
Abstract
N-acetylaspartate (NAA) quantification by 1H-magnetic resonance spectroscopy has been commonly used to assess in vivo neuronal loss in neurodegenerative disorders. Here, the authors used ex vivo and in vivo1H-magnetic resonance spectroscopy in rat and primate models of progressive striatal degeneration induced by the mitochondrial toxin 3-nitropropionate (3NP) to determine whether early NAA depletions could also be associated with neuronal dysfunction. In rats that were treated for 3 days with 3NP and had motor symptoms, the authors found a significant decrease in NAA concentrations, specifically restricted to the striatum. No cell loss or dying cells were found at this stage in these animals. After 5 days of 3NP treatment, a further decrease in striatal NAA concentrations was observed in association with the occurrence of dying neurons in the dorsolateral striatum. In 3NP-treated primates, a similar striatal-selective and early decrease in NAA concentrations was observed after only a few weeks of neurotoxic treatment, without any sign of ongoing cell death. This early decrease in striatal NAA was partially reversed after 4 weeks of 3NP withdrawal. These results demonstrate that early NAA depletions reflect a reversible state of neuronal dysfunction preceding cell degeneration and suggest that in vivo quantification of NAA 1H-magnetic resonance spectroscopy may become a valuable tool for assessing early neuronal dysfunction and the effects of potential neuroprotective therapies in neurodegenerative disorders.
Considerable interest in understanding the role and behavior of N-acetylaspartate (NAA) has arisen in recent years because of the fact that this compound contributes to the major resonance seen on 1H-magnetic resonance spectroscopy (1H-MRS). In addition, because of its almost exclusive localization in neurons in the adult brain, NAA seen in 1H-MRS has been used as a marker of neuronal density (Tallan, 1956; Urenjak et al., 1992). Accordingly, regional decreases in NAA concentrations have been reported in many experimental and clinical conditions that are associated with actual neuronal cell loss, for example, excitotoxic lesions, ischemia, epilepsia, and neurodegenerative diseases (Ebisu et al., 1994; Higuchi et al., 1997; Vermathen et al., 1997; Lopez-Villegas et al., 1997). However, more recent data have also shown that NAA depletions could be reversed after therapeutic interventions (Kalra et al., 1998; Ellis et al., 1997; Holshouser et al., 1995; Vion-Dury et al., 1994) or even spontaneously (De Stefano et al., 1995), suggesting that NAA concentration might also represent a reversible spectroscopic marker for neuronal dysfunction. Alternatively, because intramitochondrial NAA neosynthesis has also been shown to be affected by mitochondrial toxins (Bates et al., 1996), NAA depletions could be more simply related to a deficit in the mitochondrial oxidative phosphorylation process.
To study this question further, we used a well-characterized model of progressive striatal degeneration in rats and primates that is induced by systemic administration of 3-nitropropionic acid (3NP), an irreversible inhibitor of the mitochondrial complex II succinate dehydrogenase. An interesting feature of this model resides in the temporal evolution of the neurodegenerative processes, which progresses from an early stage of neuronal dysfunction without detectable cell death to a more advanced stage of actual neurodegeneration characterized by the presence of necrosis and apoptotic features within the caudate-putamen complex (for review, Brouillet et al., 1999). In the late stage of the neurotoxic treatment, the 3NP-induced striatal lesion recapitulates most of the histologic features of Huntington disease (HD), that is, bilateral lesions displaying selective loss of striatal projection neurons and relative sparing of intrinsic neurons, astrocytes, afferent fibers, and fibers of passage (Beal et al., 1993; for review, Brouillet et al., 1999). Therefore, only the striatal projection neurons, which constitute more than 95% of the total striatal neurons, are significantly affected in this lesion model, whereas striatal astrocytes remain largely spared and able to trigger compensatory mechanisms (Hassel and Sonnewald, 1995). In addition, because 3NP readily crosses the blood-brain barrier, the systemic administration of this mitochondrial toxin results in a ubiquitous inhibition of cerebral succinate dehydrogenase (SDH) (Brouillet et al., 1998). Therefore, by comparing two cerebral regions that are differentially vulnerable to 3NP, it is possible to study the effect on regional NAA levels of either a mitochondrial blockade alone (in a region resistant to 3NP, such as the cerebral cortex) or a similar mitochondrial blockade combined with the occurrence of premorbid neurodegenerative events (in a region highly vulnerable to 3NP, such as the striatum).
In the present study, we examined the biochemical, anatomic, and behavioral correlates of NAA depletions in 3NP-treated rats at different stages of the neurotoxic treatment, using an ex vivo1H-MRS approach that yielded highly resolved spectra and provided actual quantification of numerous biologically relevant brain metabolites. To further assess the potential clinical relevance of these findings, we also observed nonhuman primates to determine whether NAA depletions could also be reversible after 3NP withdrawal. For this purpose, we took advantage of the 3NP primate model, which is characterized by a striatal degenerative process that is more progressive (15 weeks) than the one observed in the rat model (6 days) and an in vivo localized 1H-MRS approach (Dautry et al., 1999) to perform a follow-up study of striatal NAA concentrations in a group of 3NP-treated baboons at various stages of the neurotoxic treatment (before any treatment, at an early stage of the neurotoxic treatment [i.e., between 8 and 10 weeks of 3NP treatment], and 4 weeks after termination of the 3NP treatment).
MATERIALS AND METHODS
Chronic 3NP treatment in rats
Male Lewis rats (body weight, 300–350 g; n = 30) were obtained from IFFA Credo (L'Arbresle, France). Because vulnerability of animals to 3NP toxicity increases with age (Brouillet et al., 1999), all rats were of the same age (10 weeks) at the beginning of the neurotoxic infusion. The 3NP (Sigma, Saint-Quentin Fallavier, France) solution was prepared as previously described (Brouillet et al., 1998). The neurotoxic treatment consisted of continuous infusion of 3NP for 3 days (n = 9) or 5 days (n = 6) using osmotic minipumps (model 2ML4, Alzet Inc., Palo Alto, CA, U.S.A.) that were implanted subcutaneously under ketamine-xylazine anesthesia in the back of the animals. The 3NP concentration in the minipump was adjusted to the initial weight of each animal so that all rats received the same 3NP regimen corresponding to 38 mg/kg/d. Control animals (n = 9 at 3 days; n = 6 at 5 days) were age-matched Lewis rats implanted with empty minipumps.
After 3 days or 5 days of the neurotoxin treatment, the rats were killed and their brains were rapidly removed. Because 3NP is active systemically and induces symmetrical bilateral lesions in the striatum, we used the same animal for biochemical and histochemical analysis: the brain was cut along the rostrocaudal axis on a cold plate; one brain half was immediately frozen in isopentane (−25°C) for SDH histochemistry and the terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling (TUNEL) method, and the other brain half was rapidly cut in 2-mm-thick slices from which the striatum and cerebral cortex (mainly corresponding to the somatosensory area) were dissected for extraction of brain metabolites and ex vivo1H-MRS analysis. All experimental procedures were performed in strict accordance with the recommendation of the European Ethical Community (86/609/EEC) and the French National Committee (87/848) for the Care and Use of Laboratory Animals.
