed bars) rats. Values are means ± SD. *p < 0.05 versus nondiabetic rats.
In contrast, nitroglycerin produced similar dose-related dilatation of the basilar artery in nondiabetic and diabetic rats. Nitroglycerin (0.1 and 1.0 μM) dilated the basilar artery by 15 ± 9 and 27 ± 11%, respectively, in nondiabetic rats and by 13 ± 11 and 24 ± 16%, respectively, in diabetic rats (p > 0.05) (Fig. 1). Thus, impaired responses of the basilar artery to acetylcholine and bradykinin in diabetic rats cannot be explained by nonspecific impairment of vasodilatation during diabetes mellitus.
Responses following L-arginine
Topical application of L-arginine (0.1 mM) produced only minimal changes in baseline diameter of the basilar artery in nondiabetic (−2 ± 3%) and diabetic (−5 ± 7%) rats.
L-arginine (0.1 mM) did not alter dilatation of the basilar artery in nondiabetic rats in response to acetylcholine (Fig. 2) and bradykinin (Fig. 3). In addition, topical application of L-arginine (0.1 mM) did not enhance dilatation of the basilar artery in diabetic rats in response to acetylcholine (Fig. 2) and bradykinin (Fig. 3). Furthermore, L-arginine (0.1 mM) did not alter dilatation of the basilar artery in response to nitroglycerin in nondiabetic and diabetic rats (Fig. 4).
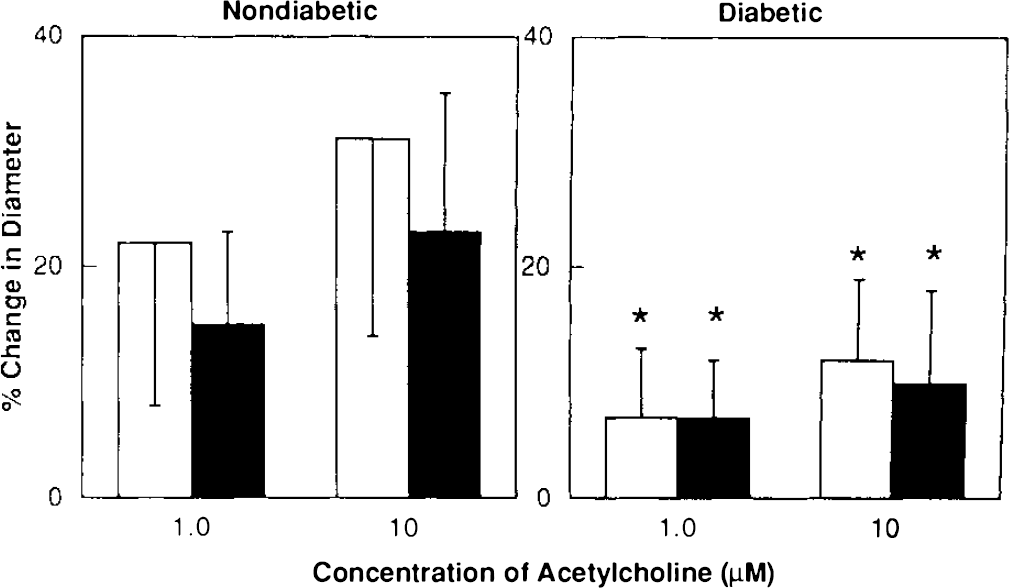
Responses of the basilar artery to acetylcholine in nondiabetic and diabetic rats before (open bars) and after (closed bars) application of L-arginine (0.1 mM). Values are means ± SD. *p < 0.05 versus nondiabetic rats.
