
Abstract
Excitotoxicity is implicated in the pathogenesis of several neurologic diseases, such as chronic neurodegenerative diseases and stroke. Recently, it was reported that excitotoxicity has a relationship to apoptotic neuronal death, and that the mitochondrial toxin, 3-nitropropionic acid (3-NP), could induce apoptosis in the striatum. Although striatal lesions produced by 3-NP could develop through an excitotoxic mechanism, the exact relationship between apoptosis induction and excitotoxicity after 3-NP treatment is still not clear. The authors investigated the role of excitotoxicity and oxidative stress on apoptosis induction within the striatum after intraperitoneal injection of 3-NP. The authors demonstrated that removal of the corticostriatal glutamate pathway reduced superoxide production and apoptosis induction in the denervated striatum of decorticated mice after 3-NP treatment. Also, the N-methyl-
Excitotoxicity related to mitochondrial dysfunction and free radical-induced oxidative damage has been implicated in the pathogenesis of several neurobiologic disorders, such as ischemic neuronal injury and chronic neurodegenerative diseases (Beal, 1992; Dugan et al., 1995). Traditionally, excitotoxic neuronal cell death has been considered to be necrotic in nature, but recent studies suggest that apoptotic cell death may have a role in the pathogenesis of excitotoxin-induced cell death. It has been reported that stimulation of N-methyl-
3-Nitropropionic acid (3-NP), an irreversible inhibitor of succinate dehydrogenase, is a widely distributed plant and fungal mitochondrial toxin known to induce damage to the striatum in humans and animals. Clinically, 3-NP has produced delayed dystonia in humans and abnormal choreiform movements in primates (Ludolph et al., 1991). Therefore, experiments using 3-NP were conducted as an animal model of Huntington disease. Because mitochondrial energy toxins cause histotoxic hypoxia, it is of interest that lesions with mitochondrial toxins share several attributes with ischemic brain damage, such as excitotoxicity. 3-NP may also produce neuropathologic changes similar to those induced by hypoxic and ischemic diseases (Gould and Gustine, 1982). It has also been reported that oxidative stress plays a substantial role in the neurotoxicity of 3-NP-induced neuronal injury. Lesions produced by systemic administration of 3-NP increased the production of hydroxyl-free radicals in the striatum as assessed by the conversion of salicylate to 2,3- and 2,5-dihydroxybenzoic acid, and 3-NP neurotoxicity was attenuated in copper/zinc superoxide dismutase transgenic mice (Beal et al., 1995).
We have demonstrated that apoptosis occurred in the rat striatum after intraperitoneal injection of 3-NP and that apoptotic neuronal death was the main part of striatal damage (Sato et al., 1997). Thus, we speculated that apoptosis induced by 3-NP in the striatum might be related to excitotoxicity, but this has not been clearly elucidated in vivo. Although MK-801 could not prevent apoptosis induced by 3-NP treatment in a cell culture system (Pang and Geddes, 1997), there is a possibility that 3-NP toxicity in vivo, in an intact system, could involve apoptosis induction by means of excitotoxicity. Therefore, the involvement of excitotoxicity and oxidative stress in apoptosis induced by treatment with 3-NP in vivo requires comprehensive elucidation.
To confirm the role of excitotoxicity and the oxidative stress mechanism of apoptosis in the striatum induced by 3-NP in vivo, we investigated the involvement of excitotoxicity and whether free radicals are related to this process. Also, we evaluated initial striatal damage and type of cell death by observing the striatum each day after 3-NP treatment for different durations.
MATERIALS AND METHODS
Animals and materials
Male CD1 mice (3 months old, 35 to 40 g; Charles River, Wilmington, MA, U.S.A.) were used in each experiment. All procedures were in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and were approved by Stanford University's Administrative Panel on Laboratory Animal Care and the University of California, San Francisco, Committee on Animal Research.
3-NP (Aldrich Chemical Co., Milwaukee, WI, U.S.A.) was dissolved in saline to a concentration of 25 mg/mL (pH 7.4) and passed through an 0.2-μm filter to remove any bacterial contamination. 3-NP solution was administered intraperitoneally twice daily at a dose of 60 mg/kg in each experiment. To evaluate striatal damage and the pattern of cell death, the 3-NP solution was administered intraperitoneally for 1 to 5 days. Each day four mice per treatment group were euthanized, and the brains were collected for histologic evaluation. The mice were treated with 3-NP or saline for 5 days and then used for nicotinamide-adenine dinucleotide phosphate (NADPH) diaphorase staining. One day after the last injection, the animals were anesthetized with xylazine (4 mg/kg) and chloral hydrate (350 mg/kg) and then decapitated.
To evaluate the effects of decortication in apoptosis induction after 3-NP treatment, the mice (n = 12) were anesthetized with chloral hydrate (350 mg/kg) and xylazine (4 mg/kg) and fixed in a stereotaxic frame. The scalp was incised on the midline, and the skull was exposed and carefully removed with a dental drill under surgical microscopy. The dorsolateral aspects of the left frontoparietal lobe were removed down to the white matter by suction. The decorticated mice were kept for 1 month before being used in further experiments to allow the extracellular glutamate pool to decrease. Six decorticated mice were used for histologic evaluation and six were used for in situ detection of superoxide anion radical (O2{-). To elucidate which subtype of glutamate receptor was involved in inducing apoptosis by means of 3-NP, an NMDA glutamate receptor antagonist, MK-801 (1 mg/kg per day) (RBI, Natick, MA, U.S.A.), a non-NMDA glutamate receptor antagonist, 2,3-dihydroxy-6-nitro-7-sulphamoyl-benzo(F)quinoxaline (NBQX) (30 mg/kg per day) (RBI), and saline (200 μL/d) as a control were injected intraperitoneally 15 minutes before each 3-NP injection. After the treatment six mice in each group were used for histologic evaluation, and four in each group were used for DNA extraction and gel electrophoresis. One day after the last injection, the animals were anesthetized and then decapitated.