Behavioral evaluation in rats
3NP-treated animals and their corresponding controls were evaluated daily for motor deficits with use of a quantitative neurologic scale. Motor deficits were first determined based on the presence and severity of various motor symptoms, including dystonia (intermittent dystonia of one hindlimb = 1; intermittent dystonia of the two hindlimbs = 2; permanent dystonia of hindlimbs = 3), gait abnormalities (uncoordinated and wobbling gait = 4; lying on one side but showing uncoordinated movements when stimulated = 5), and near-death recumbency (almost complete paralysis with rapid breathing = 6). In addition, the ability of the animals to grasp a cage grid with the forepaws for few seconds (able = 0; unable = 1) and the ability to remain on a small platform (9 × 5 cm) for more than 10 seconds (able = 0; unable = 1) were assessed. A final neurologic score was calculated for each animal as the sum of indices of motor symptoms, grasping, and equilibrium capacities (normal animal = 0; maximal neurologic impairment = 8).
Ex vivo1H-MRS and metabolite quantitation in rats
On the brain half that was used for 1H-MRS, absolute concentration levels of NAA, aspartate, glutamate, inositol, succinate, glutamine, alanine, taurine, creatine, and gamma-aminobutyric acid (GABA) were obtained from the striatum and cortex, separately. The striatal and frontal cortex areas were dissected at 4°C from a 2-mm-thick slice, and metabolites were extracted by sonication in 500 μL of ice-cold 0.1-N perchloric acid solution. Protein concentration for each sample was determined in quadruplicate from 5 μL aliquots using a standard colorimetric technique (Bicinchoninic Acid Protein Assay Reagent, Pierce, Rockford, IL, U.S.A.). Samples were then centrifuged at 15,000 g for 15 minutes at 4°C, and the supernatants were collected and neutralized with 5-N potassium hydroxide. A second centrifugation, at 15,000 g (15 minutes, 4°C), was performed, and 400 μL of the supernatants were lyophilized. Tissue extracts were resuspended in D2O and adjusted to pH 1.3 with DCl in a final volume of 400 μL. Each sample was placed in a 5-mm nuclear magnetic resonance tube equipped with a coaxial insert containing a reference solution of formic acid (41 mM). Measurements were obtained at 25°C on a Bruker AMX-300WB spectrometer (Ettlingen, Germany) operating at 300 MHz. A fully relaxed one-pulse sequence with water presaturation was used (90° flip angle, 4000 Hz spectral width, 4 K complex points, 10.3 seconds repetition time, 800 signal averages). Proton spectra (Fig. 1) were processed with a 1-Hz line-broadening filter and a convolution difference filter (broadening factor = 30 Hz; substraction factor = 0.6) to remove broad baseline signals (Barker et al., 1993). The peak area of each metabolite of interest, Smet, was normalized by the peak area of the formic acid external reference, Sref. The ratio Smet/Sref was converted to the absolute amount of metabolite using a calibration curve previously established with solutions of glycine at various known concentrations. For each sample, the absolute concentration of metabolites was finally expressed in mmol/L · kg protein.
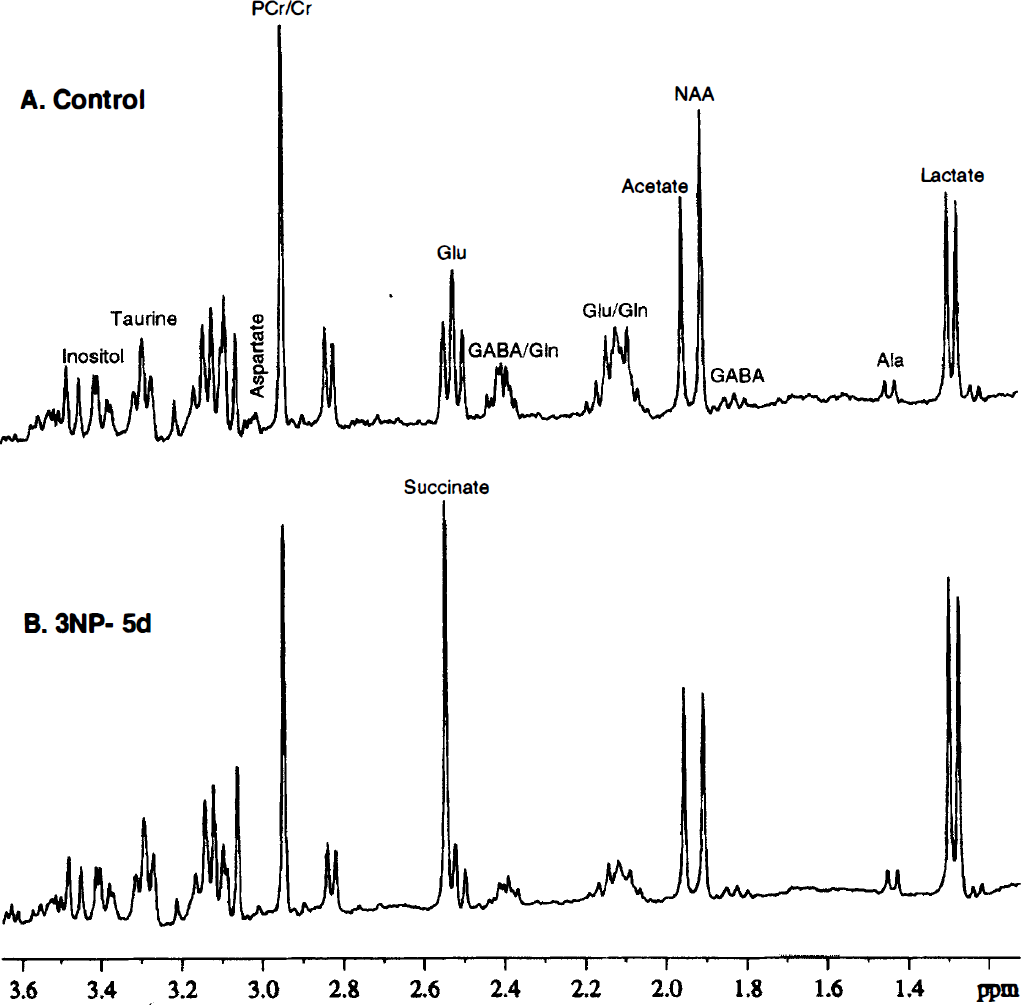
High-resolution 1H-nuclear magnetic resonance spectra of perchloric acid extracts in one control
Semiquantitative in situ measurement of SDH inhibition in rats
The second brain half, which was frozen in isopentane (−25°C), was used, and semiquantitative SDH histochemistry was performed as previously described (Brouillet et al., 1998). The frozen brains were sectioned at 20 μm, mounted on glass microscope slides, air-dried for 30 to 60 minutes, and kept at −20°C for 1 to 2 days before histochemical staining. SDH activity was revealed using succinate as a specific substrate and nitro blue tetrazolium as an electron acceptor. All sections were first incubated for 10 minutes in 0.1-M phosphate-buffered saline (PBS; 0.9% NaCl) at 37°C to activate SDH and then rinsed in a large volume of PBS and incubated for 30 minutes at 37°C in a reaction medium containing 0.3-mM nitro blue tetrazolium, 0.05-M phosphate buffer (pH 7.6), and 0.05-M sodium succinate. Adjacent sections were incubated in the medium described above but lacked succinate to determine the (nonspecific) staining that was unrelated to SDH activity. Sections were then rinsed in cold PBS for 5 minutes, immersed for a few seconds in deionized water, and air-dried at room temperature. Images of each section were digitized and regions of interest (i.e., striatum and somatosensory cortex) were analyzed using a video analysis system (IMSTAR, Paris, France) as previously described (Brouillet et al., 1998). SDH activity was expressed as the optical density unit rate per 30 minutes.
