
Abstract
Axons and oligodendrocytes are vulnerable to cerebral ischemia. The absence of quantitative methods for assessment of white matter pathology in ischemia has precluded in vivo evaluation of therapeutic interventions directed at axons and oligodendrocytes. The authors demonstrate here that the quantitative extent of white matter pathology was reduced by restoration of cerebral blood flow after 2 hours of middle cerebral artery occlusion. Focal ischemia was induced in anesthetized rats by intraluminal thread placement, either transiently (for 2 hours) or permanently. At 24 hours after induction of ischemia, axonal damage was determined by amyloid precursor protein (APP) immunohistochemistry, and the ischemic insult to oligodendrocytes was assessed by Tau-1 immunostaining in the same sections. In adjacent sections, ischemic damage to neuronal perikarya was defined histologically. The hemispheric extent of axonal damage was reduced by 70% in the transiently occluded animals from that in permanently occluded animals. The volumes of oligodendrocyte pathology and of neuronal perikaryal damage were reduced by 62% and 58%, respectively, in the transiently occluded animals. These results demonstrate that this methodologic approach for assessing ischemic damage in axons and oligodendrocytes can detect relative alterations in gray and white matter pathology with intervention strategies.
White matter and gray matter are vulnerable to ischemic damage in human stroke, and functional deficits are generally a reflection of damage to both neuronal perikarya and their myelinated axons. In both axons and oligodendrocytes (the cells responsible for myelination), the temporal profile of pathologic changes detected by either electron microscopy (Pantoni et al., 1996; Petito, 1986) or immunohistochemistry (Dewar and Dawson, 1995, 1997; Irving et al., 1997; Yam et al., 1998) in experimental models of focal ischemia indicates that damage to these cellular elements occurs in response to the local ischemic insult and is not a consequence of impaired perikaryal function. The results of these in vivo studies are consistent with in vitro investigations which demonstrate that isolated myelinated fiber tracts, which do not contain neuronal perikarya, exhibit both functional and structural impairment in response to 60 minutes of anoxia (Waxman et al., 1992). Similarly, cultured oligodendrocytes are vulnerable to a variety of insults, including oxygen/glucose deprivation (McDonald et al., 1998), exposure to glutamate (Oka et al., 1993), and oxidative stress (Kim and Kim, 1991; Laskiewicz et al., 1999; Richter-Landsberg and Vollgraf, 1998). Pretreatment with the spin-trap agent α-phenyl-tert-butyl-nitrone before middle cerebral artery occlusion (MCAO) reduces the ischemic insult to oligodendrocytes (Irving et al., 1997). In view of the susceptibility of neuronal perikarya, axons, and oligodendrocytes to an ischemic insult, it is axiomatic that improvements in functional outcome after stroke will be enhanced if protection of both gray and white matter is achieved.
Definition of the ischemic lesion by the most comprehensive and sensitive methodology, quantitative histopathology, relies heavily on the identification and anatomic mapping of eosinophilic neuronal perikarya (Osborne et al., 1987), with little or no assessment of axonal or oligodendrocyte damage. The insensitivity of conventional histological markers (e.g. hematoxylin and eosin staining) for detecting axonal and oligodendrocyte damage compared with neuronal damage has contributed to a neglect of white matter pathology in experimental studies of ischemia in vivo. We have therefore developed sensitive immunohistochemical methods that allow us to examine both axonal and oligodendrocyte pathology.
Immunolabeling of amyloid precursor protein (APP) is a sensitive marker of ischemically damaged axons (Dietrich et al., 1998; Stephenson et al., 1992; Yam et al., 1997), and we have shown that APP accumulation in swollen and bulbous axons clearly delineates the margin of a focal ischemic lesion in white matter, thus permitting a quantitative assessment of axonal damage (Yam et al., 1997, 1998). Immunoreactivity of the microtubule-associated protein tau increases rapidly in oligodendrocytes in response to focal ischemia in vivo (Dewar and Dawson, 1995; Irving et al., 1997), and this technique allows a quantitative assessment of oligodendrocyte pathology. In the present study we used these immunohistochemical techniques to provide quantitative assessments of axonal and oligodendrocyte pathology compatible with our conventional quantitative histopathologic assessment of neuronal damage. We used tissue reperfusion as a proof of the concept that the combination of these methods provides a sensitive approach to determine the efficacy of an intervention strategy to reduce both white and gray matter damage after a focal ischemic insult in the rat.