Histologic evaluation and in situ detection of DNA-fragmented cells
After euthanasia, the brains were removed and sectioned at a 20-μm thickness on a cryostat. At 500-μm intervals, sections were stained with cresyl violet to measure the lesion volume. The striatum and unstained area were measured in each section by an image analysis system. The striatum and lesion volume were calculated by multiplying each measured area by the distance between sections. Sections from the level of the midstriatum were identified for histologic evaluation after hematoxylin and eosin (H&E) and cresyl violet staining. Sections 200 μm rostral to those used for histologic evaluation were stained using the terminal deoxynucleotidyl transferase-mediated uridine 5'-triphosphate-biotin nick end labeling (TUNEL) method to detect DNA fragmentation in situ. The procedure was carried out as described by Gavrieli et al. (1992) with slight modifications. The sections were air dried and fixed for 30 minutes in 3.7% formaldehyde in 0.1 mol/L phosphate-buffered saline (PBS). The slides were incubated in 0.3% Triton X-100 in PBS for 30 minutes. After rinsing the sections in PBS, they were equilibrated for 15 minutes in terminal deoxynucleotidyl transferase buffer containing 2.5 mmol/L cobalt chloride and 200 mmol/L potassium cacodylate (pH 7.2) (Boehringer Mannheim, Indianapolis, IN, U.S.A.). The labeling reaction was performed at 37°C for 1 hour with terminal deoxynucleotidyl transferase (300 U/mL; GibcoBRL, Gaithersburg, MD, U.S.A.) and 40 μmol/L of biotinylated 14-dATP (GibcoBRL) in terminal deoxynucleotidyl transferase buffer. The sections were washed twice for 15 minutes in a PBS solution containing 150 mmol/L sodium chloride, 15 mmol/L sodium citrate, and 2% bovine serum albumin (pH 7.4). Thereafter, the sections were incubated with a complex of avidin and biotinylated horseradish peroxidase (Vectastatin Elite ABC Kit, Vector Labs, Burlingame, CA, U.S.A.) for 30 minutes. The samples were then washed with 0.175 mol/L sodium acetate, and TUNEL labeling was visualized using diaminobenzidine (0.25 g/L) and nickel chloride (10 g/L). The sections were dehydrated in an ascending ethanol series, immersed in xylene, and coverslipped with Permount (Fisher Scientific, Pittsburgh, PA, U.S.A.).
DNA extraction and gel electrophoresis
To confirm and compare the presence of apoptosis among the MK-801, NBQX, and control groups, we studied fragmentation patterns of DNA isolated from the striatum using agarose gel electrophoresis. Immediately after decapitation, striatal tissue was removed from four animals in each of the three groups. The tissue was homogenized in proteinase K (500 μg/mL), 10 mmol/L Tris HCl (pH 8.0), 0.1 mol/L EDTA (pH 8.0) and 0.5% sodium dodecyl sulfate and incubated overnight at 55°C. DNA was extracted with a 25:24:1 ratio of phenol to chloroform to isoamyl alcohol, precipitated overnight in 95% ethanol and 68 mmol/L NaCl at −20°C, and resuspended in 10 mmol/L Tris HCl and 1 mmol/L EDTA (pH 8.0). The DNA was incubated for 30 minutes at 37°C in 0.1 μg/μL DNase-free RNase (Boehringer Mannheim), and 5 μg/lane were loaded onto a 1.8% agarose gel containing ethidium bromide (0.3 g/mL) and electrophoresed at 100 V for 2 hours. The DNA was visualized and photographed using ultraviolet transillumination (300-nm wavelength).
In situ detection of superoxide anion radical production
The production of O2·- in the striatum after 3-NP injection was investigated by in situ detection of oxidized hydroethidine (HEt) (Murakami et al., 1998). Hydroethidine is selectively oxidized to ethidium by O2·- but not by other reactive oxygen species (Bindokas et al., 1996). Decorticated mice were anesthetized after 3-NP treatment for 2 days, and then 200 μL of hydroethidine (stock solution of hydroethidine: 100 mg/mL in dimethyl sulfoxide finally diluted to 1 mg/mL with PBS) were intravenously administered through the jugular vein under surgical microscopy. The animals were killed 2 hours after the hydroethidine injection by transcardial perfusion with 200 mL of 10 U/mL heparin in saline and 200 mL of 3.7% formaldehyde in PBS under anesthesia. After postfixation overnight in 3.7% formaldehyde, brain sections 50-μm thick were cut at the level of the midstriatum using a vibratome and placed on glass slides. They were then incubated with 2.5 × 10−3 mg/mL Hoechst 33258 (Molecular Probes, Eugene, OR, U.S.A.) in PBS for 15 minutes in a dark chamber, rinsed in distilled H2O, and mounted with Aquamount (Shandon, Pittsburgh, PA, U.S.A.). These sections were observed with a microscope under fluorescent light (HBO/100, Zeiss, Thornwood, NY, U.S.A.) and photographed using double exposure to produce images of both oxidized hydroethidine and Hoechst 33258. Intensity and expression patterns of the oxidized hydroethidine were observed and compared between the denervated and intact striatum in decorticated mice.
Nicotinamide-adenine dinucleotide phosphate diaphorase staining
Mice treated with 3-NP and control mice treated with saline for 5 days were killed under anesthesia by transcardial perfusion. After postfixation overnight in 3.7% formaldehyde, brain sections 50-μm thick at the level of the midstriatum were cut using a vibratome, placed on glass slides, and then dried at room temperature. A mixture of 0.5 mL of 0.1 mol/L Tris-HCL, pH 8.0, 0.1 mL of 10 mmol/L EDTA, 0.02 mL of 10% Triton X-100 in distilled water, 0.1 mL of 2 mmol/L nitroblue tetrazolium, and 0.0 5 mL of 10 mmol/L NADPH in 0.02 mol/L sodium carbonate and bicarbonate buffer per milliliter was used, and the samples were incubated in a dark moisture chamber overnight at 4°C. After counterstaining with methyl green, the sections were dehydrated in an ascending ethanol series, immersed in xylene, and coverslipped with Permount (Fisher Scientific).
Caspase-3 (CPP32) immunohistochemistry
Sections adjacent to those used for TUNEL labeling were evaluated for CPP32 immunoreactivity. The sections were air dried, fixed for 30 minutes in 3.7% formaldehyde in PBS, and incubated in 0.3% Triton X, 100 mL in PBS, for 30 minutes. After rinsing in PBS, the sections were blocked using 10% normal horse serum in PBS for 1 hour. After another PBS rinse, the sections were incubated with a primary antibody, CPP32-p20, which is polyclonal goat antibody against a p20 fragment of the 32-kDa cysteine protease (Santa Cruz Biotechnology, Santa Cruz, CA, U.S.A.) in PBS with 2% horse serum at 4°C for 72 hours. After they were rinsed three times in PBS, the sections were incubated with biotinylated antigoat immunoglobulin G (Boehringer Mannheim) in PBS with 1% goat serum for 1 hour. Thereafter, the sections were incubated with a complex of avidin and biotinylated horseradish peroxidase (Vectastatin Elite ABC Kit) for 30 minutes. The samples were then washed with 0.175 mol/L sodium acetate, and CPP32-p20 labeling was visualized using diaminobenzidine (0.25 g/L) and nickel chloride (10 g/L). The sections were dehydrated in an ascending ethanol series, immersed in xylene, and coverslipped with Permount (Fisher Scientific).