TUNEL method
Evaluation of the number of TUNEL-positive cells was performed on frozen rat sections adjacent to those used for SDH in situ histochemistry. Sections were postfixed with 4% paraformaldehyde in PBS for 30 minutes, incubated in a solution of 0.1% sodium citrate and 1% Triton-X100 for 5 minutes, rinsed in PBS, incubated in proteinase K (10 μg/mL in PBS, pH 7.4) for 5 minutes, reimmersed in 4% paraformaldehyde for 15 minutes, and rinsed three times in PBS before TUNEL reaction. TUNEL reaction (30 minutes at 37°C) was performed using a commercial kit and according to the manufacturer's instructions (Meylan; Roche, France). Incorporation of fluorescein dUTP was analyzed under epifluorescence with an Olympus Provis microscope (Rungis, France). Positive controls consisted of sections from normal brain treated with DNAse I (0.5 μg/mL, 10-mM Tris-HCl, pH 7.5, 10 minutes at room temperature) or rat brains previously lesioned with 3NP. The TUNEL reactions in 3NP-treated rats were performed twice, in parallels with positive controls (sections from control animals, pretreated with DNAse I).
Chronic 3NP treatment in primates
Four baboons (Papio anubis; body weight, 10–13 kg) received chronic 3NP treatment as previously described (Brouillet et al., 1995). The 3NP treatment consisted of two daily intramuscular injections of 3NP (given in the legs at 9:00 A.M. and 4:00 P.M.; starting dose: 6 mg/kg per injection), which were progressively increased at weekly intervals (weekly increment: 1 mg/kg per injection). Serial in vivo1H-MRS and MRI investigations were performed every fortnight until a significant depletion in striatal NAA concentrations could be detected. On average, −17% of NAA was observed after 8 to 10 weeks of 3NP treatment. At that time point, one baboon was killed (pentobarbital, 120 mg/kg intravenously) and its brain was processed for histology, whereas the 3NP treatment was stopped in the three remaining animals. Four weeks later, 1H-MRS and MRI were performed again on these three animals. Immediately after this last nuclear magnetic resonance experiment, the three animals were killed and their brains were processed for histology.
In vivo1H-MRS in primates
In vivo1H-MRS experiments were performed using a Bruker 3T whole-body scanner. Baboons were anesthetized with ketamine-xylazine (15 mg/kg and 1.5 mg/kg, respectively), and their heads were positioned into a transmit-receive pseudo-Helmholtz probe with use of an MRI-compatible head holder, which allows a reproducible positioning of the animal in the scanner. Transverse T1-weighted images were first acquired to localize the spectroscopic volume of interest and to detect the presence of any striatal lesion. Localized water-suppressed proton spectra (Point Resolved Spectroscopy, TR/TE = 2500/135 milliseconds, 128 scans, 2048 data points, 2500 Hz bandwidth) were then acquired in a 5.2-mL volume of interest encompassing the rostral half of both caudate-putamen nuclei (volume of interest dimensions were 32.5 × 13.3 × 11.5 mm in the x, y, and z directions, respectively; Fig. 2B). T2 values of NAA were determined by collecting metabolite spectra at different TEs (60, 90, 135, 180, 225, and 270 milliseconds) and using a monoexponential curve for fitting. Water transverse relaxation in the spectroscopic volume of interest was investigated by fully relaxed single-scan spectra at different TEs (30, 45, 68, 101, 200, 500, 800, and 1500 milliseconds) and analyzed using a biexponential fit. The calculated brain water signal intensity at TE = 0 was used as an internal reference for calculation of the metabolite concentrations and corrected for cerebrospinal fluid contamination (Ernst et al., 1993). Nuclear-magnetic-resonance-visible water was assumed to represent 71.3% (average of water content in gray and white matter) of brain tissue volume, and brain tissue density was taken as 1.05 mg/mL (Kreis et al., 1993). Data processing was performed as previously described (Dautry et al., 1999). Absolute NAA concentrations in the striatum of primates were obtained and expressed as mmol/L · kg wet tissue.
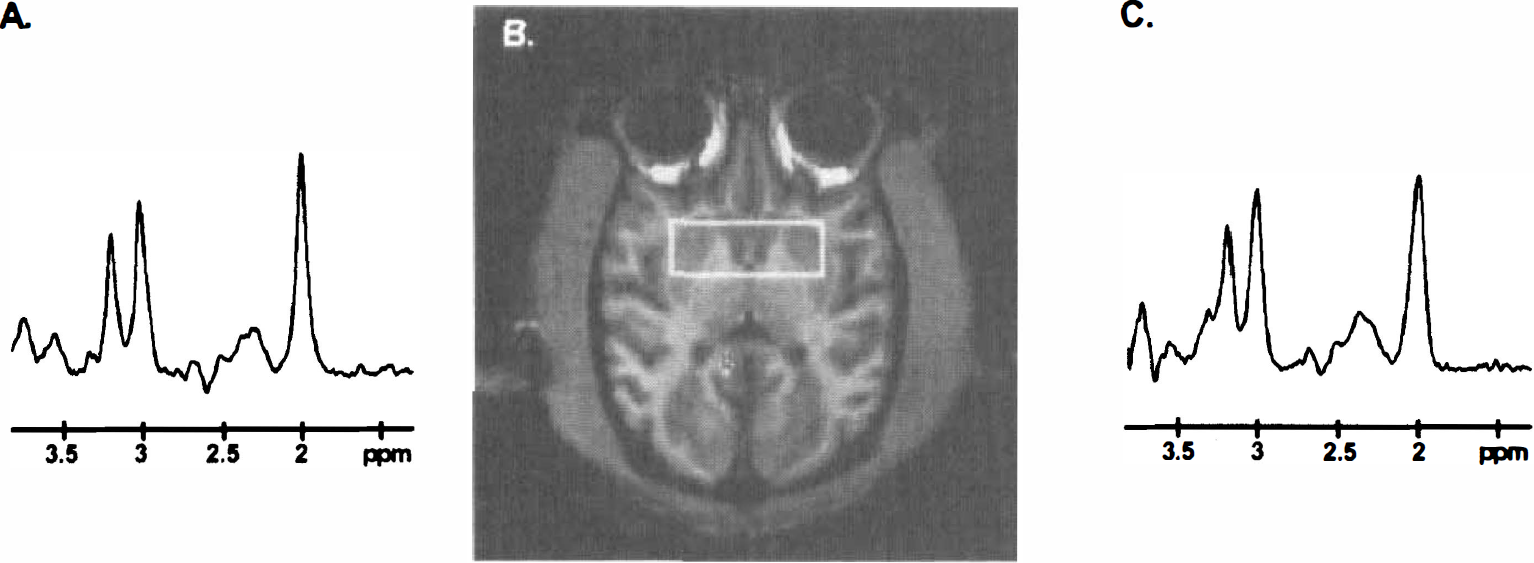
Localized water-suppressed proton spectra obtained in one control
Histology in nonhuman primates