METHODS
Focal cerebral ischemia
Fifteen Sprague-Dawley male adult rats weighing 270 to 320 g were used. Eight rats underwent transient MCAO lasting 2 hours and then were allowed to survive another 22 hours. In seven rats the MCA was permanently occluded for 24 hours. Anesthesia was induced in a perspex box saturated with a mixture of 5% halothane in 70% nitrous oxide: 30% oxygen. Rats were then intubated with a 16-gauge cannula and mechanically ventilated with approximately 1.5% halothane and a nitrous oxide and oxygen mixture (70%: 30%). A femoral artery was cannulated to allow monitoring of arterial blood pressure and sampling of arterial blood (for measurement of arterial blood gas status and plasma glucose concentration). Rectal temperature was maintained in the normal range throughout the surgical procedure.
Focal cerebral ischemia was induced with the intraluminal vascular occlusion technique (Longa et al., 1989). In eight animals the thread was removed after 2 hours of occlusion (transient MCAO); it remained in place in the remaining seven animals (permanent MCAO). The muscles and skin of the animals were sutured and the animals were administered atropine sulfate (1 mg/kg) and saline (2 ml) subcutaneously. Anesthesia was discontinued and the animals were given access to water and soft food. At 24 hours from the induction of the ischemia, the animals were killed by transcardiac perfusion fixation with 4% paraformaldehyde in 50 mmol/L phosphate buffer (pH 7.4). One animal of the seven in the permanent MCAO group died before 24 hours, probably as a result of cerebral swelling. Two animals of the eight with transient MCAO had gross macroscopic evidence of subarachnoid hemorrhage at the MCA and in the surrounding of the circle of Willis; these animals were therefore excluded from the study before processing of the brains. The brains from six animals who underwent permanent MCAO and six who underwent transient MCAO were embedded in paraffin, and coronal sections (5 microns) were cut throughout the cerebral hemispheres.
Histology and immunohistochemistry
Sections at eight predetermined coronal levels (Osborne et al., 1987) were stained with hematoxylin and eosin for assessment of ischemic damage to neuronal perikarya. Adjacent sections were processed for double-label immunohistochemistry as follows. Sections were stained first with an antibody against tau to label ischemic oligodendrocytes (Dewar and Dawson, 1995; Irving et al., 1997) and second with an antibody against APP to label ischemically damaged axons. Dewaxed sections were microwaved for 10 minutes in 10 mmol/L citric acid (pH 6.0), allowed to cool, and incubated in 3% H2O2 in methanol for 30 minutes and then for 1 hour in 50 mmol/L phosphate-buffered saline (PBS, pH 7.2) containing 0.5% bovine serum albumin and 10% normal horse serum. After incubation overnight at 4°C with the monoclonal antibody Tau-1 (diluted 1:5,000 in PBS), the sections were incubated with secondary antibody (biotinylated horse antimouse, 1:100, Vector Laboratories, U.K.) followed by the avidin-biotin complex (ABC kit, Vector Laboratories). Tau-1 immunoreactivity was revealed using VIP (Vector Laboratories) as the chromagen, producing a pink-purple stain. After rinsing in distilled water, the staining protocol was repeated using an antibody against APP (Clone 22C 11; Boehringer, Mannheim, Germany) diluted 1:500 in PBS. APP immunostaining was revealed with SG as a chromagen (Vector Laboratories), producing a blue-gray stain. At the end of the procedure, the sections were dehydrated, cleared, and mounted for light microscopic analysis. Negative controls for Tau-1 and APP antibodies, where the primary antibody was omitted from the procedure, were included in the protocol, and minimal staining was detected. All further analysis was performed by an investigator masked to the duration of ischemia in each animal.
Quantification of damage to axons, oligodendrocytes, and neuronal perikarya
Sections doubled-labeled for tau and APP were captured digitally using an image analysis system (MCID, M4 Imaging Research, Canada) and printed at a fixed magnification (×20). The sections were then viewed using light microscopy. The distribution of APP-positive axons (which exhibited a bulbous or swollen morphology, Fig. 1B) was plotted onto the relevant digitized images at the eight predetermined coronal planes. To quantify the extent of axonal pathology, a 2-mm spaced grid printed on transparent acetate was superimposed on the digitized images, and the number of grid intersections with the plotted APP-positive axons was determined. An APP score for the cerebral hemisphere was calculated by summing the scores from the eight individual coronal planes.