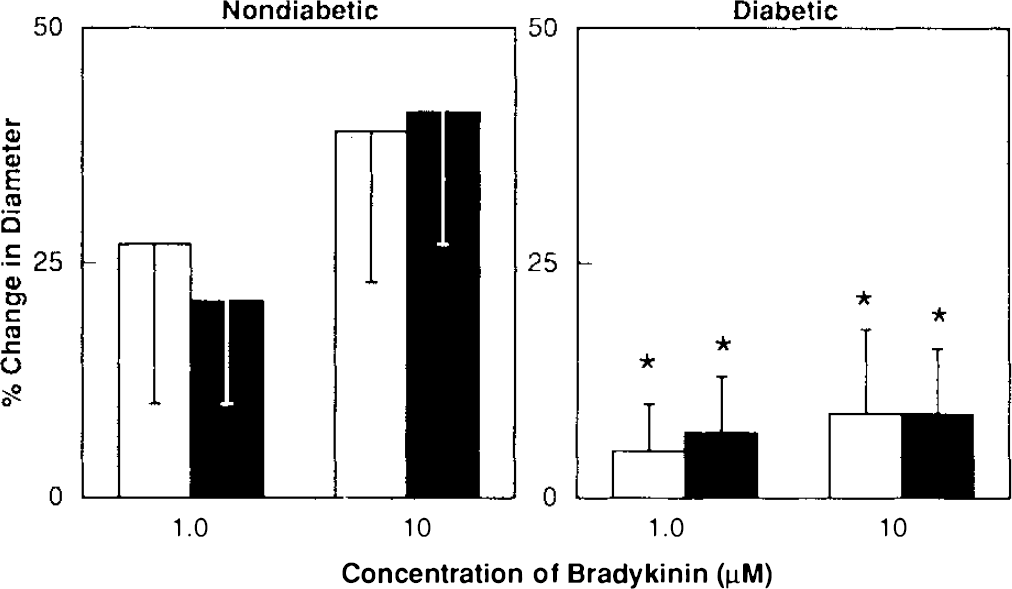
Responses of the basilar artery to bradykinin in nondiabetic and diabetic rats before (open bars) and after (closed bars) application of L-arginine (0.1 mM). Values are means ± SD. *p < 0.05 versus nondiabetic rats.
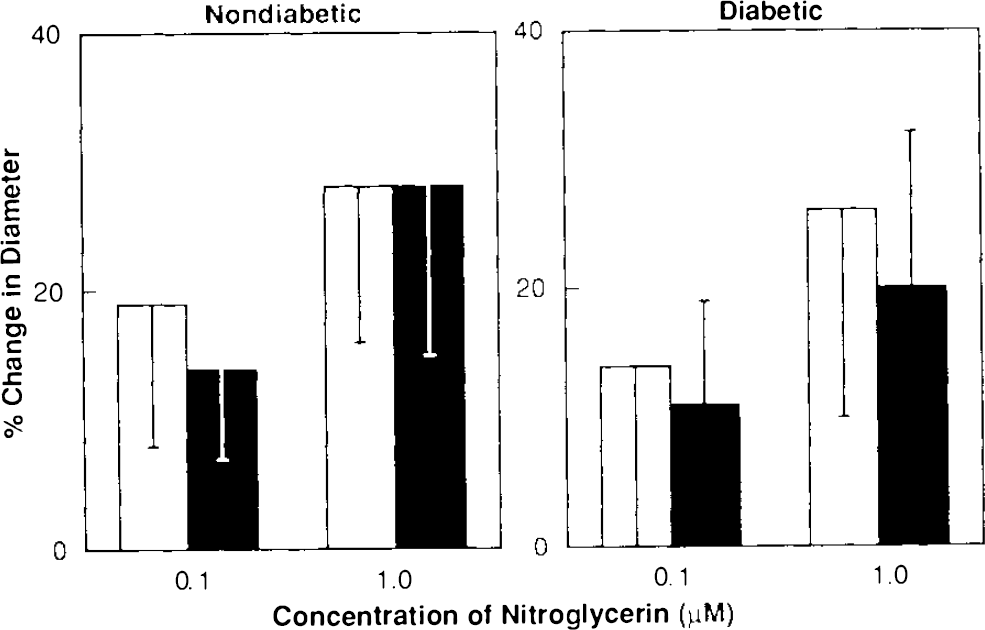
Responses of the basilar artery to nitroglycerin in nondiabetic and diabetic (closed bars) rats before (open bars) and after (closed bars) application of L-arginine (0.1 mM). Values are means ± SD.
Topical application of L-arginine (3.0 mM) produced only a minimal change in baseline diameter of the basilar artery in nondiabetic rats (−6 ± 5%), but produced a marked decrease in baseline diameter of the basilar artery in diabetic rats (−15 ± 8%).