Within 3 to 12 hours after the last 1H-MRS scan, baboons were anesthetized (ketamine-xylazine mixture, 15/1.5 mg/kg intramuscular), and then received an overdose of pentobarbital (120 mg/kg intravenously). Finally, animals were perfused transcardially with ice-cold 1% paraformaldehyde in phosphate buffer preceding 4% paraformaldehyde for 8 to 9 minutes. The brain was immediately removed from the skull, cut into 2-cm-thick coronal blocks, postfixed for 6 to 8 hours in 4% paraformaldehyde at 4°C, and cryoprotected in a series of graded sucrose solutions (12%, 16%, and 18%) in PBS. The blocks were frozen on dry ice and sections (40 μm) were cut on a freezing microtome (Microm; Francheville, France). The sections from each block were kept in anatomic series, and every 10th section was stained with gallocyanin. The remaining sections were cryoprotected with a solution of 30% glycerol and 30% ethylene glycol in 0.05-M PBS and stored at −20°C. To differentiate between alterations in neuronal densities and changes caused by neuronal dysfunction, two different antibodies were used: a mouse monoclonal antibody (NeuN, diluted 1:1000, Chemicon International Inc., Temecula, CA, U.S.A.), which is specific of vertebrate neuron-specific nuclear protein expressed by most neurons, and an antirat calbindin-D28k rabbit serum (diluted 1:3000, Swant, Bellinzona, Switzerland), which labels a calcium binding protein expressed by a subpopulation of striatal projection neurons. In addition, other calcium-binding proteins or markers expressed by striatal interneurons were studied using sera raised against calretinin (diluted 1:5000, Swant), parvalbumin (diluted 1:3000, Swant), or choline acetyltranferase (diluted 1:10,000, Chemicon International Inc.). All antibodies were used according to the following protocol: after 48 hours of incubation in the first antibody at room temperature, sections were processed according to the avidin-biotin method (Hsu et al., 1981) with tyramine amplification (Berghorn et al., 1994), except for the NeuN antibody.
TUNEL labeling was also performed in selected baboon brain sections taken at the level of the caudate-putamen complex. For this purpose, brain sections were mounted on gelatin-coated slides, air-dried, rehydrated and incubated in a solution of 0.1% sodium citrate and 1% Triton-X100 for 30 minutes, rinsed in PBS, incubated in proteinase K (1 mg/mL in PBS, pH 7.4, 5 minutes), then incubated in 4% paraformaldehyde for 15 minutes, and finally rinsed three times in PBS before TUNEL reaction. TUNEL reaction and positive controls (DNAse I) were processed as for the rat sections (30 minutes at 37°C).
Statistical analysis
Results were expressed as means ± standard deviations. Statistical analysis included paired or unpaired Student t tests and one-way analysis of variance preceding a Protected Least Significant Difference Fisher test.
RESULTS
SDH activity, TUNEL-positive cells, and motor deficits in 3NP-treated rats
In rats, a general decrease in SDH activity was observed in the whole brain section of the animals treated with 3NP for 3 days, as compared with the control group (Figs. 3B and 3C). Semiquantitative measurements confirmed that 3NP treatment induced a highly significant SDH inhibition (Fig. 4A, P < .001 vs. controls) both in the striatum (−57.2% ± 5.3%) and cerebral cortex (−52.7% ± 9.0%). At this stage of neurotoxic treatment, there was no difference in the degree of SDH inhibition between the two structures. The ratio of SDH activity between the striatum and cortex determined in the rats treated with 3NP for 3 days (1.06 ± 0.08) was similar in controls (1.04 ± 0.02), suggesting that no ongoing degeneration was taking place in the striatum of the rats treated with 3NP for 3 days. In both structures, SDH inhibition was associated with an increase in succinate concentrations (striatum, 19.9 ± 2.2 mmol/L · kg; cortex, 15.2 ± 2.2 mmol/L · kg protein) as determined by ex vivo1H-MRS (Fig. 4B). Remarkably, the succinate concentration in the striatum was significantly higher than in the cortex (P < 0.001, Student paired t test), suggesting that for a similar level of irreversible inhibition of SDH, 3NP produced a stronger inhibition of the oxidative metabolism in the striatum, compared with the cerebral cortex. In the 5-day 3NP-treated group, a further decrease in SDH activity was observed in the striatum (−70.5% ± 7.7%) but not in the cerebral cortex (Figs. 3D and 4A). The striatum/cortex ratio of SDH activity in rats treated with 3NP for 5 days (0.57 ± 0.05) was significantly decreased (P < 0.0001) as compared with controls (1.01 ± 0.03), suggesting the occurrence of an actual neuronal loss in the striatum of the rats treated with 3NP for 5 days. At this time point, SDH inhibition in both structures was also associated with succinate accumulation, which was further increased in the striatum but not in the frontal cortex (Fig. 4B), as compared with 3-day 3NP-treated extracts (analysis of variance, P < .0002, PLSD Fisher). The presence of dying cells with DNA fragmentation was tested in parallel at the two different time points (i.e., after 3 days and 5 days of 3NP treatment using the TUNEL method). No TUNEL-positive nuclei were observed in the striatum of control animals or animals treated with 3NP for 3 days (Figs. 3E and 3F), contrasting with the numerous TUNEL-positive nuclei observed in the lateral striatum of the rats treated with 3NP for 5 days (Fig. 3G). The presence of an ongoing process of degeneration in rats treated with 3NP for 5 days accounted, at least partially, for the stronger decrease in SDH activity observed at this stage in the striatum as compared with the cerebral cortex (Fig. 3D).