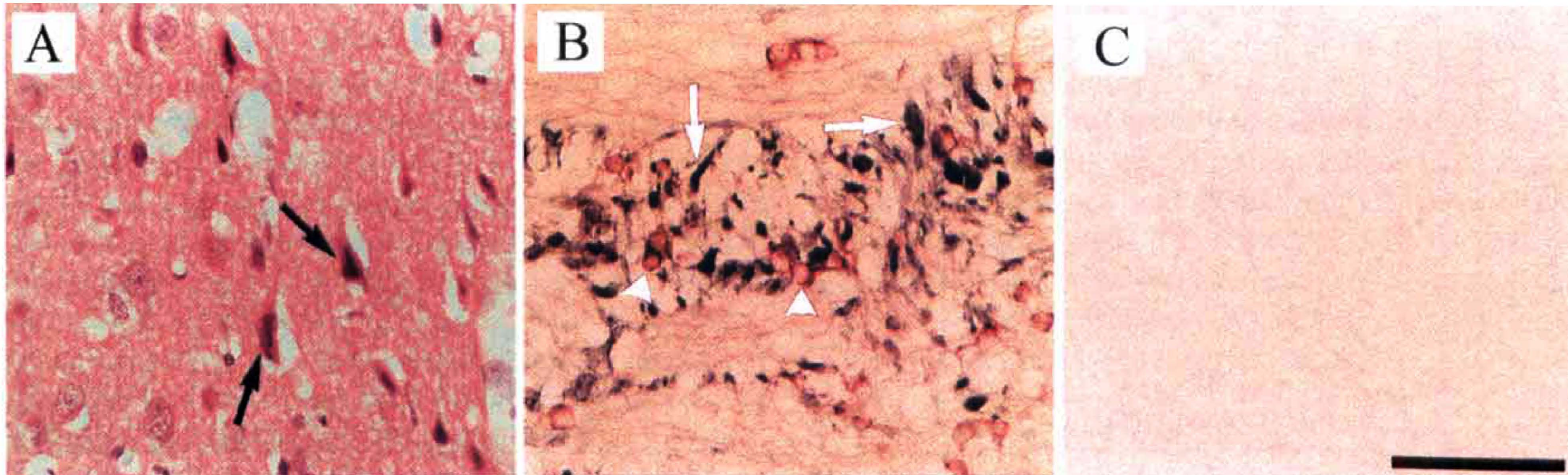
Pathologic features of ischemic damage in neuronal perikarya in the cerebral cortex and axons and oligodendrocytes in the internal capsule. (
Sections double-labeled for tau and APP were viewed using light microscopy, and the distribution of tau-positive oligodendrocytes (Fig. 1B) was plotted onto copies of the relevant digitized images as described above. Areas of tau-positive oligodendrocytes were transposed onto scale diagrams of the eight coronal planes (Osborne et al., 1987). The areas of tau-positive oligodendrocytes on the scale diagrams were measured using an image analyzer (MCID, M4 Imaging Research), and the hemispheric volume of tau-positive oligodendrocytes was computed (Osborne et al., 1987). Hematoxylin and eosin-stained sections were viewed by light microscopy, and areas where neuronal perikarya displayed the morphologic features of ischemic damage (Fig. 1A) were delineated on the scale diagrams of the eight predetermined coronal planes. The areas of ischemic neuronal perikarya were measured using image analysis and the volumes were computed (Osborne et al., 1987). The intrarater variability for assessing the area of tau-positive oligodendrocytes and the APP score at a single level was similar to that described previously for assessing areas of ischemic damage to neuronal perikarya (Osborne et al., 1987).
Statistical analysis
Data are presented as mean ± standard deviation. To test our a priori hypothesis, total hemispheric measures of axonal, oligodendrocyte, and neuronal perikaryal damage in the transient and permanent MCAO animals were compared using Student's t-test. For post hoc descriptive purposes, the extent of axonal, oligodendrocyte, and neuronal perikarya damage at each coronal plane in the transient and permanent MCAO animals were compared using Student's t-test.
RESULTS
Pathologic features of axons, oligodendrocytes, and damaged neuronal perikarya
Neuronal perikarya. Ipsilateral to the MCAO within the cerebral cortex, caudate nucleus, and hypothalamus, neuronal perikarya clearly exhibited the characteristic morphologic features of ischemic damage (i.e., shrinkage and triangulation of the nucleus and cytoplasm and increased basophilia of cytoplasm ‘Fig. 1A’). In both the permanent and transient MCAO groups, sharp boundaries between ischemic and nonischemic neuronal perikarya were easily identifiable throughout the rostrocaudal extent of the ipsilateral hemisphere on hematoxylin and eosin-stained sections. However, in white matter, boundaries between ischemic and nonischemic tissue could not be reliably identified.