Quantitation and statistical analysis
TUNEL-labeled cells per square millimeter, which displayed densely labeled, small particles in the cytoplasm (apoptotic bodies) and different types of chromatin condensation around the margin of the nucleus forming either crescentic caps or rings, were quantified with a light microscope. They were counted in the middle and lateral part of the lesion with high magnification (x400) and were averaged in both striatum. NADPH diaphorase-positive neurons were counted in the lesion, the area surrounding the lesion, and a comparable region in the control mice using high magnification (x400). Cells with oxidized hydroethidine signals that extended to the cytosol were counted photographically using a fluorescent microscope, and the ratio of those cells to the total cells with Hoechst staining was calculated using high magnification (x400). The percentage of lesion volume was calculated as lesion volume per striatal volume × 100 to exclude any edematous condition. The statistical significance of difference was evaluated by analysis of variance as a percent of lesion volume and TUNEL-labeled cells per square millimeter among the control, MK-801, and NBQX groups.
RESULTS
Neurologic outcome after 3-NP treatment
During the 3-NP treatment, the neurologic outcome was variable. The animals showed increased movement for 20 to 30 minutes, and then they showed normal movement. Decreased movement came after 3-NP treatment for 3 days. After treatment for 4 days, the mice took on a stooped posture and became ataxic. The mice became recumbent after treatment for 5 days. One mouse had only decreased movement, but no lesion was found in the striatum after it was killed. Mice with ataxic movement and without a recumbent position after 3-NP treatment for 4 days showed consistent lesions in the lateral striatum, although the lesion size varied. Mice with a recumbent position and ataxic movement after 3-NP treatment for 5 days had consistent and large lesions in the striatum (data not shown). After MK-801 injection, the mice were hyperactive for 5 to 10 minutes and then finally showed normal movement during 3-NP treatment. Neurologic outcome after NBQX injection with 3-NP was similar to the 3-NP effects mentioned above.
Histologic characteristics in the striatum during daily 3-nitropropionic acid treatment
After 3-NP treatment for 1 to 3 days, no striatal damage was seen by cresyl violet staining or H&E staining, and no TUNEL-labeled cells were detected by in situ TUNEL labeling. Also, no lesion was seen in the hippocampus. Indeed, even after striatal lesions were fully developed, no lesion could be detected in the hippocampus (data not shown).