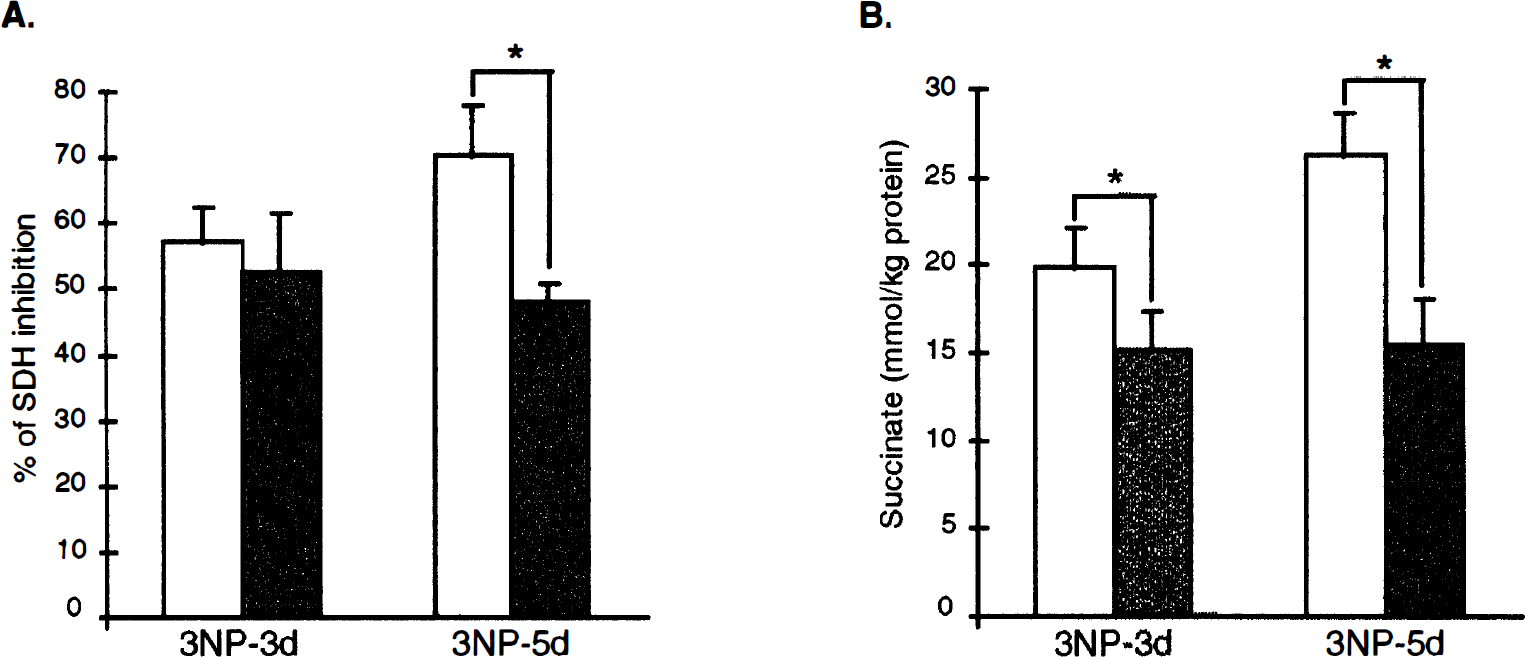
Semiquantitative measurements of 3NP-induced SDH inhibition
Behavioral observations in the same groups showed the presence of abnormal movements after only 3 days of 3NP treatment (gait abnormalities, bradykinesia, and transient leg dystonia), resulting in a progressive decrease in locomotor activity (neurologic score: 2.77 ± 0.22). After 5 days of 3NP treatment, a significant worsening of the motor syndrome was observed (neurologic score: 3.50 ± 0.22, P < 0.04), compared with 3 days of 3NP treatment.
Ex vivo1H-MRS studies in striatum of 3NP-treated rats: 3 days versus 5 days
In the group treated with 3NP for 3 days, there was a significant decrease in concentrations of NAA (−15%), aspartate (−55%), glutamate (−48%), and glutamine (−40%) in the striatal extracts, whereas the concentration of alanine was strongly increased (+53%, Table 1). In the striatal extracts of the 5-day group, NAA and aspartate concentrations were further decreased, whereas depletions in glutamate and increases in alanine remained stable, as compared with the 3-day 3NP-treated extracts. A partial but significant recovery in glutamine concentration was observed in the extracts between 3 and 5 days of 3NP treatment. In addition, the concentrations of inositol and taurine in the 5-day 3NP-treated extracts were significantly increased (+21 and +37%, respectively), compared with control values. Finally, GABA and creatine levels remained unaffected by either 3 or 5 days of 3NP treatment, suggesting that striatal cell loss was not severe in 5-day group.
Quantitative analysis by ex vivo ′H-NMR of striatal and cortical metabolites in normal rats and rats treated for 3 or 5 days with 3 NP
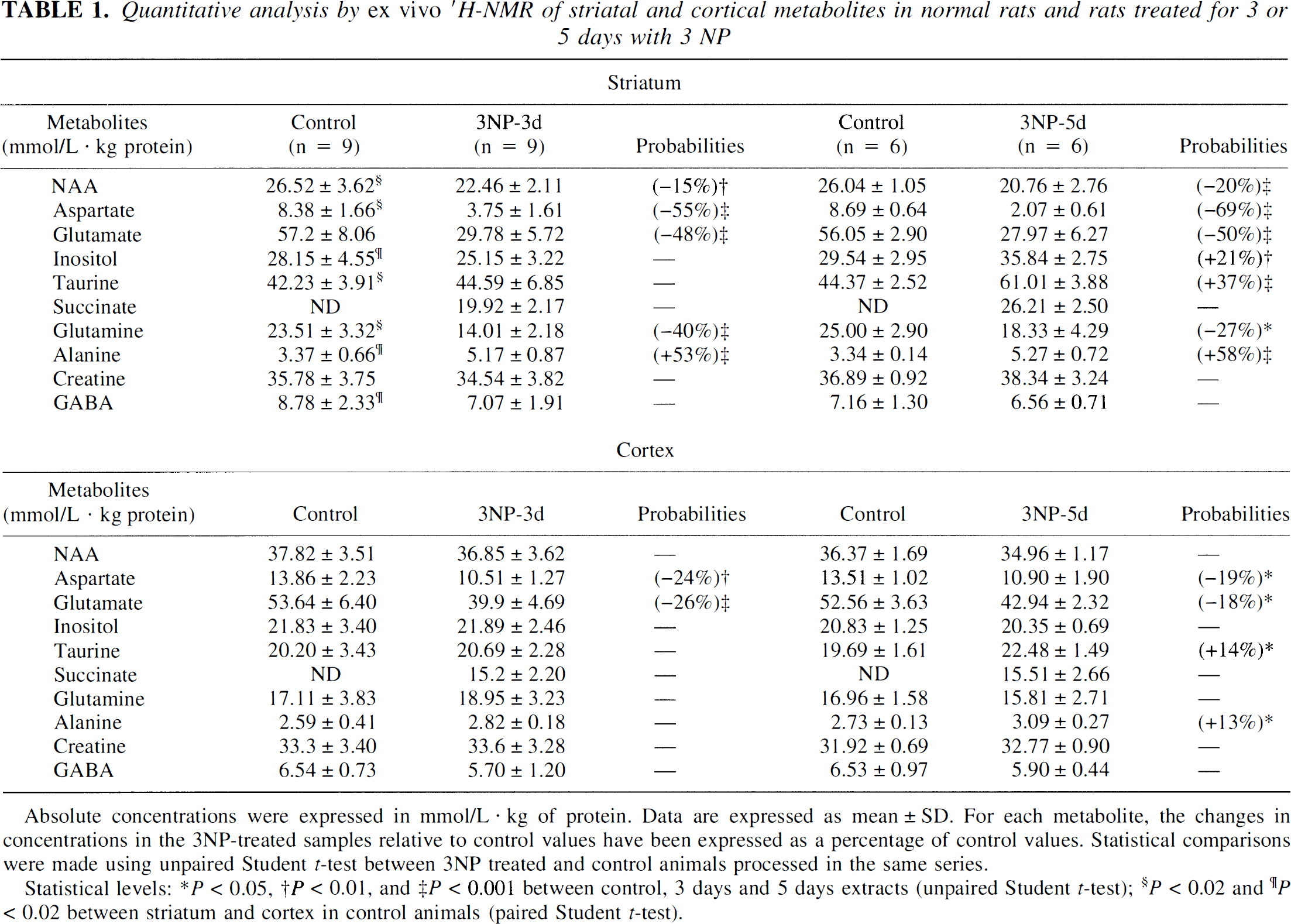
Absolute concentrations were expressed in mmol/L · kg of protein. Data are expressed as mean ± SD. For each metabolite, the changes in concentrations in the 3NP-treated samples relative to control values have been expressed as a percentage of control values. Statistical comparisons were made using unpaired Student t-test between 3NP treated and control animals processed in the same series.