L-arginine (3.0 mM) significantly impaired dilatation of the basilar artery in nondiabetic rats in response to acetylcholine (Fig. 5) and bradykinin (Fig. 6). This paradoxical attenuation of acetylcholine- and bradykinin-induced dilatation of the basilar artery in nondiabetic rats could not be explained by a nonspecific effect of L-arginine on vasodilatation, since responses to nitroglycerin (Fig. 7) were not altered in nondiabetic rats following application of L-arginine (3.0 mM), and application of D-argi-nine (3.0 mM) did not alter dilatation of the basilar artery to acetylcholine in nondiabetic rats. Acetylcholine (1.0 and 10 μM) dilated the basilar artery by 22 ± 9 and 35 ± 2%, respectively, before and by 22 ± 8 and 40 ± 13%, respectively, after application of D-arginine (n = 3).
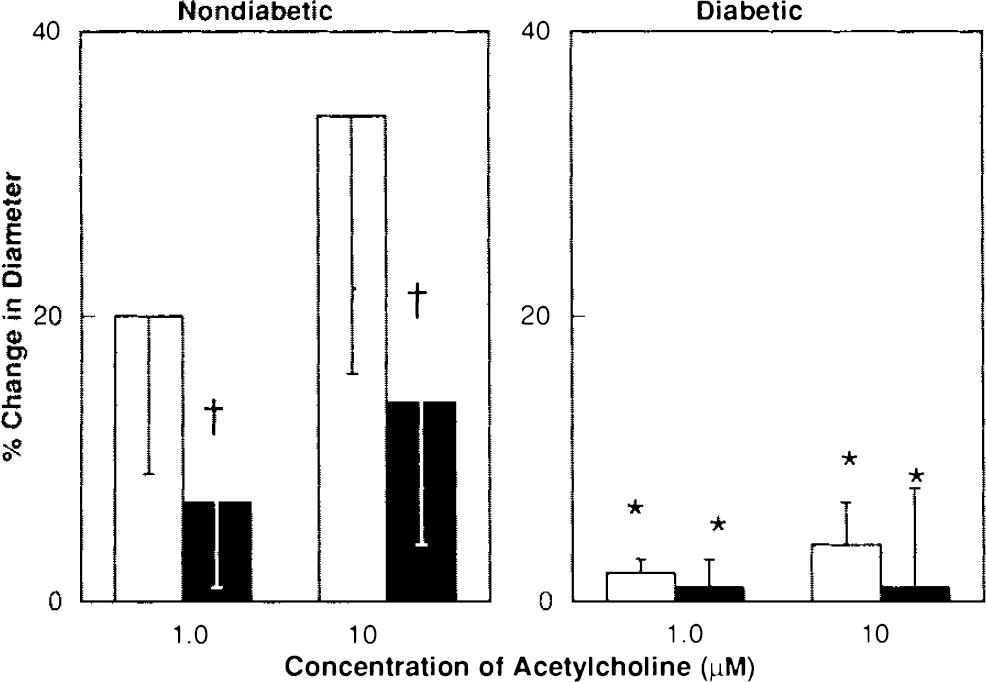
Responses of the basilar artery to acetylcholine in nondiabetic and diabetic (closed bars) rats before (open bars) and after (closed bars) application of L-arginine (3.0 mM). Values are means ± SD. *p < 0.05 versus nondiabetic rats, †p < 005 versus response before L-arginine (3.0 mM).
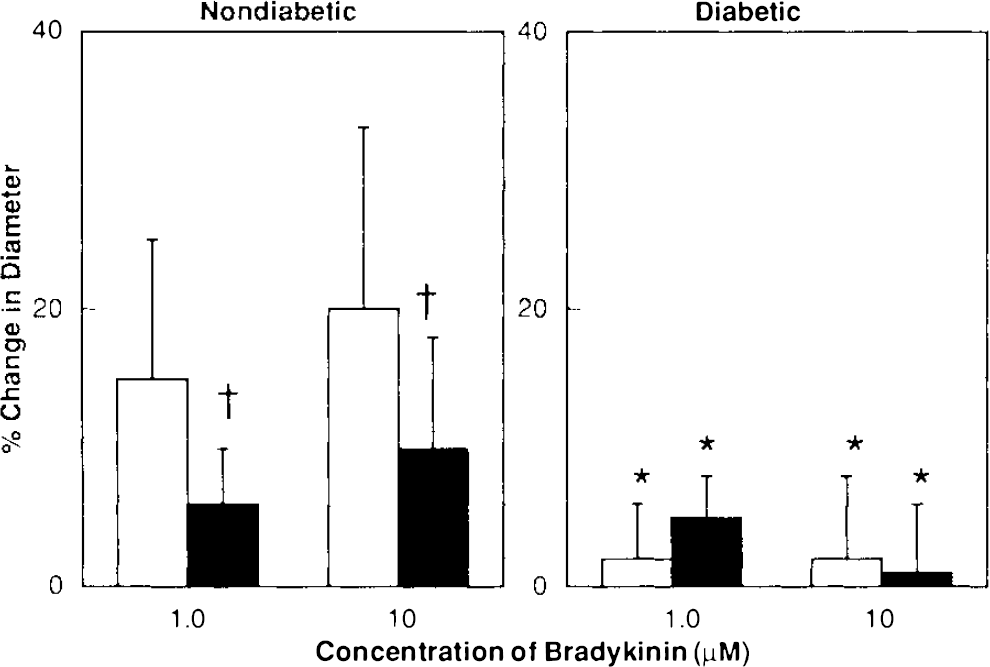
Responses of the basilar artery to bradykinin in nondiabetic and diabetic rats before (open bars) and after (closed bars) application of L-arginine (3.0 mM). Values are means ± SD. *p < 0.05 versus nondiabetic rats, †p < 0.05 versus response before L-arginine (3.0 mM).