After 3-NP treatment for 4 days, lesions with an unstained pale area were detected in the striatum by cresyl violet and H&E staining. However, at this time point, lesion characteristics varied and included a diffuse and irregular pattern of cell loss. In the striatum of some mice treated for 4 days, there were small, round, darkly stained cells that were scattered and mixed within normal neuronal cells without an unstained pale area using cresyl violet staining under lower magnification (Fig. 1A). Under higher magnification, those cells appeared with cellular and nuclear shrinkage, and darkly stained chromatin condensation scattered in normal striatal cells (Fig. 1B). In those cases, the results are defined as early lesions. After treatment for 4 days, TUNEL staining was variable, just as the lesion appearance was variable in sections stained with cresyl violet. Usually, diffuse and lightly stained cells were observed (data not shown). In early lesioned striatum, TUNEL-labeled cells were observed (Fig. 1C) even if an unstained pale area could not be detected using cresyl violet staining. At higher magnification, chromatin condensation and small particles in the cytoplasm that resembled apoptotic bodies were detected by TUNEL staining in such early lesioned striatum (Figs. 1D and 1E). However, there were no diffuse and lightly stained cells, as were frequently found in the fully developed lesions after 3-NP treatment for 5 days (see below).
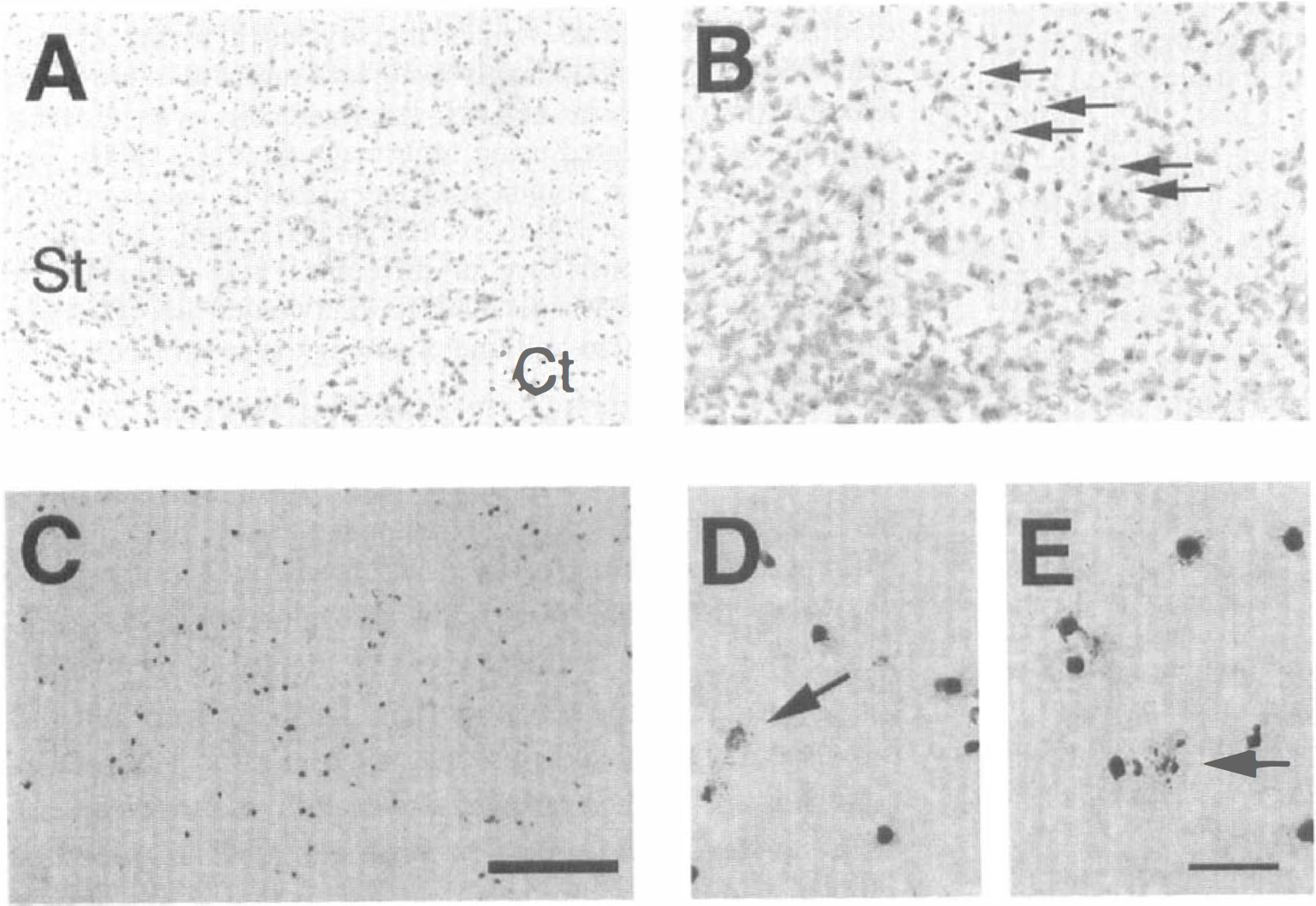
Histologic findings in early lesioned striatum (St) after 3-nitropropionic acid (3-NP) treatment for 4 days. There was no unstained pale area with cresyl violet staining except in the small, round and darkly stained cells that are scattered and mixed with the normal neuronal cells
After 3-NP treatment for 5 days, the lateral portion of the striatum contained large unstained pale regions with substantial cell loss (Fig. 2A). Although histologic examination showed substantial cell loss in the lesioned striatum by methyl green counterstaining, the NADPH diaphorase neurons were relatively spared, as indicated by a dark blue pattern and the frequency of NADPH diaphorase-positive neurons in the lesioned striatum (5.0 ± 0.4/mm2, 5.9 ± 0.6/mm2, 5.4 ± 0.2/mm2, NADPH-positive cells in lesioned striatum, surrounding area of 3-NP-treated mice, and striatum of saline-treated mice, respectively; mean ± SD) (Figs. 2C and 2D). However, NADPH diaphorase staining was light in the core of some samples. By H&E staining of the lesioned striatum, cellular shrinkage and chromatin condensation in the nuclei, findings of the apoptotic process (Fig. 2E), as well as cellular swelling and lysis, typical findings of necrosis (Fig. 2F), were observed. In TUNEL staining, there was an increase in the number of TUNEL-labeled cells in the lesioned striatum (Fig. 2B), which were occasionally accompanied by apoptotic bodies (Fig. 2G), whereas some necrotic cells had light, diffuse nuclear and cellular staining without apoptotic bodies (Fig. 2H).