Statistical levels: P < 0.05
P < 0.01
P < 0.001 between control, 3 days and 5 days extracts (unpaired Student t-test)
P < 0.02
P < 0.02 between striatum and cortex in control animals (paired Student t-test).
Ex vivo1H-MRS studies in 3NP-treated rats: striatum versus cortex
In the 3NP-treated rats, one striking difference between the striatum and cortex was that cortical levels in NAA, inositol, and glutamine remained totally unaffected by 3NP, both in the 3-day and the 5-day extracts, as compared with controls (Table 1, bottom panel), whereas all striatal concentrations in these metabolites were strongly affected by 3NP at both time points for NAA and glutamine. Furthermore, despite a similar degree of SDH inhibition observed in both structures after 3 days of 3NP treatment, the decreases in aspartate and glutamate were less important in the cortex than in the striatum. In addition, whereas a significant recovery in the concentrations of these two metabolites was observed in the cortical extracts, striatal aspartate and glutamate levels remained severely affected in the 5-day extracts. Similarly, whereas a moderate increase in alanine and taurine concentrations was observed in the cerebral cortex of the animals treated with 3NP for 5 days, a marked increase in the absolute concentrations of these two metabolites was found in the striatum at the same time point.
In vivo1H-MRS studies in baboons
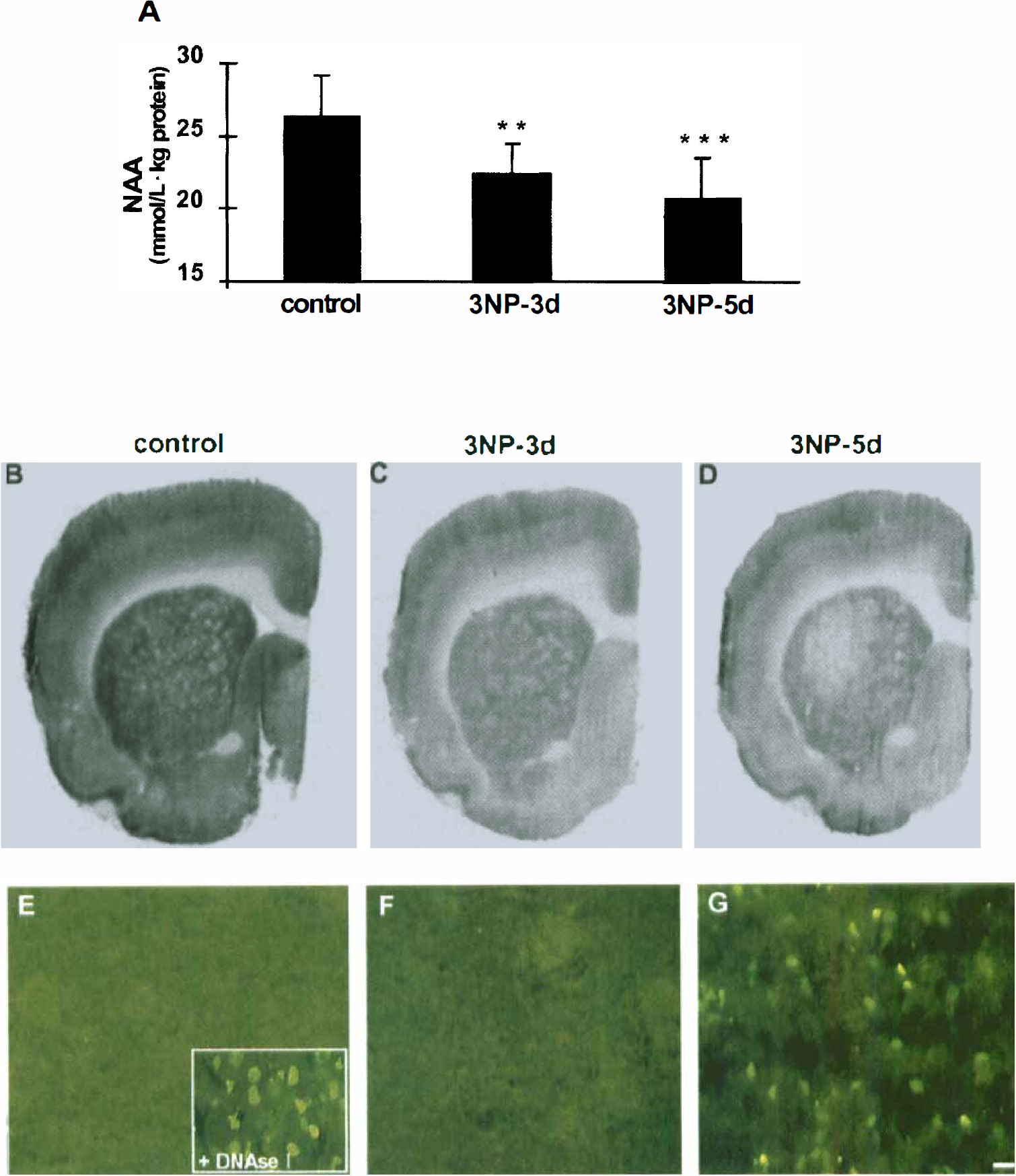
As shown in Figure 5A, a significant decrease in absolute striatal NAA concentrations (−17%, P < .03, analysis of variance PLSD Fisher, n = 4,) was observed in baboons after 8 to 10 weeks of 3NP treatment, compared with the control values determined in the same animals (spectra representation are shown in Figs. 2A [control] and 2C [3NP-treated]). Choline and phospho-creatine/creatine concentrations measured in the striatum remained unchanged from control values at this time point. This early loss of striatal NAA was not correlated with the presence of striatal lesions at MRI examination in any animal. After 4 weeks of 3NP withdrawal, the decrease in striatal NAA was no longer significant (Fig. 5A, 3NP withdrawal). Again, this time point was not associated with any detectable striatal lesion on the T1-weighted MRI examination.