Axons. In sections immunostained for APP, ischemic damage to axons was observed as intense APP immunoreactivity in swollen or bulbous axons (Fig. 1B) within ipsilateral subcortical white matter, fiber tracts permeating the striatum, the internal capsule, and the medial forebrain bundle. The anatomically circumscribed zones of APP immunoreactivity allowed the boundary of the ischemic lesion to be delineated in all of these myelinated fiber tracts in both the permanent and transient MCAO groups.
Oligodendrocytes. In sections immunostained for tau, intensely stained cells with the morphologic appearance of oligodendrocytes were present throughout ipsilateral gray and white matter of the permanent and transient MCAO groups (Fig. 1B). Double immunolabeling using antibodies that specifically label astrocytes glial fibrillary acidic protein or microglia (MRF1) (Tanaka et al., 1998) demonstrated that the tau-positive cells after ischemia were distinct from these two cell types. In both the permanent and transient MCAO groups, boundaries could be identified between areas containing numerous intensely stained tau-positive oligodendrocytes within the territory of the MCA and areas with few and faintly stained oligodendrocytes located outside the ischemic territory.
Modification of pathology in axons, oligodendrocytes, and neuronal perikarya
The extent of axonal, oligodendrocyte, and neuronal perikaryal damage was reduced in the transient MCAO group compared with the permanent MCAO group (Figs. 2, 3). In animals with transient MCAO, hemispheric axonal pathology, as reflected by the total APP score, was significantly lower (P = 0.0015) than in animals with permanent MCAO. In animals with transient MCAO, the volume of tau-positive oligodendrocytes was significantly (P = 0.03) less than in animals with permanent MCAO. In animals with transient MCAO, the volume of ischemically damaged neuronal perikarya was significantly (P = 0.02) less than in animals with permanent MCAO (Fig. 2). Thus, restricting the period of occlusion to 2 hours significantly attenuated the hemispheric extent of