
Abstract
Ischemia is implicated in periventricular white matter injury (PWMI), a lesion associated with cerebral palsy. PWMI features selective damage to early cells of the oligodendrocyte lineage, a phenomenon associated with glutamate receptor activation. We have investigated the distribution of glutamate in rat periventricular white matter at post-natal day 7. Immuno-electron microcopy was used to identify O4(+) oligodendroglia in control rats, and a similar approach was employed to stain glutamate in these cells before and after 90 mins of hypoxia-ischemia. This relatively brief period of hypoxia-ischemia produced mild cell injury, corresponding to the early stages of PWMI. Glutamate-like reactivity was higher in oligodendrocytes than in other cell types (2.13±0.25 counts/μm2), and declined significantly during hypoxia-ischemia (0.93±0.15 counts/μm2: P < 0.001). Astrocytes had lower glutamate levels (0.7±0.07 counts/μm2), and showed a relatively small decline during hypoxiaischemia. Axonal regions contained high levels of glutamate (1.84±0.20 counts/μm2), much of which was lost during hypoxia-ischemia (0.72±0.20 counts/μm2: P >0.001). These findings suggest that oligodendroglia and axons are the major source of extracellular glutamate in developing white matter during hypoxia-ischemia, and that astrocytes fail to accumulate the glutamate lost from these sources. We also examined glutamate levels in the choroid plexus. Control glutamate levels were high in both choroid epithelial (1.90±0.20 counts/μm2), and ependymal cells (2.20±0.28 counts/μm2), and hypoxia-ischemia produced a large fall in ependymal glutamate (0.97±0.08 counts/μm2: P >0.001). The ependymal cells were damaged by the insult and represent a further potential source of glutamate during ischemia.
Introduction
Periventricular white matter injury (PWMI) is the leading cause of cerebral palsy and chronic neurologic disability in survivors of prematurity (Back and Rivkees, 2004). Although the pathogenic events that trigger PWMI remain controversial, the late precursor cells and immature oligodendrocytes that predominate in developing periventricular white matter are particularly susceptible to ischemia. These cells suffer rapid necrotic death when exposed to ischemic conditions in cell culture (Fern and Moller, 2000), and in isolated optic nerve (Wilke et al, 2004), and are selectively lost after ischemia in vivo (Back et al, 2002a; Follett et al, 2000; Lin et al, 2004; Liu et al, 2002); (Ness et al, 2001). The sensitivity of late precursor cells (pre-oligodendrocytes) and immature oligodendrocytes to ischemic injury is associated with a limited capacity to resist oxidative stress (Back et al, 1998, 2005; Baud et al, 2004a; Bernardo et al, 2003; Haynes et al, 2003), and with the expression of Ca2+-permeable non-N-methyl-D-aspartate (NMDA)-type glutamate receptors on the somata (Fern and Moller, 2000; Follett et al, 2000; Wilke et al, 2004). Recent findings have revealed that the processes of immature oligodendrocytes also express NMDA-type glutamate receptors that mediate early disintegration of these structures under ischemic conditions (Káradóttir et al, 2005; Salter and Fern, 2005). The central role of ionotropic glutamate receptors in injury of these cells places the mechanism underlying ischemic glutamate release in developing white matter near the top of an important excitotoxic cascade.
Activation of ionotropic glutamate receptors on oligodendroglial cells during ischemia requires an elevation in extracellular glutamate that must be associated with net glutamate loss from at least one cellular compartment. The potential sources of cellular glutamate available for release during ischemia include astrocytes, oligodendrocytes, axons, and cells from neighboring structures such as the choroid plexus. Of these sources, ischemic glutamate release from astrocytes has been well characterized in gray matter where it may involve reverse uptake, swelling-mediated release, vesicular release, or release via hemi-channels (Anderson and Swanson, 2000; Bezzi et al, 2004; Ye et al, 2003). While there is evidence for significant release of astrocyte glutamate in isolated white matter during modeled ischemia (Wilke et al, 2004), it is not known how glutamate levels change in these cells during hypoxia-ischemia in vivo. Little is known about the role of oligodendroglial cells in glutamate homeostasis, but there is evidence from cell culture and whole-mount pre-parations for both release and uptake of glutamate during ischemic conditions (Fern and Moller, 2000; Wilke et al, 2004). The fate of axonal glutamate during ischemia is also unclear, although the significance of this pool has been emphasized in various reviews (Volpe, 2001) and there is recent evidence for some loss from axons during ischemia in isolated optic nerve (Wilke et al, 2004).