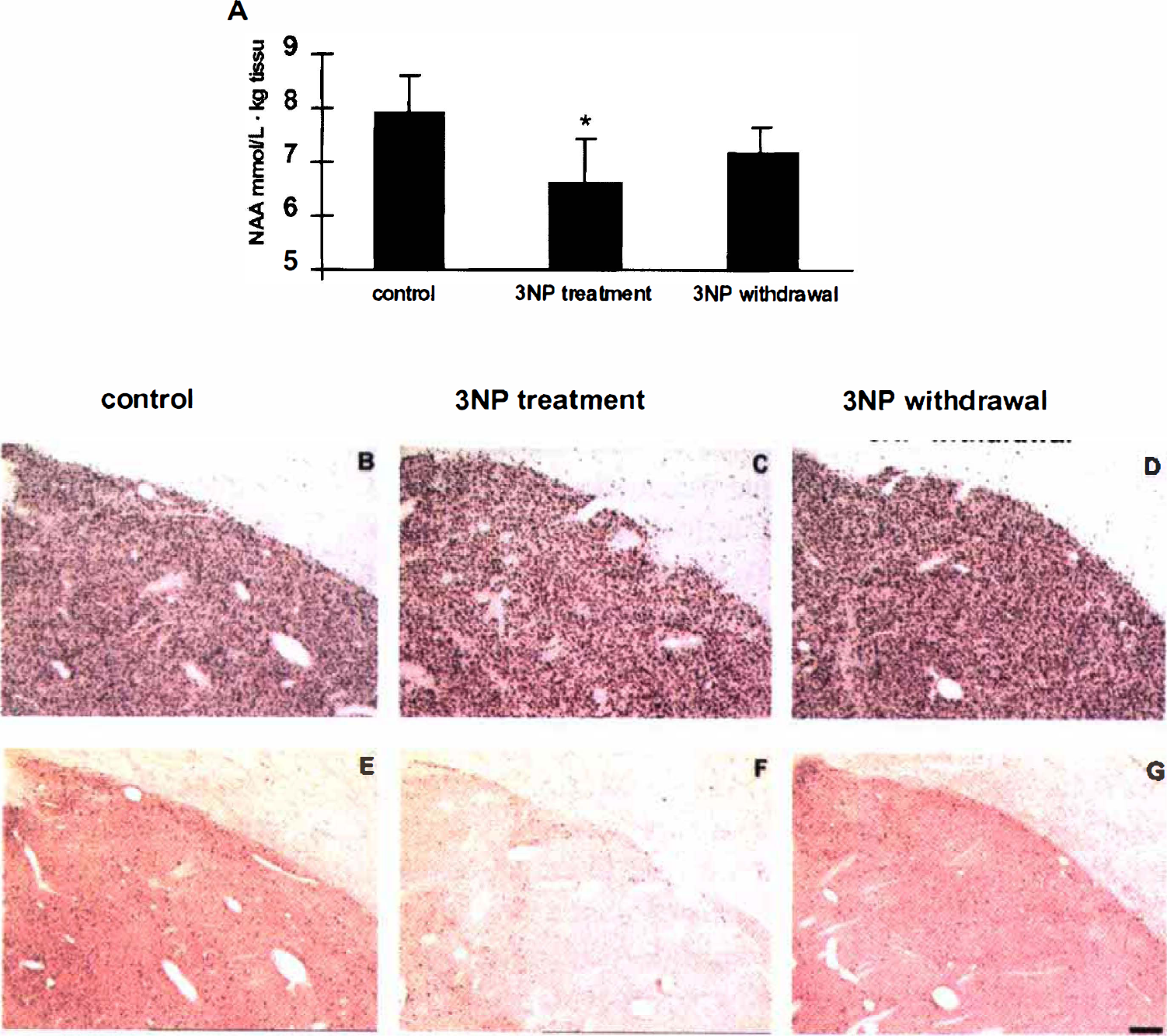
Immunohistology in the striatum of baboons
In the baboon that was killed after presenting a significant decrease in striatal NAA concentration, no major structural changes were observed post mortem in the dorsolateral putamen on sections immunolabeled for NeuN, a neuronal marker (Fig. 5B–Fig. 5D), or processed by the TUNEL method (data not shown). However, when calbindin-D28K (a selective marker of the striatal projection neurons) was used (Fig. 5E–Fig. G), a major decrease in immunoreactivity could be noted in the dorsolateral putamen and caudate nucleus (Fig. 5F), compared with control data (Fig. 5E). In marked contrast to these alterations, which were specific to the medium-sized spiny neurons, no changes were observed in other striatal cell types, such as the calretinin-immunopositive, parvalbumin-immunopositive, and choline-acetyltransferase-immunopositive interneurons (data not shown). In the three baboons studied after 4 weeks without 3NP treatment, a partial recovery in calbindin-D28K immunoreactivity was observed in both the dorsolateral putamen and the caudate nucleus (Fig. 5G).
DISCUSSION
Recent studies on cell death and regenerative mechanisms in experimental models of neurodegenerative disorders have suggested that the functional status of neurons could follow a continuum of different states, ranging from health to metabolic distress, neuronal dysfunction, and finally ongoing cell death. These different states are likely associated with varying degrees of energy metabolism impairment, free radicals production, electrophysiologic dysfunction, loss of calcium homeostasis, and, possibly, selective activation of cell death biochemical pathways, which eventually lead to actual neurodegeneration characterized by irreversible structural damages, such as plasma membrane disruption and nuclear DNA fragmentation (Isacson, 1993). From this neuronal health concept, the idea of neuroprotection has emerged, and numerous investigators are now focusing on discovering reagents that are able to rescue neurons from death. However, the success of such a neuroprotective strategy will be highly dependent on our ability to identify the correct time window for therapeutic intervention, which itself is linked to the identification of specific in vivo markers for the premorbid state of neuronal dysfunction (Bazzett et al., 1995). The present study identifies NAA as one such neuron-specific in vivo marker that can be depleted in a cerebral region, reaching a state of premorbid neuronal dysfunction.
Early NAA depletions and metabolic distress
It has been recently proposed that NAA depletions may be caused by the direct inhibition of the mitochondrial oxidative phosphorylation (Bates et al., 1996). According to this hypothesis, the mitochondrial blockade by various toxins produces a loss of ATP, which directly affects the energy-dependent glutamate-aspartate translocase that is responsible for the transport of glutamate into the mitochondrial matrix. Because glutamate is the major source of aspartate converted into NAA by the transaminase L-aspartate N-acetyl transferase, it is proposed that inhibition of the mitochondrial oxidative phosphorylation process affects NAA production. However, the present rat and primate studies suggest a more complex explanation. We found that, in 3NP-treated rats, significant striatal NAA depletion could be observed in absence of dying cells, evidenced by the lack of striatal TUNEL-positive cells, the preservation of the striatum/cortex ratio of SDH activity, and the preservation of GABA concentration. In 3NP-treated primates, a significant NAA depletion was also found at the early stage of intoxication in the absence of obvious striatal lesions, as assessed by MRI and histologic evaluation. It is unlikely that the NAA depletion produced by 3NP treatment is directly related to the partial blockade of SDH because no NAA depletion was found in the cerebral cortex of 3NP-treated rats, although this cerebral region displayed a level of SDH inhibition similar to that found in striatum. Therefore, the partial blockade of the respiratory chain is not by itself sufficient to induce NAA depletion. Moreover, the significant decrease in aspartate (the direct precursor in the NAA synthesis) observed in the cortical extracts was not associated with significant NAA depletion. Therefore, the selective depletion of NAA in the striatum might be more related to an inherent vulnerability of striatal neurons to mitochondrial challenges. The higher accumulation of succinate and greater depletions in aspartate and glutamate levels observed for a similar degree of SDH inhibition in the striatum, compared with the cortex, suggest that, in the striatum, the oxidative metabolism (tricarboxylic acid cycle and respiratory chain) is more prone to be functionally affected, compared with the cerebral cortex. Although the exact mechanism underlying this phenomenon is still unknown, it is consistent with the preferential vulnerability of the striatum to 3NP-induced neurodegeneration (Beal et al., 1993; Brouillet et al., 1995; Brouillet et al., 1998; for review, Brouillet et al., 1999).