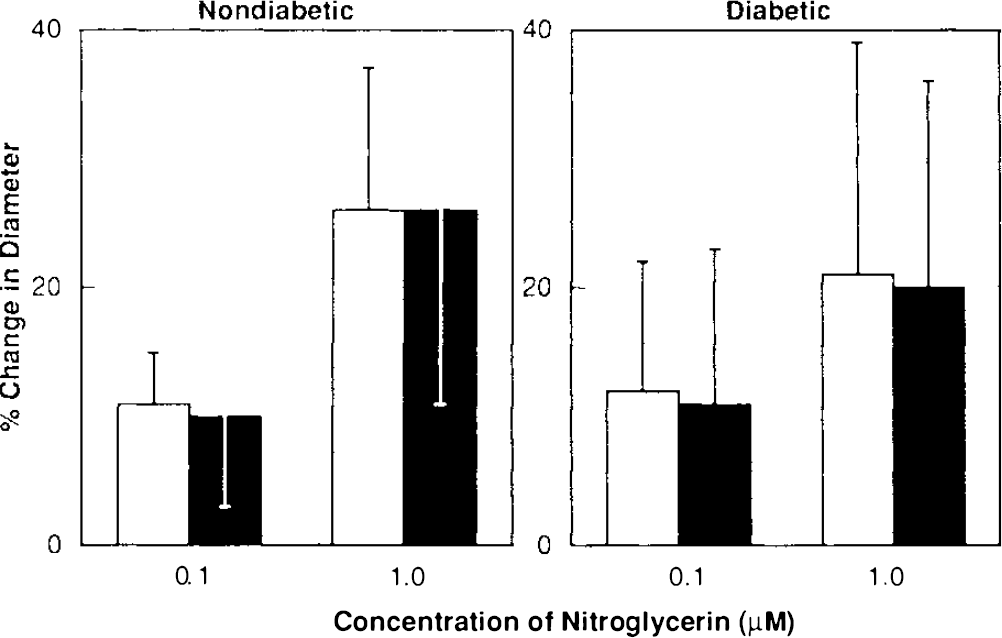
Responses of the basilar artery to nitroglycerin in nondiabetic and diabetic rats before (open bars) and after (closed bars) application of L-arginine (3.0 mM). Values are means ± SD.
Topical application of L-arginine (3.0 mM) did not enhance dilatation of the basilar artery in diabetic rats in response to acetylcholine (Fig. 5) and bradykinin (Fig. 6). Furthermore, L-arginine (3.0 mM) did not alter dilatation of the basilar artery in response to nitroglycerin in diabetic rats (Fig. 7).
Taken together, our findings with L-arginine suggest that an alteration in the availability of L-arginine to nitric oxide synthase may not account for impaired dilatation of the basilar artery during diabetes mellitus.
DISCUSSION
The major new finding of the present study is that treatment of the basilar artery with L-arginine does not restore impaired dilator responses to acetylcholine and bradykinin observed during diabetes mellitus. Thus, impaired dilatation of the basilar artery during diabetes mellitus does not appear to be related to an alteration in the availability of L-arginine to nitric oxide synthase, but may be related to alterations in other cellular pathways, i.e., chronic production of oxygen radicals, production of advanced glycosylation end products, increased synthesis/release of sorbitol via the polyol pathway, and/or alteration in the enzyme nitric oxide synthase.