axonal, oligodendrocyte, and neuronal perikaryal pathology. In animals with transient MCAO, the area of axonal pathology was significantly smaller than in the permanent MCAO group in two of the more caudal coronal planes examined (coordinates 5.15 and 3.75 mm anterior to the interaural line) (Fig. 3). In animals with transient MCAO, the area of tau-positive oligodendrocytes was significantly smaller than in the permanent MCAO group in three of the most rostral planes examined (coordinates from 10.5 to 7.19 mm anterior to the interaural line). In animals with transient MCAO, the area with ischemically damaged neuronal perikarya was significantly smaller than in the permanent MCAO group in the five most rostral planes examined (coordinates from 10.5 to 5.15 mm).
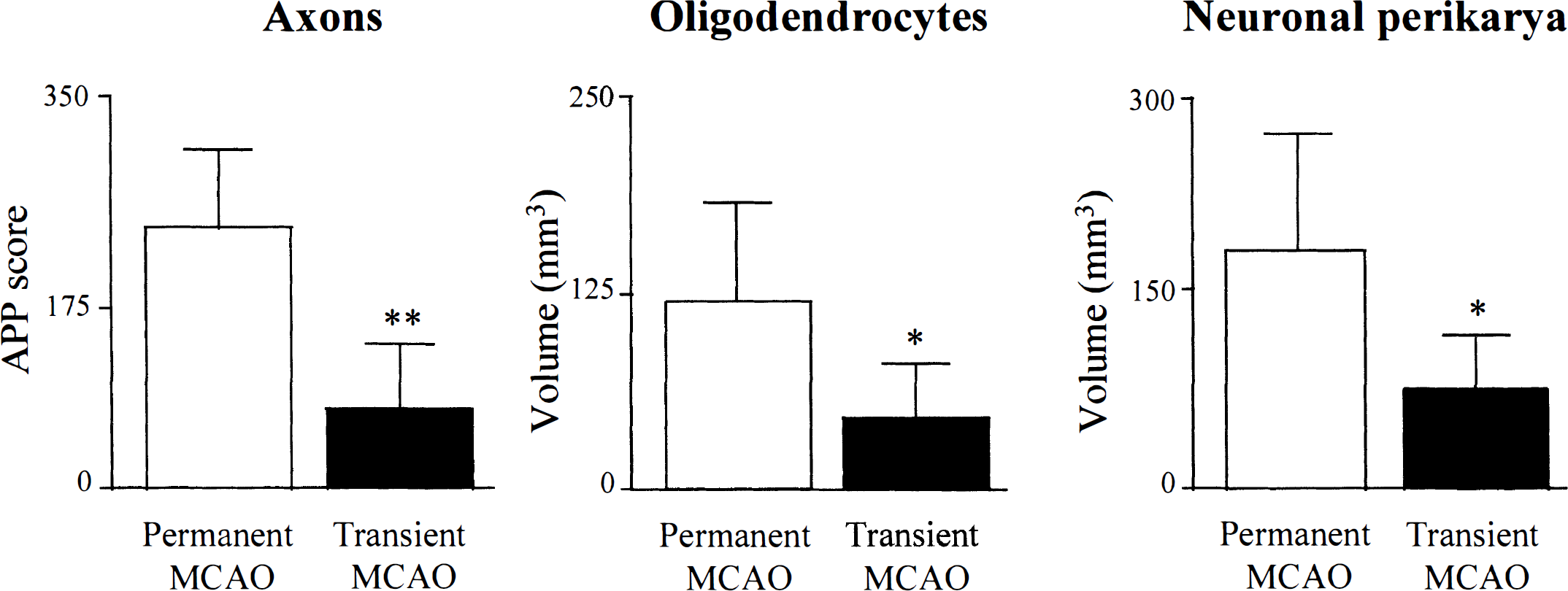
Transient occlusion of the middle cerebral artery is associated with significantly less damage to axons, oligodendrocytes, and neuronal perikarya than permanent occlusion. Data are the total hemispheric amyloid precursor protein scores and volumes of oligodendrocyte pathology and neuronal necrosis derived from area measurements at the eight predetermined coronal planes. Data are expressed as mean ± SD, n = 6 per group. **P < 0.002; *P < 0.05, two-tailed unpaired Student's t-test.
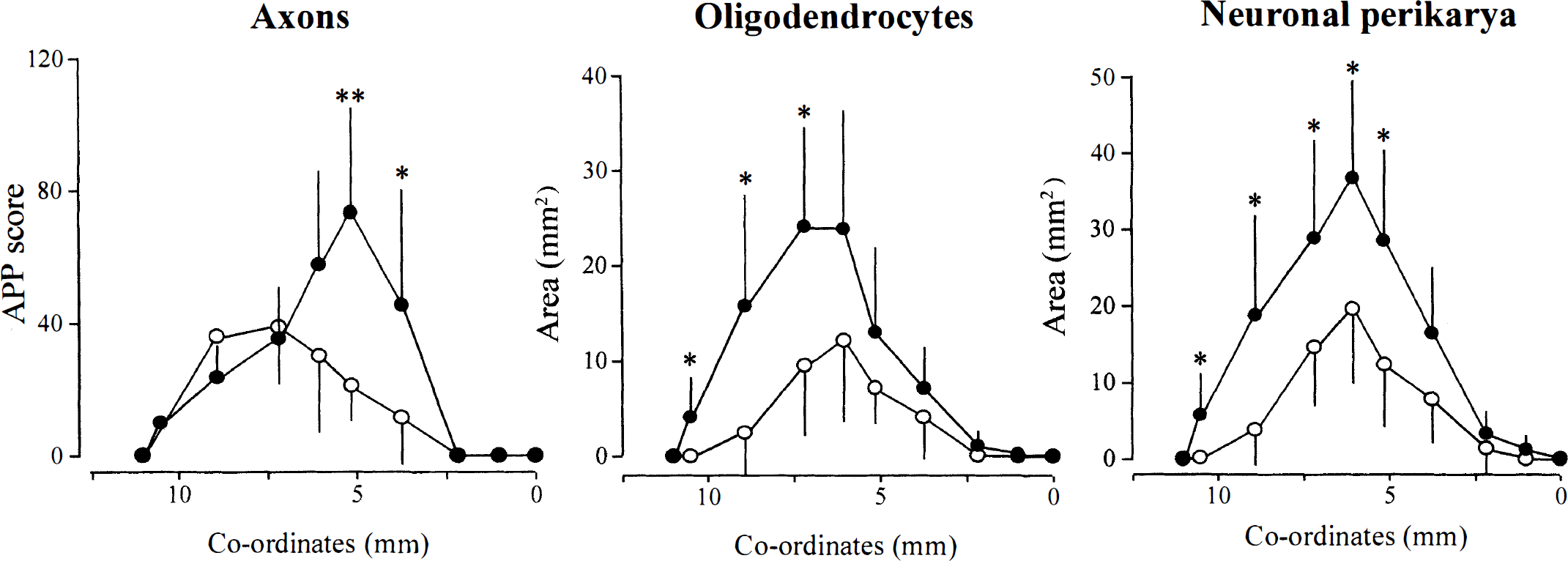
Rostrocaudal extents of axon, oligodendrocyte, and neuronal perikarya damage in the transient and permanent occlusion groups. Amyloid precursor protein (APP) scores, areas of tau-positive oligodendrocytes, and areas of neuronal necrosis are shown at each of the eight coronal planes analyzed. Coordinates (mm) are from the interaural line (Konig and Klippel, 1963). Post hoc analyses demonstrated that the APP score in the transient occlusion group was significantly lower than in the permanent occlusion group at two of the more caudal coronal planes, whereas the areas of oligodendrocyte and neuronal pathology were significantly lower at more rostral coronal planes. Data are mean ± standard deviation, n = 6 per group. **P < 0.01; *P < 0.05, two-tailed unpaired Student's t-test.