To define cellular glutamate depletion under pathophysiologic conditions in vivo, we have probed the relative levels of intracellular glutamate in various cell compartments in post-natal day 7 (P7) rat periventricular white matter, using immunoelectron microcopy (immuno-EM). By comparing changes before and after perinatal hypoxia-ischemia we have determined which compartments are glutamate depleted by these conditions. At P7, rat periventricular white matter is rich in oligodendrocytes (Craig et al, 2003) and sustains degenerative changes related to excitotoxic injury (Follett et al, 2000). Glutamate stores were detected in immature oligodendrocytes, axons, and astrocytes under control conditions and hypoxia-ischemia triggered a depletion of glutamate that was most pronounced in oligodendrocytes and axons. The particular susceptibility of the perinatal brain to excitotoxic injury during ischemia is therefore associated with a preferential loss of glutamate staining from oligodendroglia and axons with no increase in staining within astrocytes.
Materials and Methods
Animal Surgical Procedures
We employed a modification of the widely studied Rice–Vannucci model of perinatal hypoxia-ischemia where the common carotid artery is permanently ligated before exposure to hypoxic conditions (Vannucci et al, 1999). Since we found that ligation of the carotid artery can result in suboptimal fixation of the ischemic cerebral hemisphere, the left common carotid artery was reversibly ligated in Sprague-Dawley rat pups at P7 operated under isoflurane anesthesia. Post-natal day 0 was counted as the first day of life and a total of four animals were examined. After a 2 h recovery period from the carotid ligation, pups were placed for 1.5 or 2.5 h in containers (submerged in a 37°C water bath to maintain normothermia) through which flowed humidified 8% O2/92% N2. To restore flow in the carotid artery, reversible ligation was achieved by arterial occlusion with a silastic ligature that was released under deep anesthesia immediately before perfusion of the animal with EM fixative for glutamate localization (below). Restoration of flow was verified by visual inspection with a dissecting microscope and was consistently achieved up to 6 h after the ligature was released.
Immuno-Electron Microcopy for Glutamate Localization
At the conclusion of the exposure to hypoxia, animals were deeply anesthetized for transcardiac perfusion, 0.05 cc of heparin (10,000 U/ml; Elkin-Sinn Inc., Cherry Hill, NJ, USA, NDC 0641-2470-41) was injected into the left ventricle and was followed by 50 ml of freshly made 3% glutaraldehyde (Electron Microscopy Sciences, Hatfield, PA, USA, no. 16320, glutaraldehyde 50% solution, EM Grade) in 0.01 mol/L phosphate buffer (PB), pH 7.4. Brains were stored in phosphate-buffered saline (PBS) at 2°C to 4°C until processing: Brains were cut into single blocks running from the cortical surface to the ventricular surface (including the choroid plexus), ~ 2 to 3 mm lateral from the mid-line at the level of the infundibulum. The brain tissue was then post-fixed with 2% osmium tetroxide and dehydrated in ethanol and propylene oxide. Blocks were infiltrated in epoxy resin and ultra-thin sections running from the cortical surface to the ventricular surface were cut before 10 mins exposed to sodium metaperiodate, washing with PB, and blocking with PB + bovine serum albumin (BSA) and submerging in PB + TWEEN 20 (0.1%) + rabbit anti-glutamate polyclonal (Chemicon International, Temecula, CA, USA, 1:10) for 3 h. Sections were then washed in PBS/BSA before submersion in goat anti-rabbit 30 nm (1:100) gold conjugate for 1 h. After a further wash the sections were submerged in 0.5% gluteraldehyde in PB + TWEEN 20 for 5 mins then counterstained with uranyl acetate and lead citrate before carbon coating. Sections were examined with a JEOL 100CX electron microscope. Control sections where primary antibody was omitted from the normal protocols were performed, yielding no background staining. In further controls, the 1° ab was quenched with 300 μmol/L glutamate for 30 mins before proceeding with the normal protocol. This also produced no staining of sections.