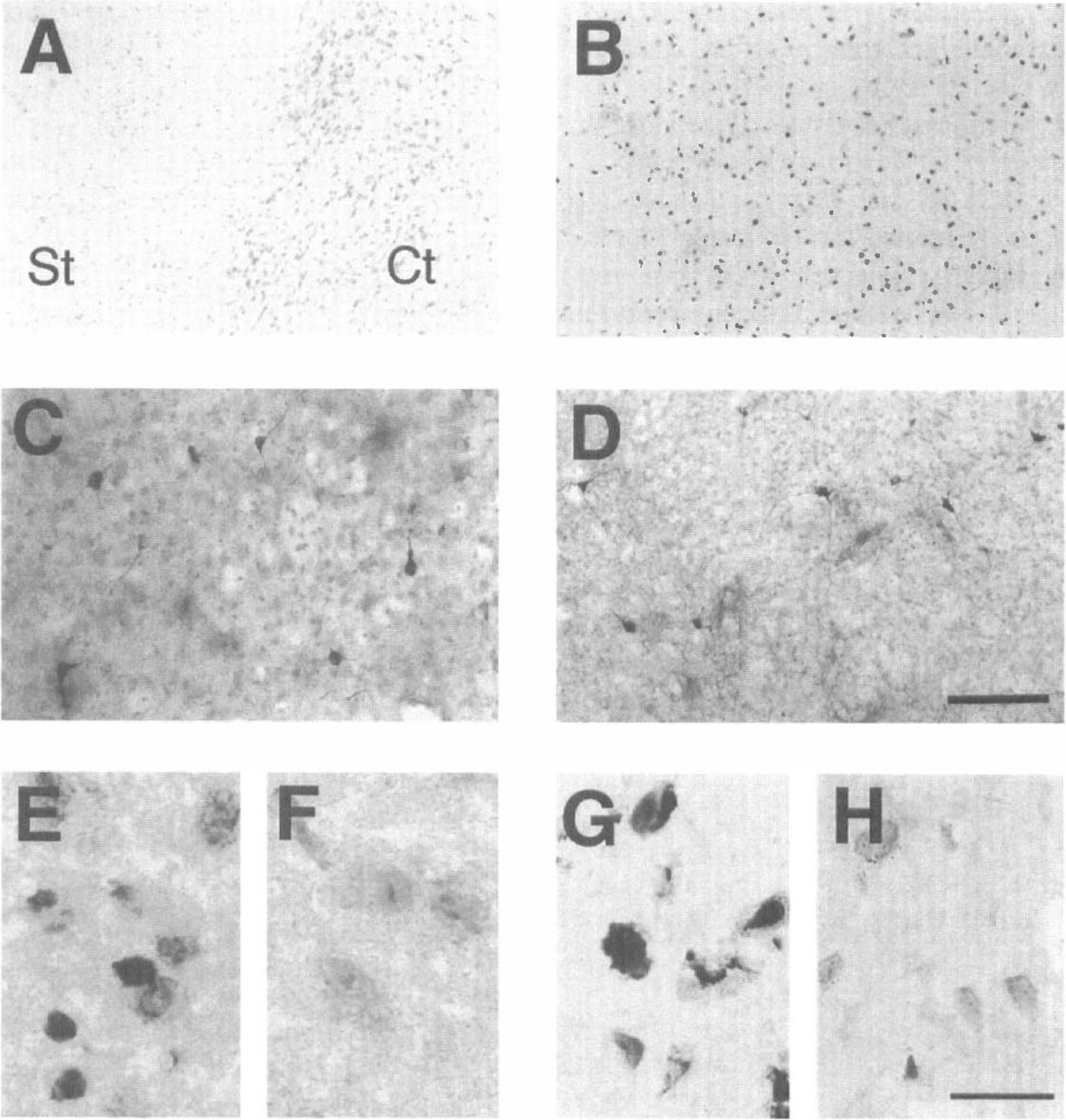
Histologic findings in the striatum (St) after 3-nitropropionic acid (3-NP) treatment for 5 days. Fully developed lesions showed large unstained pale areas with cresyl violet staining
Comparison of TUNEL in situ detection, production of superoxide anion radical and caspase immunoreactivity in intact and denervated striatum of decorticated mice
The lateral portion of the intact striatum in decorticated mice after cresyl violet staining was pale after 5 days of 3-NP treatment (Fig. 3A). To determine the relationship between excitotoxicity and the induction of apoptosis after 3-NP treatment, we evaluated DNA fragmentation by TUNEL staining in the lesioned striatum of decorticated mice. TUNEL-labeled cells (1790.0 ± 99.2/ mm2, mean ± SD) were detected in the intact striatum but not in the denervated striatum. They were densely labeled in the nuclei and showed characteristic features of apoptotic morphology such as nuclear and cellular shrinkage and chromatin condensation, and occasional fragmentation of the nuclei (Figs. 3B and 3C). To determine the role of O2·– in the induction of apoptosis during the excitotoxic process, we evaluated O2·– by in situ detection with hydroethidine. It was visible as small red particles in the cytosol and perinuclear area. In the denervated striatum, fewer oxidized hydroethidine signals were visible as small red particles in the cytosol compared to the intact striatum (2.3% ± 0.2, 31.2% ± 0.9; percent of cells with oxidized hydroethidine signal in the cytosol to total cells, denervated and intact striatum, respectively, mean ± SD) (Figs. 3D and 3E). For determining CPP32 immunoreactivity related to oxidative stress and one of the apoptotic pathways, we evaluated CPP32 immunoreactivity in 3-NP-treated decorticated mice. CPP32 immunohistochemistry of the intact striatum detected densely labeled nuclei and/or coarse granular immunodeposits in the cytoplasm of neuronal cells and in the nuclei of shrunken cells. CPP32 immunoreactivity was weakly labeled, and coarse immunodeposits were not detected in the denervated lesioned striatum (Figs. 3F and 3G).
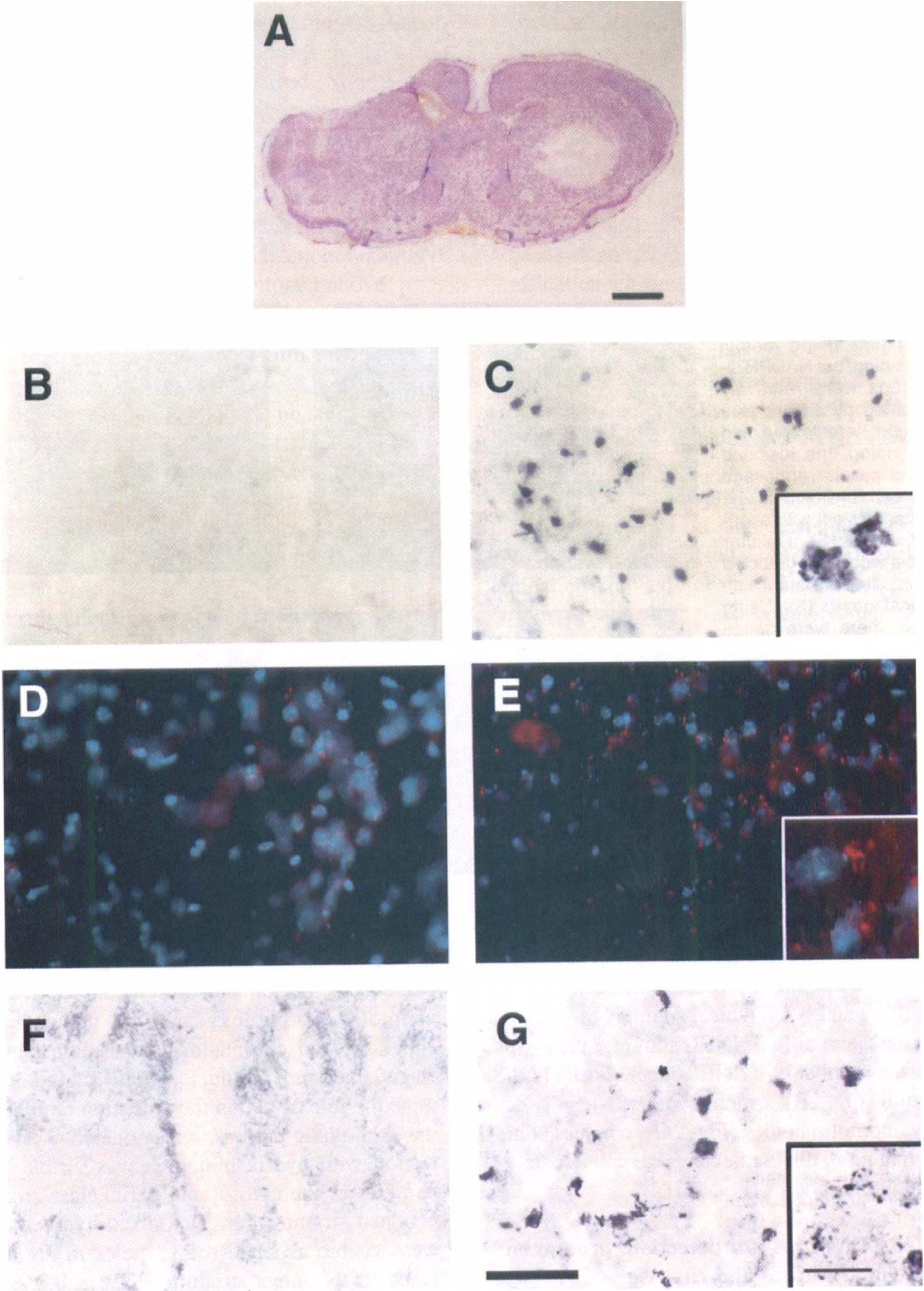
Representative findings of superoxide anion radical (O2·–) production, caspase-3 (CPP32) immunoreactivity, and apoptosis through intact glutamate corticostriatal pathway in decorticated mice after 3-nitropropionic acid (3-NP) treatment for 5 days. The intact striatum showed a pale area with cresyl violet staining, but in the denervated striatum no pale area was seen
N-methyl-d-aspartate receptor antagonist prevented 3-nitropropionic acid-induced striatal damage and apoptosis