Anatomic distributions of axonal, oligodendrocyte, and neuronal perikaryal pathology
A consistent feature in animals with permanent MCAO was the extensive axonal pathology in the internal capsule, including both dorsal and ventral aspects of this major myelinated fiber tract (see Fig. 4). The greatest differences in axonal pathology between the two experimental groups was observed in the internal capsule and subcortical white matter (Table 1). In the transient MCAO animals, axonal pathology was present only in the ventral aspect of the internal capsule. In contrast, axonal pathology was consistently observed in the medial forebrain bundle of both experimental groups. In animals with permanent MCAO, tau-positive oligodendrocytes were consistently observed in the internal capsule and medial forebrain bundle, and this neuroanatomic distribution was not different in the transient MCAO group. In contrast to permanent MCAO, transient MCAO was associated with minimal neuronal perikaryal and oligodendrocyte pathology in the cerebral cortex (see Fig. 4).
Frequency of axonal and oligodendrocyte pathology after permanent or transient MCA occlusion in myelinated fiber tracts

The presence or absence of APP-positive axons and tau-positive oligodendrocytes was noted at each of the neuroanatomical locations listed. The number of animals exhibiting the feature is shown at each neuroanatomical location examined. The internal capsule and medial forebrain bundle were examined at coordinate 5.15 and the subcortical white matter at coordinate 7.19 (see Fig. 4). Note that in the transient MCAO group (trans) fewer of the animals have axonal and oligodendrocyte pathology in the dorsal internal capsule and the subcortical white matter compared to the permanent MCAO group (perm). In contrast, similar numbers of animals in each group display axonal and oligodendrocyte pathology in the region of the medial forebrain bundle. Perm, permanent; Trans, transient.
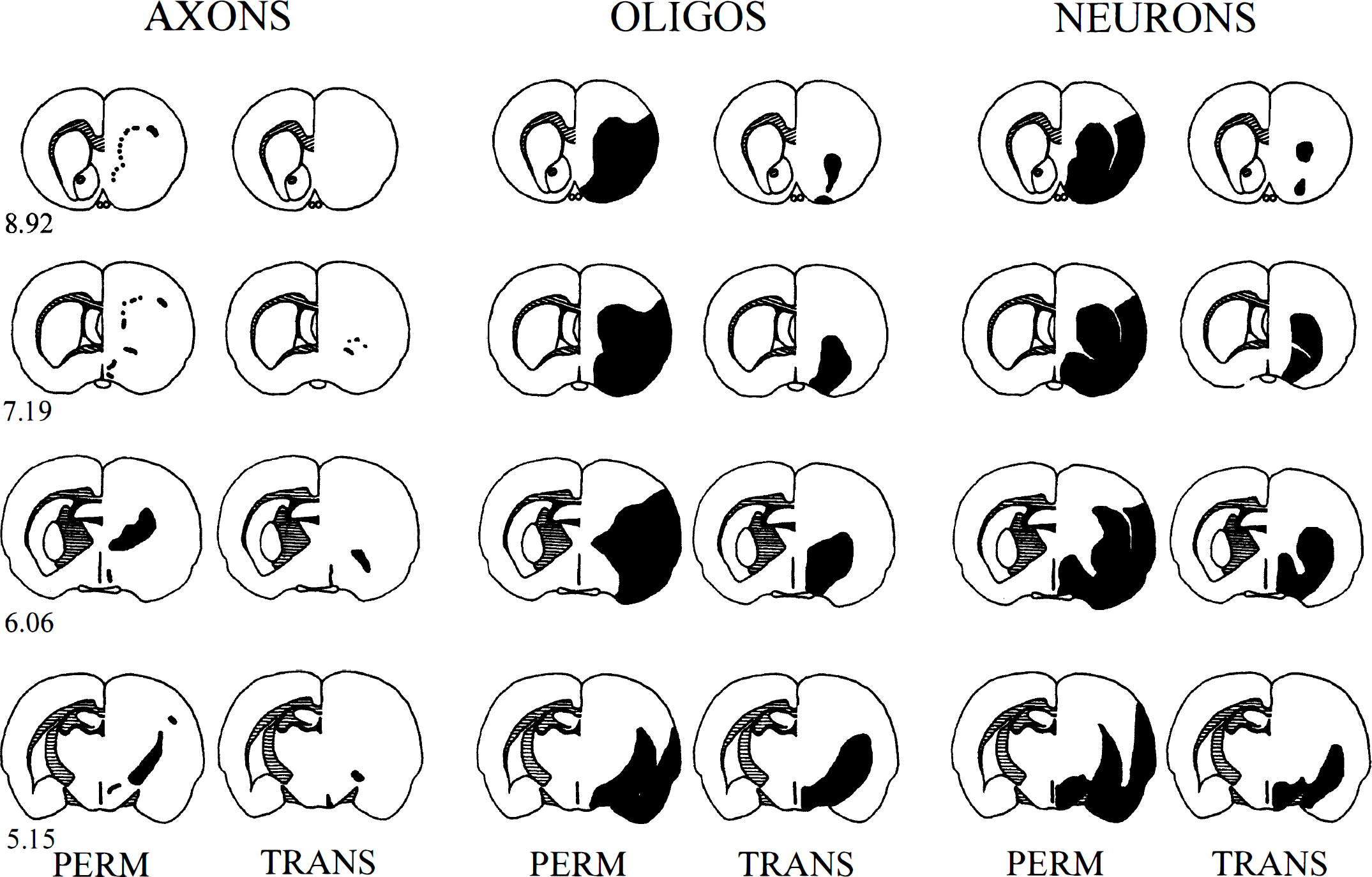
Relative neuroanatomic distributions of axonal, oligodendrocyte, and neuronal pathology at four of the eight coronal planes used for quantitative analyses from a representative animal in the permanent and transient middle cerebral artery occlusion groups. The coordinates for each coronal plane are shown on the left. Black shading in the right hemisphere represents the areas containing amyloid precursor