Gold particle counts were performed by eye using images scanned into Photoshop (Adobe). The individual performing the counts (RF) was blinded to the nature of each image. Individual whole identified cells (which all contained a significant nucleus) and axonal regions were traced by hand to determine cross-section area using Image-J software. The particles counts were restricted to the cytoplasmic compartment including the nucleus and mitochondria. Gold particle counts were normalized to cell area and glutamate levels are reported as particle density (glutamate-like reactivity (GLR)) in units of particle count per square micron.
Immuno-Electron Microcopy for O4 Antibody Localization
For optimal preservation of O4 antibody immunoreactivity for EM localization, 0.05 cm3 of heparin (10,000U/ml; Elkin-Sinn Inc., NDC 0641-2470-41) was injected into the left ventricle at the time of transcardiac perfusion followed by 50 ml of freshly made 4% paraformaldehyde in 0.01 mol/L PB, pH 7.4, containing 1% glutaraldehyde (Electron Microscopy Sciences, no. 16320, glutaraldehyde 50% solution, EM Grade), 3% sucrose and 0.02% calcium chloride. The perfusate was delivered at a flow rate 15 ml/min with a Masterflex pump (no. 7520-00; Cole Parmer, Vernon Hills, IL, USA) with Easy-Load system (no. 7518-10). After removal of the brain it was stored in PBS at 2°C to 4°C. Brains were sectioned at 100 μm with a Leica, Nu Bloch, Germany, VTS 1000 vibrating microtome. To optimize penetration of the O4 antibody, tissue sections were permeabilized by the freeze-thaw method as previously described (Meshul and McGinty, 2000). Tissue was processed for O4 immunohistochemistry, as previously described (Back et al, 2002a; Lin et al, 2004; Liu et al, 2002), and the primary antibody visualized by immunoperoxidase staining with diaminobenzidine. The brain tissue was post-fixed with 2% osmium tetroxide and dehydrated in ethanol. Sections were flat embedded in epoxy resin and ultra-thin sections running from the cortical surface to the ventricular surface were cut. Sections were counterstained with uranyl acetate and lead citrate.
Note on Nomenclature
Cells of the oligodendrocyte lineage undergo a sequence of developmental stages that can be defined by morphologic and immuno-staining criteria. In this manuscript, the late precursor O4(+)/01(–) cells of the oligodendrocyte lineage are termed pre-oligodendrocytes while O4(+)/O1(+) cells that are committed to myelination are referred to as immature oligodendrocytes. Oligodendroglia refers to any cells of the oligodendrocyte lineage.
Results
To reliably stain for glutamate in cells exposed to hypoxia–ischemia, we established conditions that produced a mild cellular insult, corresponding to the early phases of PWMI. Since 2.5 h of hypoxiaischemia produces significant cerebral injury in the P7 rat after a 7-day survival (Vannucci et al, 1999), we, thus, analyzed the response to a relatively brief period of hypoxia–ischemia (1.5 h). Figure 1A confirmed that 1.5 h of hypoxia–ischemia did not produce overt cerebral injury, whereas 2.5 h of hypoxia–ischemia generated a spectrum of mild to severe cerebral injury (Figures 1B and 1C) similar to that we have previously reported (Back et al, 2002a). Staining for glial fibrillary acid protein (GFAP) confirmed that 1.5 h of hypoxia–ischemia did not stimulate a reactive gial response (Figure 1D). By contrast, 2.5 h of hypoxia–ischemia elicited a spectrum of reactive gliosis that coincided with the degree of overt cerebral injury (Figures 1E and 1F).