To determine whether a particular subtype of the glutamate receptor is regulated for the induction of apoptosis, MK-801 and NBQX were used as pretreatments to block receptor activation, and the lesions and apoptosis were evaluated (Figs. 4A to 4F). The lateral portion of the striatum was pale bilaterally after 3-NP treatment for 5 days (Fig. 4A). After 3-NP treatment for 5 days, TUNEL staining was visualized with dark-field phase-contrast microscopy to evaluate the distribution of apoptotic and necrotic neurons in the lesioned striatum. A bright yellow represented apoptotic neurons and a faint yellow represented necrotic neurons using a yellow filter with dark-field phase-contrast microscopy (Kondo et al., 1997). In the control group, apoptotic neurons were widely distributed in the lateral striatum. Most of the bright yellow apoptotic neurons were observed in the peripheral area of the lesioned striatum. Necrotic neurons were also observed (Fig. 4B). In the MK-801 group, there were few or no apoptotic neurons observed with dark-field phase-contrast microscopy. Necrotic neurons were also few or not observed (Fig. 4C). In the NBQX group, distribution and number of apoptotic and necrotic neurons were similar to the control group (Fig. 4D). After 3-NP treatment for 5 days, the lesion volume and TUNEL-labeled cells were quantified and compared among the three groups (Figs. 4E and 4F). The MK-801 group showed significantly reduced volume percent of lesion compared to the control and NBQX groups, but the NBQX group was not significantly different from the control group (39.7% ± 11.2, 1.26% ± 1.26, 31.2% ± 4.5 for control, MK-801, and NBQX groups, respectively, mean (±SD) (Fig. 4E). In the MK-801 group, there were less TUNEL-labeled cells than in the control and NBQX groups, but there was no significant difference between the control and NBQX groups (1770.6 ± 101.4/mm2, 106.6 ± 106.6/mm2, 1194.6 ± 516.6/mm2 for control, MK-801 and NBQX groups, respectively, mean (± SD) (Fig. 4F).
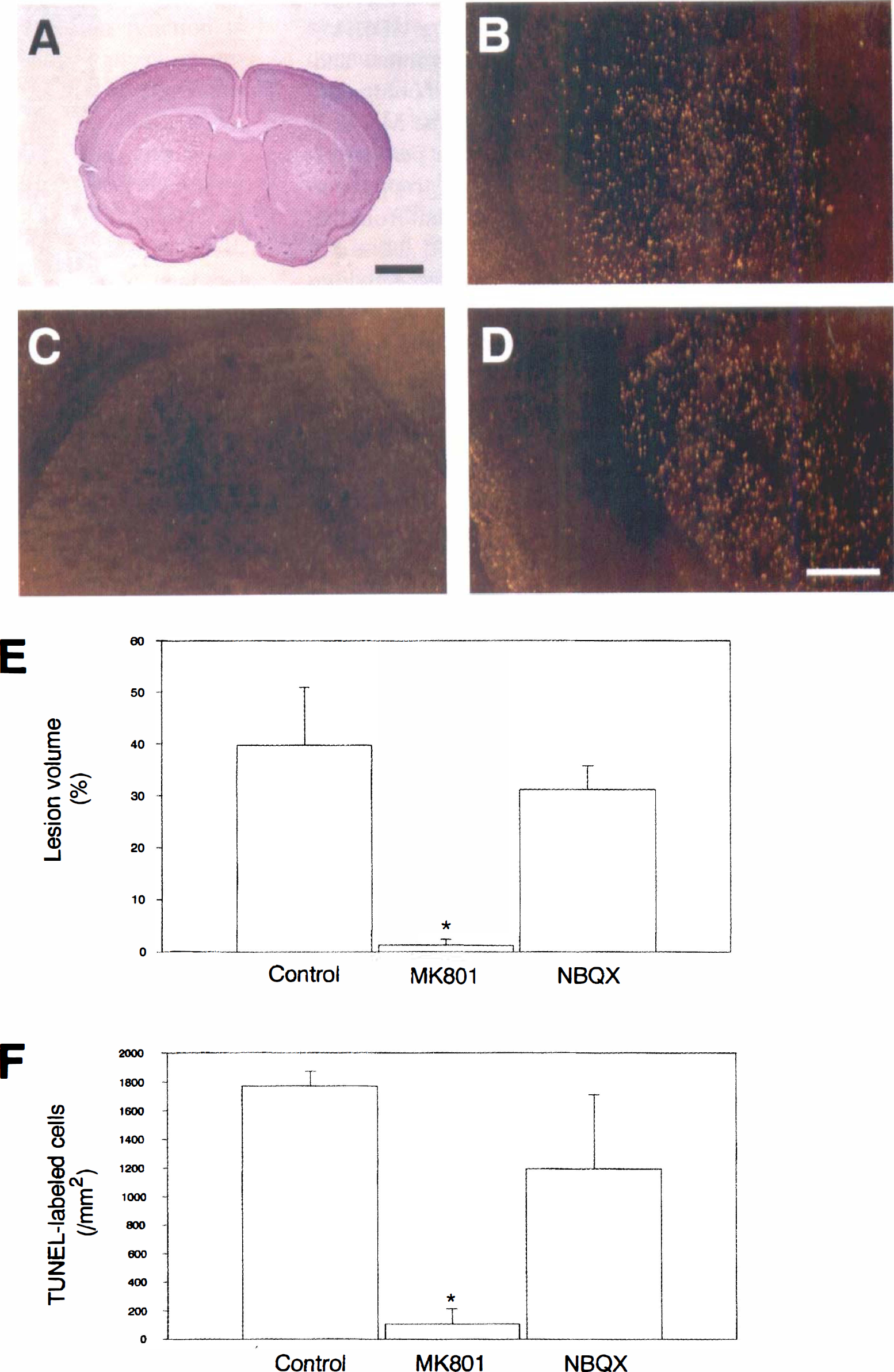
The role of the N-methyl-
MK-801 prevented DNA internucleosomal fragmentation
To confirm the effect of MK-801 and NBQX on the induction of apoptotic neuronal death, we evaluated gel electrophoresis of DNA isolated from the striatum after 3-NP treatment with MK-801, NBQX, and saline pre-treatment for 5 days. DNA from the control and NBQX groups showed signs of internucleosomal fragmentation as evidenced by a ladder pattern caused by cleavage into segments that were multiples of approximately 200 base pairs in length, which is consistent with apoptosis. Internucleosomal fragmentation was also accompanied by some random fragmentation as detected by an overlying continuous smear of DNA, which is consistent with necrosis. However, in the MK-801 group there was no evidence of random or internucleosomal fragmentation (Fig. 5).