protein-positive axons (AXONS), tau-positive oligodendrocytes (OLIGOS), and necrotic neurons (NEURONS). The hatched shading in the left hemisphere illustrates the myelinated fiber tracts of the subcortical white matter, anterior commissure, and internal capsule. Note the presence of damaged axons in both the dorsal and ventral aspects of the internal capsule (6.06 and 5.15) and the subcortical white matter (8.92 and 7.19) in the permanent occlusion animal but the absence of axonal damage in the dorsal aspect of the internal capsule and subcortical white matter in the transient occlusion animal. In both permanent and transient animals, there is axonal damage in the medial forebrain bundle (6.06 and 5.15). Tau-positive oligodendrocytes were present in both white matter tracts and gray matter in the cerebral cortex. The reduction in the extent of oligodendrocyte pathology in the transient animal occurs predominantly in the gray matter of the cerebral cortex. There is also significantly less neuronal perikaryal damage in the cortex of the transient versus the permanent animal.
Physiologic variables
At MCAO, there were no significant differences between the transient and permanent groups in mean arterial pressure (87 ± 3 and 87 ± 2 mm Hg, respectively), rectal temperature (37.1°C ± 0.1°C and 37.1°C ± 0.1°C, respectively), or plasma glucose (8.0 ± 1.5 mmol/L and 7.0 ± 0.5 mmol/L, respectively), or at any time during the monitoring period.
DISCUSSION
Our results provide compelling evidence that the methodology described can demonstrate protection of both gray and white matter after focal cerebral ischemia in vivo. Two hours of ischemia was selected to provide proof of the concept that our methods could detect reductions in the extent of white matter pathology. In the rat, 2 hours of transient MCAO produces consistent damage to gray matter, but the volume of gray matter damage is significantly less than with permanent MCAO (Garcia et al., 1995; Kaplan et al., 1991; Kawamura et al., 1994). The difference in the volume of gray matter damage between 2 hours of transient intraluminal thread and permanent thread placement at the origin of the MCA in the present study is similar to that reported previously (Garcia et al., 1995; Kawamura et al., 1994). Our data also demonstrate that there is a significant difference between the two groups in the extent of white matter pathology.