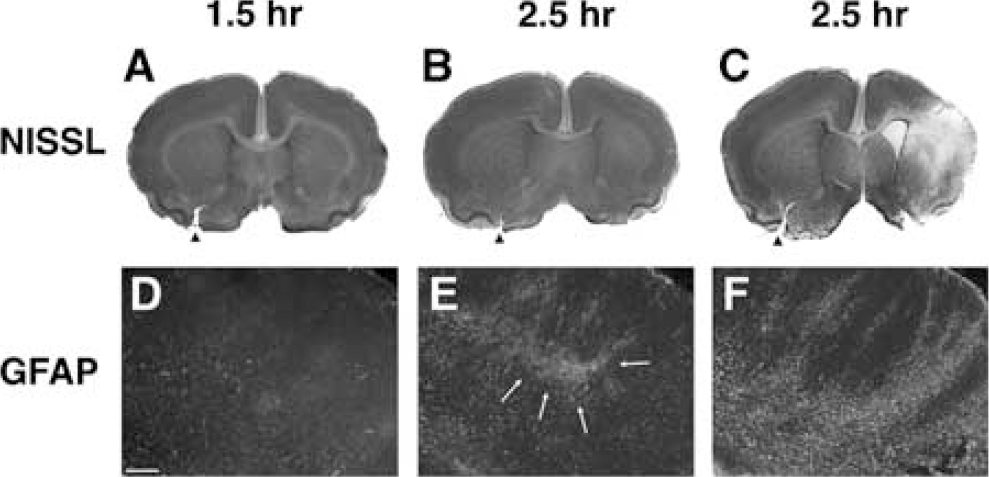
Relative cerebral injury in response to 1.5 or 2.5 h of hypoxia–ischemia after a 7-day survival period and 100% survival. Twenty animals from two separate litters (n = 10/L) received unilateral carotid ligation at P7 and were randomized to a 1.5 or 2.5 h period of exposure to 8% oxygen (see Materials and methods). Injury in three representative animals as assessed by Nissl stain (
Ultrastructural Features of O4(+) Cells of the Oligodendrocyte Lineage
To define perturbations in glutamate content in early stages of the oligodendrocyte lineage relative to other classes of cellular elements in ischemic periventricular white matter, we localized GLR in four classes of cellular elements in the cerebral white matter of the P7 rat: oligodendrocytes, astrocytes, choroid epithelia, and ependymal cells; in addition to axonal regions. Axon regions were chosen to largely avoid glial processes but contained a significant proportion of extracellular space that will make GLR values in these regions an underestimation of true axonal GLR. Since there has been limited analysis of the ultrastructural features of immuno-histochemically verified early oligodendrocyte cells (LeVine and Goldman, 1988), we first defined the ultrastructural features of these cells by immuno-EM with the O4 monoclonal antibody (see Materials and methods), which labels both preoligodendrocytes and immature oligodendrocytes. In P7 rat cerebral white matter, >90% of the cells labeled by the O4 antibody are immature oligodendrocytes and the remainder are pre-oligodendrocytes (Craig et al, 2003).
Consistent with the distribution of the O4 antibody by light microscopy, cell labeling localized to the plasma membrane of the somata (Figures 2A to 2C) and processes (Figures 2D and 2E). Typically, the nucleus was eccentrically positioned in the cell with a mass of cytoplasm at the opposing pole. The nucleus of the O4(+) cells varied in overall intensity, which was related to the distribution and extent of clumping of the electron-dense chromatin. Three general categories of nuclear intensity were distinguished that were similar to those previously defined for oligodendrocytes (Mori and Leblond, 1970). The overall intensity of one population of nuclei was light in appearance and contained very evenly dispersed chromatin (Figure 2A). The chromatin of a second population of medium-intensity nuclei displayed greater marginalization at the nuclear membrane and more clumping of the chromatin (Figure 2B). The third population of dark-intensity nuclei contained distinct clumps of chromatin at the margin and interior of the nucleus. A survey of 24 O4(+) cells indicated that the majority was medium-intensity cells (54%), whereas light (29%) and dark (17%) oligodendroglia were the minority.