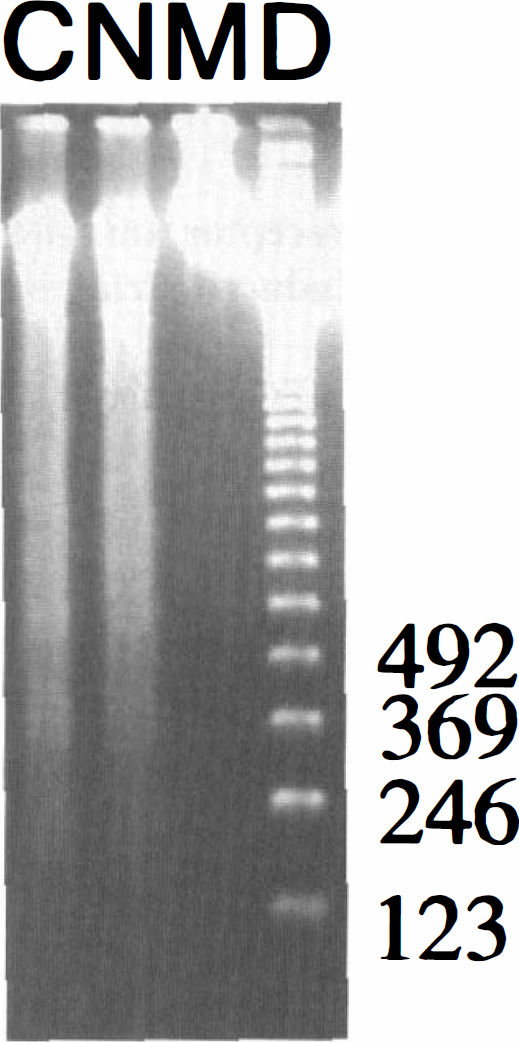
Representative findings of DNA gel electrophoresis after 3-nitropropionic acid (3-NP) treatment with MK-801 and 2,3-dihydroxy-6-nitro-7-sulfamoyl-benzo(F)quinoxaline (NBQX) for 5 days. DNA gel electrophoresis from the striatum of the control and NBQX groups showed a ladder pattern with 200 base pair segments in length. It was also accompanied by random fragmentation as shown by an overlying continuous smear pattern. However, in the MK-801 group, there was neither random nor internucleosomal DNA fragmentation. C, control group; N, NBQX group; M, N-methyl-
DISCUSSION
Although numerous studies have evaluated neuronal cell death by 3-NP, it remains uncertain whether excitotoxicity in 3-NP-induced lesions is related to neuronal death such as necrosis or apoptosis, or both, and what mechanisms are involved. In the present study, we have demonstrated that activation of the NMDA receptor is necessary for striatal apoptosis after systemic 3-NP injection.
Our previous study demonstrated that degeneration within the striatum after 3-NP treatment in rats was mainly caused by apoptosis because more than 40% of the cells within the striatum were TUNEL-labeled. It also showed that agarose gel electrophoresis of DNA extracted from the animals that were treated with 3-NP showed evidence of internucleosomal DNA degradation, as indicated by the ladder pattern of DNA fragments. However, sometimes there also was a random fragmentation pattern that might have been necrosis (Sato et al., 1997). These results suggest that the striatal lesion induced by 3-NP has characteristics of both apoptosis and necrosis, although apoptosis was more prominent.
The present study in mice is consistent with our previous results in rats in which apoptosis occurred after 3-NP systemic injection (Sato et al., 1997). Furthermore, in the present study, early lesioning in the striatum was detected in some mice during daily 3-NP treatment, especially in mice treated for 4 days. At high magnification, there were cells with cellular and nuclear shrinkage and chromatin condensation. TUNEL-labeled cells were scattered in the same area. TUNEL-labeled cells were densely labeled in their nuclei, accompanied by chromatin condensation, and sometimes formed a crescentic cap and apoptotic body-like fragmentation around the nuclei, which is consistent with apoptotic neuronal cell death. Although whether TUNEL-labeled cells are exactly apoptotic cells is still controversial, apoptosis and necrosis could be morphologically distinguished. This issue has been discussed in our previous study on kainic acid-induced neuronal cell death in transgenic mice that over-express human copper/zinc superoxide dismutase (Kondo et al., 1997). The present findings with TUNEL staining suggest that apoptotic neuronal death probably developed initially, followed by necrosis.
It has been reported that the intensity of energy deficiency caused by failure of mitochondrial function is strongly related to the development of apoptosis or necrosis in neuronal cell death after excitotoxicity. Glutamate-induced cell death could be either apoptotic or necrotic depending on the intensity of stimuli and is dependent on mitochondrial membrane potential caused by intensity of energy failure (Ankarcrona et al., 1995). In the present study, we used a small, divided dose of 3-NP. Therefore, our results suggest that apoptotic neuronal death initially involved in striatal damage caused by mild energy failure created more energy failure with subsequent 3-NP treatment and then neuronal necrosis occurred.
Decortication to remove the glutamatergic input into the striatum fully prevented apoptotic neuronal death after 3-NP treatment, compared to the intact striatum in decorticated mice. Although there is no report about effects of decortication on apoptosis induced by 3-NP, our results are consistent with previous reports showing that decortication reduced striatal lesions induced by 3-NP (Beal et al., 1993). In the present study, NADPH diaphorase neurons were relatively spared in the lesioned striatum, particularly in the peripheral area, after 3-NP treatment. This suggests that the lesion in the striatum might occur because of an excitotoxic mechanism, although in previous reports, such as the 3-NP rat model, and under different conditions, NADPH diaphorase neurons were not spared (except for chronic injection of 3-NP) in the lesioned striatum (Beal et al., 1993).