Responses to acetylcholine and bradykinin
Several investigators have shown that acetylcholine produces marked relaxation of the basilar artery in rats (Lai et al., 1989; Soltis and Bohr, 1987), rabbits (Nakagomi et al., 1988; Fujiwara et al., 1986), cats (Hamel et al., 1988), and humans (Kanamaru et al., 1989; Whalley et al., 1987) using in vitro methodologies. We (Mayhan, 1990b) and others (Faraci, 1990; Faraci, 1991) examined responses of the basilar artery in vivo in rats to acetylcholine. These studies found that acetylcholine produced a dose-related dilatation of the basilar artery that was inhibited by NG-monomethyl-L-arginine (L-NMMA), an enzymatic inhibitor of nitric oxide synthase (Mayhan, 1990b; Faraci, 1990; Faraci, 1991). Thus, dilatation of the basilar artery in rats in response to acetylcholine appears to be related to the release of nitric oxide or a nitric oxide-containing compound.
Bradykinin also produces relaxation of the human (Whalley et al., 1987), feline (Hamel et al., 1988), and canine (Katusic et al., 1989) basilar artery in vitro. Unlike that found for pial arterioles, in which bradykinin produces dilatation via the production of oxygen radicals (Kontos et al., 1984; Rosenblum, 1987), the mechanism of relaxation of the human (Whalley et al., 1987) and canine (Katusic et al., 1989) basilar artery appears to be related to the production of an endothelium-derived relaxing factor (EDRF), presumably nitric oxide. Treatment with indomethacin or inhibitors of oxygen radicals does not alter relaxation of the human or canine basilar artery in response to bradykinin (Whalley et al., 1987, Katusic et al., 1989). We have examined the effects of bradykinin on dilatation of the rat basilar artery in vivo (Mayhan, 1990b). We found that bradykinin produced a dose-related dilatation of the basilar artery in rats that was not affected by treatment with indomethacin, but was inhibited by treatment with L-NMMA (Mayhan, 1990b). Thus, these findings (Mayhan, 1990b) also suggest that dilatation of the basilar artery in response to bradykinin is not related to products released via the cyclooxygenase pathway, but is related to the release of nitric oxide or a nitric oxide-containing compound.
Several investigators have shown that endothelium-dependent dilatation of cerebral blood vessels is altered during diabetes mellitus (Fujii et al., 1992; Pelligrino and Albrecht, 1991; Pelligrino et al., 1992; Dandona et al., 1978). However, for the most part, these studies have not examined mechanisms by which diabetes affects responses of the cerebral circulation, and no studies have examined the effects of diabetes on the vertebrobasilar system. We have shown that endothelium-dependent dilatation of the basilar artery in response to acetylcholine and bradykinin is profoundly impaired in diabetic compared to nondiabetic rats (Mayhan, 1992). Impaired responses of the basilar artery to acetylcholine and bradykinin appear to be specific for endothelium-dependent agonists since nitroglycerin produces similar dose-related dilatation of the basilar artery in nondiabetic and diabetic rats.
Mechanisms of impaired reactivity during diabetes
Several mechanisms have been suggested to account for impaired endothelium-dependent relaxation of peripheral blood vessels during diabetes mellitus, including production of a cyclooxygenase constrictor substance with subsequent activation of the prostaglandin H2/thromboxane A2 receptor (Tesfamariam et al., 1989; Shimizu et al., 1993); a deficit in substrate (L-arginine) utilization/availability (Pieper and Peltier, 1995); increased chronic production of oxygen radicals, which serve to inactivate nitric oxide (Pieper et al., 1993; Langenstroer and Pieper, 1992; Pieper et al., 1992; Hattori et al., 1991; Pieper and Gross, 1988); increased production of advanced glycosylation end products (Tilton et al., 1993; Corbett et al., 1992; Bucala et al., 1991; Brownlee et al., 1988; Wu, 1993); and increased levels of sorbitol (Taylor et al., 1994; Knudsen et al., 1989; Tesfamariam et al., 1993; Carrington et al., 1993; Wakasugi et al., 1991; Tesfamariam, 1994). Thus, mechanisms responsible for impaired endothelium-dependent reactivity of peripheral arteries appear to involve several cellular processes.