Early striatal NAA depletions are associated with a premorbid state of neuronal dysfunction
In rats, apart from the region-selective NAA depletions, another main difference between striatal and cortical extracts after 3 days of 3NP treatment was the marked and sustained increases in alanine observed in the striatum. Alanine is a glia-derived metabolite known to be involved in the metabolic trafficking between neurons and astrocytes and known to serve as a means of replenishing neuronal amino acid pools (Forissier and Baverel, 1981; Shank et al., 1985; Westergaard et al., 1993; Tsacopoulos et al., 1994; Gamberino et al., 1997). Accordingly, one may speculate that the marked increase of alanine in the striatum reflects a glial activation taking place in this structure, possibly in response to the severe impairment in energy metabolism occurring in the striatal neurons. There was no such phenomenon observed in the cortical extracts, further indicating that, even if cortical neurons do suffer from the metabolic distress induced by 3NP, this impairment is not sufficient in itself to increase the metabolic coupling between glial cells and neurons. Consistent with the behavioral observations obtained in these animals and indicating the presence of abnormal movements, all of the present data demonstrate that the early loss in striatal NAA correlates with severe neuronal dysfunction, not only with metabolic impairment. In addition, the lack of DNA strand breaks (TUNEL-positive cells) in the striatum of rats treated with 3NP for 3 days, as well as the ubiquitous decrease in SDH activity and the normal GABA concentration in the structure, argues against the idea that cell loss could have significantly contributed to the NAA depletion observed at this early stage of treatment. It is, therefore, reasonable to consider that NAA depletions at this stage are mainly, if not exclusively, associated with neuronal dysfunction, preceding the selective degeneration of the striatal projection neurons and characterized by neuronal distress, impaired neurotransmission, and metabolic compensation by local astrocytes. This idea is further reinforced by the data obtained in the rats treated with 3NP for 5 days. This late stage of 3NP treatment is characterized by a further decrease in striatal NAA concentrations and a worsening of the behavioral deficit, and it is associated with the presence of TUNEL-positive neurons in the dorsolateral striatum. These changes are also correlated with significant increases in inositol and taurine and a further loss of SDH activity in the striatal extracts. Whereas the presence of TUNEL-positive nuclei could be interpreted in terms of DNA damage preceding cell death, alterations in inositol and taurine are interpreted as indirect indices of severe neuronal insult. Moreover, these two metabolites are thought to be osmoregulatory and antiexcitotoxic molecules (Thurston et al., 1989; Huxtable, 1992; Oja and Saransaari, 1996; French et al., 1986), and an increase in their concentrations correlates with various pathologic conditions, including edema and excessive activation of the N-methyl-D-aspartate receptors (Menendez et al., 1993; Shibanoki et al., 1993). These conditions are largely involved in the cell death mechanisms induced by 3NP (Brouillet et al., 1999). It is, therefore, possible that part of the large NAA depletion observed in rats treated with 3NP for 5 days, compared with rats treated for 3 days, is caused by ongoing striatal degeneration added to the NAA depletion that results from neuronal dysfunction. However, it should be pointed out that, even if the presence of TUNEL-positive nuclei and profound decrease in SDH activity could be illustrated in the dorsolateral striatum of the rats treated with 3NP for 5 days, these features of structural cell damage were not associated with major GABAergic cell loss, as evidenced by normal GABA concentrations. This suggests that, even in the rats treated with 3NP for 5 days, the contribution of neuronal loss to the observed NAA depletion must be minimal. This further validates the assertion that the early NAA depletion observed after 3 days of 3NP reflects a premorbid state of a possibly reversible neuronal dysfunction.
Early striatal NAA depletions are partially reversible in chronically 3NP-treated primates
To further assess the hypothesis that NAA depletion could serve as an index of premorbid neuronal dysfunction, the potential reversibility of early NAA depletions was tested in baboons that were chronically treated with 3NP, using in vivo1H-MRS. Indeed, whereas the serial assessment of striatal NAA showed that, in agreement with a previous study (Dautry et al., 1999), NAA concentrations were significantly diminished after 8 to 10 weeks of 3NP treatment in the four baboons, as compared with control values (i.e., before 3NP treatment), this decrease was no longer significant after 4 weeks of 3NP withdrawal. This finding is consistent with results obtained from the rats previously reported in the present study, and it suggests that, in the early phase of the 3NP treatment, striatal NAA depletions in the primate model are not caused by cell loss but mainly by neuronal dysfunction. Consistent with this hypothesis, no striatal lesion could be detected in any baboon, either in vivo using T1- or T2-weighted MRI or post mortem using NeuN immunocytochemistry or TUNEL labeling. However, in the one animal examined immediately after the last 3NP injection (i.e., after 10 weeks of continuous 3NP treatment), the depletion of −17% in NAA was associated with a major decrease in calbindin immunoreactivity but with no change in NeuN immunolabeling. This major decrease in calbindin immunoreactivity, reflecting neuronal dysfunction, is consistent with an impairment in the intracellular calcium buffering capacity of a subpopulation of striatal GABAergic projection neurons, incidentally known to be particularly vulnerable to both 3NP treatment and the neurodegenerative process of HD (Seto-Ohshima et al., 1988; Brouillet et al., 1999). Interestingly, as was the case in the rat model, the early NAA depletions observed in the primate model were associated with a state of premorbid neuronal dysfunction previously defined for striatal neurons as being associated with a “widespread transient decrease in calbindin, not associated with neuronal cell loss” (Bazzett et al., 1995).
CONCLUSION
The present findings may have important clinical implications in the search for a treatment of neurodegenerative diseases, in particular HD and Parkinson disease. Finding markers for the chain of events preceding actual neuronal degeneration is required to define the onset of neuronal dysfunction and the optimal time window for therapeutic intervention. In the future, in vivo serial 1H-MRS assessment of NAA concentrations might become a prominent tool, making it possible to characterize these early stages of neurodegenerative disorders that are mainly associated with neuronal dysfunction and to assess the potential efficacy of neuroprotective/neurotrophic interventions. Recent studies on animal models of HD have already suggested that trophic factors, such as ciliary neurotrophic factor, could be efficient in slowing down the degenerative processes of HD (Emerich et al., 1997). In this context, the present study strongly suggests that serial assessment of NAA concentrations using 1H-MRS is essential to this aim.
Footnotes
Acknowledgments
This work was supported by Commissariat à l'énergie Atomique and Centre National de la Recherche Scientifique. The authors thank C. Jouy and F. Sergent for their outstanding care of the primate and rat colonies. The authors thank Drs. K. L. Moya and J. M. Hermel for their critical reading of the manuscript.