As to the relationship between 3-NP and excitotoxicity with the NMDA receptor, there is still some controversy. Induction of apoptosis in the striatum is presumably related to the effects of 3-NP on mitochondrial function after energy failure and, finally, to indirect excitotoxicity. A loss of adenosine triphosphate production caused by mitochondrial dysfunction can lead to membrane depolarization, removal of the voltage-dependent Mg2+ block of the NMDA receptor, and subsequent activation of the NMDA receptor (Novelli et al., 1988). The present study, in which MK-801 fully prevented apoptotic neuronal death induced by 3-NP, is consistent with that mechanism. There are reports about the effect of non-NMDA antagonists in 3-NP neurotoxicity showing that MK-801 could be more protective against 3-NP neurotoxicity by combining it with non-NMDA antagonists. NBQX, used in the present study, has a short half-life in its pharmacokinetic effect (Gill et al., 1992). This short half-life may preclude its effectiveness in our study. Nevertheless, MK-801 fully prevented neurotoxicity induced by 3-NP in this study. Hence, the NMDA receptor should be the main pathway in apoptotic neuronal death induced by 3-NP. Furthermore, it was reported that frontal cortex ablation decreases the glutamate level in the rat striatum (Kim et al., 1977). The corticostriatal glutamatergic pathway regulates striatal dopamine through presynaptic receptors that are localized on the nigrostriatal dopaminergic terminals, or presynaptic NMDA receptors, or GABAergic striatal interneurons (Smolders et al., 1996). It has been reported that 3-NP produced a dose-dependent selective toxicity to dopamine neurons in mesencephalic culture that could be attenuated by blockade of NMDA receptors (Zeevalk et al., 1995) and that the initial phase of 3-NP poisoning in vivo selectively inhibited the trichloroacetic acid cycle of GABAergic neurons where the trichloroacetic acid cycle of glia remained uninhibited, as did the trichloroacetic acid cycle associated with the large neuronal pool of glutamate, which includes glutamatergic neurons (Hassel and Sonnewald, 1995). However, another study reported that MK-801 and the non-NMDA receptor antagonist, 6-cyano-7-nitroquinoxaline-2,3-dione, could not protect against neuronal death induced by 3-NP in cultured striatal and cortical neurons and that MK-801 failed to prevent apoptotic neuronal death induced by 3-NP in rat hippocampal neuronal cultures (Pang and Geddes, 1997). The possible causes for the discrepancy between those studies and ours might be that the previous ones were in vitro experiments in which the cells would have lacked organized glutamatergic input. There could also be differences because of the doses of 3-NP that were used, especially if low doses induce apoptosis whereas higher doses induce necrosis, as in the core for glutamatergic cell death. This could also explain the report that MK-801 was not able to attenuate damage induced by intrastriatal injection of 3-NP that produced severe metabolic stress (Beal et al., 1993). In our studies (data not shown), MK-801 could not prevent damage by intrastriatal injection of 3-NP at 150 nmol, 300 nmol, or 500 nmol.
In the present study, we showed that hydroethidine oxidation, a putative index of O2·– production, was increased in the intact striatum after 3-NP treatment compared with the denervated striatum in decorticated mice. This increase in oxidized hydroethidine signals is probably attributable to O2·– production from mitochondrial injury after 3-NP treatment. Our results showed that blocking the glutamatergic corticostriatal pathway prevented O2·– production. Furthermore, apoptotic neuronal death was detected in the intact striatum in decorticated mice. The precise relationship between glutamatergic input, O2·– production, and subsequent apoptosis was not determined in the present study, but our results are consistent with previous studies. It has been reported that formation of superoxide is an obligatory step for excitotoxic cell death in rat cortical cell cultures (Patel et al., 1996) and that superoxide dismutase protects against glutamate-induced neuronal death in cortical neuronal culture (Chan et al., 1990) and delays apoptosis in sympathetic neuronal cultures deprived of nerve growth factor (Greenlund et al., 1995). Furthermore, mitochondrial manganese superoxide dismutase prevents neuronal apoptosis induced by nitric oxide-generating agents in pheochromocytoma cell culture (Keller et al., 1998b). In a previous report, we demonstrated that the ischemic penumbra area after focal ischemia in mice with manganese superoxide dismutase deficiency showed increased production of O2·– and mitochondrial derangement (Murakami et al., 1998) and that cytochrome c release from the mitochondria after transient focal cerebral ischemia in rats is related to oxidative stress and apoptosis (Fujimura et al., 1998). A study using mice with manganese superoxide dismutase deficiency might be helpful to elucidate the relationship between O2·– and striatal damage induced by 3-NP. In the present study, we did not do double-staining for hydroethidine, TUNEL, or CPP32 because of the different time course applied in each procedure. Nevertheless, these studies should be helpful to evaluate the relationship between oxidative stress and apoptotic cell death at the single-cell level.
We also examined the role of caspase in the induction of apoptosis during the excitotoxic process caused by 3-NP. In the present study, CPP32 immunoreactivity was increased in intact striatum of decorticated mice after 3-NP treatment. These findings are compatible with a previous report showing that caspase-3p32 immunoreactivity increased after reperfusion, and caspase-3p20, a specific activating form of caspase-3p32 immunoreactivity, appeared as dense, coarse granular immunodeposits in ischemia and reperfusion models (Namura et al., 1998). Recently, it was reported that 3-NP increased intracellular Ca2+ and promoted free radical production from mitochondria and caspase upregulation and finally produced apoptosis in neural cell cultures with mutant presenilin-1 (Keller et al., 1998a). Because decortication abolished the increase in CPP32 immunoreactivity, our results suggest that NMDA receptor activation is necessary for caspase upregulation caused by 3-NP.
In conclusion, excitotoxic process with NMDA receptor is required for O2·– production and apoptosis caused by 3-NP treatment. The 3-NP intraperitoneal model in mice should be a useful tool to elucidate selected conditions of mild metabolic stress that might occur in the penumbral region of infarcted tissue in pure, brief hypoxia or in neurologic diseases involving mitochondrial derangements.
Footnotes
Acknowledgments
The authors thank Juha Yrjänheikki, Liza Reola, and Bernard Calagui for their technical assistance and Cheryl Christensen for her editorial assistance.