Mechanisms that contribute to impaired dilatation of cerebral arteries and arterioles during diabetes mellitus also are not uniform. We (Mayhan et al., 1991) have shown that impaired dilatation of pial arterioles in rats during diabetes mellitus relates to the production of a cyclooxygenase constrictor substance, which presumably activates the prostaglandin H2/thromboxane A2 receptor. However, other studies (Pelligrino et al., 1994) suggest that activation of protein kinase-C accounts for impaired dilatation of rat pial arterioles during diabetes mellitus. In addition, we have shown that there are important regional differences between mechanisms that contribute to impaired responses of cerebral blood vessels during diabetes mellitus. In contrast to that observed for pial arterioles (Mayhan et al., 1991), dilatation of the basilar artery in diabetic rats could not be restored to the amount observed in nondiabetic rats with indomethacin or an antagonist of the prostaglandin H2/thromboxane A2 receptor (Mayhan, 1992). Thus, it appears that alterations in other cellular pathways must account for impaired responses of the basilar artery during diabetes mellitus.
In the present study, we investigated whether a deficit in L-arginine availability could account for impaired dilatation of the basilar artery during diabetes mellitus. A recent study suggests that impaired relaxation of the aorta in diabetic rats could be enhanced by application of L-arginine (Pieper and Peltier, 1995). These investigators (Pieper and Peltier, 1995) found that L-arginine (3 mM) potentiated relaxation of the thoracic aorta to acetylcholine, but not nitroprusside, in diabetic rats. In contrast to that study, we did not find a role for L-arginine in altered responses of the basilar artery during diabetes mellitus. Thus, it appears that there are important regional differences between mechanisms that account for altered responses of blood vessels during diabetes mellitus.
Surprisingly, we found that application of L-arginine (3.0 mM) significantly impaired dilator responses of the basilar artery to acetylcholine and bradykinin in nondiabetic rats. This effect appeared to be specific since L-arginine (3.0 mM) did not alter dilatation of the basilar artery to nitroglycerin, and D-arginine (3.0 mM) did not alter dilatation of the basilar artery to acetylcholine and bradykinin. The mechanisms by which high concentrations of L-arginine alter nitric oxide-mediated dilatation of the basilar artery are not clear. It is possible that high concentrations of L-arginine may affect transmembrane flux of calcium, the arginine transporter, and/or nitric oxide synthase. In support of this, a recent study suggests that nitric oxide may inhibit nitric oxide synthase (Assreuy et al., 1993), although it is not clear whether excess L-arginine would also produce this effect. In addition, a recent study (MacAllister et al., 1995) suggests that infusion of a high concentration of L-arginine (10 g/90 ml of saline i.v. over 10 min) in humans increases plasma levels of insulin, glucagon, and prolactin. Although vascular reactivity following infusion of L-arginine was not examined in the study of MacAllister et al. (1995), it is possible that the above mentioned hormones may affect vascular reactivity in response to endothelium-dependent agonists. In our study, we examined the effect of topical application of L-arginine on reactivity of the basilar artery. It is not clear whether topical application of L-arginine would stimulate the synthesis/release of factors that could affect vascular reactivity. Nevertheless, it appears that a high concentration of L-arginine produces modest, but selective, impairment of nitric oxide-mediated dilatation of the basilar artery.
In conclusion, this is the first study to examine the role of L-arginine in altered responses of the basilar artery to endothelium-dependent agonists during diabetes mellitus. We found that diabetes impairs endothelium-dependent dilatation of the basilar artery in response to acetylcholine and bradykinin. However, impaired responses of the basilar artery to acetylcholine and bradykinin during diabetes could not be restored by treatment with L-arginine. Thus, it does not appear that an alteration in the availability of the substrate for nitric oxide synthase, L-arginine, accounts for altered responses of the basilar artery during diabetes mellitus. We suggest that other co-factors for nitric oxide formation, e.g., nitric oxide synthase, transmembrane flux of calcium, calmodulin, nicotine-adenine-dinucleotide phosphate (NADPH), and/or alterations in other cellular processes, i.e., increased formation of oxygen radicals and/or increased formation of sorbitol, may account for impaired reactivity of the basilar artery during diabetes mellitus.
Footnotes
Acknowledgment:
This study was supported by National Heart, Lung, and Blood Institute grant HL-40781; a Grant-in-Aid from the American Heart Association, National Affiliate (91006230); a Grant-in-Aid from the American Heart Association, Nebraska Affiliate (9307792S); the University of Nebraska College of Medicine (94–08), and a Grant-in-Aid from the American Diabetes Association (07RA).