We used APP accumulation and increased tau immunoreactivity as markers of axonal and oligodendrocyte pathology, respectively, because hematoxylin and eosin staining is relatively insensitive in detecting white matter damage in the rat brain 24 hours after ischemia. APP immunochemistry is a well-established method for detecting axonal damage in models of cerebral ischemia (Dietrich et al., 1998; Kalaria et al., 1993; Stephenson et al., 1992; Yam et al., 1998). APP undergoes fast axonal transport (Koo et al., 1990), and it is presumed to accumulate at points within axons where transport has ceased, probably as a consequence of cytoskeletal derangements (Dewar and Dawson, 1997; Yam et al., 1998). This process may precede physical disconnection of the axon and thus may be reversible. Reversibility of central nervous system axonal pathology, at least in vitro, is suggested by the restoration of compound action potentials recorded in isolated optic nerves exposed to anoxia and reoxygenation, the degree of recovery during reoxygenation being related to the length of the anoxic period. However, after 60 minutes of anoxia, optic nerve function in this model recovered to only 20% to 30% of preanoxic levels (for a review of in vitro data in this model, see Stys, 1998). The situation in vivo is more complex because in addition to duration, there may also be issues relating to local differences in the severity of ischemia within a given myelinated fiber tract.
Hematoxylin and eosin staining does not readily detect oligodendrocyte damage 24 hours after focal ischemia. However, we have previously reported that ischemic oligodendrocytes exhibit increased tau immunoreactivity (Dewar and Dawson, 1995). This response occurs within 40 minutes of ischemia (Irving et al., 1997) and may therefore precede irreversible damage in these cells. Tau is a cytoskeletal protein, and alterations in its immunoreactivity may indicate cytoskeletal breakdown, similar to the pathology observed in ischemic axons. Double-label immunohistochemistry to detect pathologic changes in axons and oligodendrocytes in the same section has the advantage of not simply examining pathologic events in two functionally related cellular elements with a high degree of spatial resolution, but also allows paired statistical analysis when interventions with differential effects on axon, oligodendrocyte, and neuron damage are being examined. The major disadvantage of the approach for assessing axonal and oligodendrocyte pathology is that it is substantially more demanding technically and more time-consuming than the commonly used methods for assessing anti-ischemic efficacy of pharmacologic agents such as triphenyltetrazolium chloride. The poor staining of nonischemic white matter (Bederson et al., 1986; Bose et al., 1984, 1985; Hatfield et al., 1991) with triphenyltetrazolium chloride highlights its limitations for assessing protection of white matter.
The total hemispheric measures of all three pathologic markers (APP-positive axons, tau-positive oligodendrocytes, eosinophilic and shrunken neuronal perikarya) were significantly lower after transient compared with permanent focal ischemia. However, the analysis of areas of pathology at the selected coronal planes revealed that the differences between experimental groups did not occur at the same anatomic locations. For axonal pathology, the greatest separation of the two experimental groups was observed at more caudal coronal planes. This contrasted strikingly with neuronal perikaryal and oligodendrocyte pathology, where the separation of the two experimental groups was not confined to these planes (Fig. 3). The lesser extent of oligodendrocyte pathology after transient MCAO compared with permanent ischemia predominantly reflected a difference in the extent of tau-positive oligodendrocytes in the cerebral cortex. Postischemic reperfusion is associated with free radical-mediated exacerbation of ischemic damage in gray matter (Aronowski et al., 1997; Matsumiya et al., 1991, Xue et al., 1992). The involvement of free radicals in ischemic pathology in white matter oligodendrocytes is evidenced by the ability of pretreatment with the spin-trap agent, phenyl-N-tert-butyl-nitrone, to attenuate increased tau immunostaining (Irving et al., 1997). Despite their vulnerability to free radicals, there was no evidence that oligodendrocyte pathology was enhanced (or reduced to a lesser degree) in the transiently occluded animals, presumably because the beneficial effect of flow restoration outweighs the potentially deleterious effects of free radical generation, at least at 2 hours of occlusion.
Previous in vivo approaches to the assessment of potential anti-ischemic therapies have focused almost exclusively on mechanisms in gray matter. We have suggested that one of the fundamental biologic mechanisms underlying the failure of N-methyl-D-aspartate receptor antagonists in improving clinical outcome in stroke or head injury is the inability of these agents to protect white matter (Dewar et al., 1999). Our hypothesis is supported by the failure of MK-801 to reduce either axon or oligodendrocyte ischemic pathology in white matter (Irving et al., 1997; Yam et al., 1999). Functional recovery after an ischemic insult will be improved by protection of both gray and white matter. The methodologic approach we present here will allow the brain-protecting potential of future therapies to be determined.
Footnotes
Acknowledgments
The authors thank Mrs. J. Stewart for excellent technical support; Mrs. L. Graham for secretarial assistance; Dr. D. Hanger, Institute of Psychiatry, London, and Dr. S. Tanaka, University of Hokkaido, Japan, for gifts of Tau-1 and MRF1 antibodies; and Ms. L. Marks for assistance with MRF1 staining.