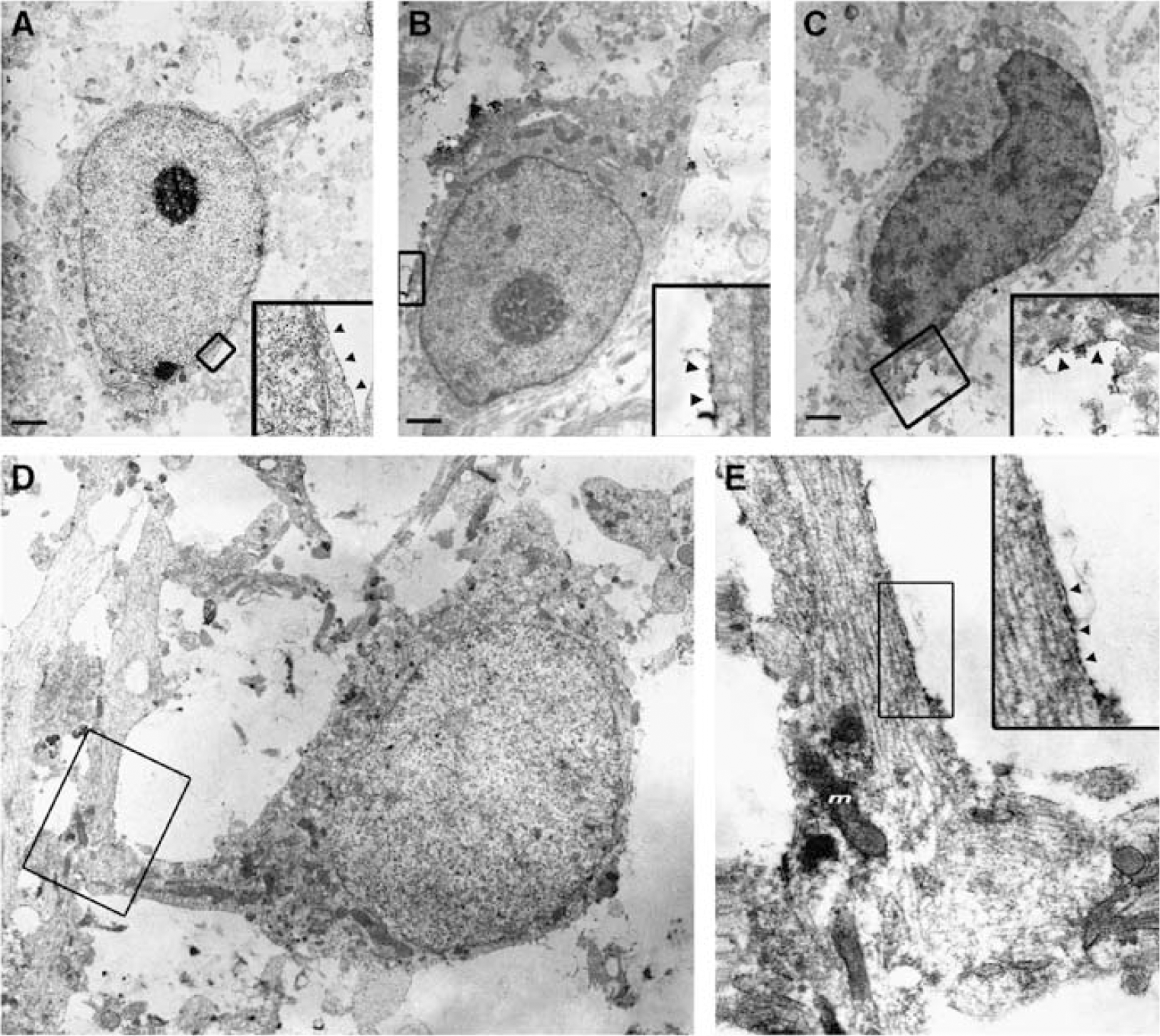
Ultrastructural features of O4(+) oligodendroglial cells. Three distinct types of O4-labeled cells, light (
The cytoplasm of all three classes of O4(+) cells contained many mitochondria, the endoplasmic reticulum was studded with ribosomes and microtubules were common, but fibrils and glycogen particles were not detected. O4 labeling was also localized along segments of adjacent axons (Figures 2D and 2E). Consistent with the early ensheathment of axons, the axon labeling localized to a single outer membrane that was closely apposed to the axonal membrane. In some instances, O4-labeled processes were visualized in contact with axons. No distinct multi-lamellar or compact myelin sheaths were seen.
Localization of Glutamate-Like Reactivity to Early Cells of the Oligodendrocyte Lineage
We found that tissue preserved for ultrastructural localization of GLR was not compatible with the colocalization of O4-antibody label. Hence, early cells of the oligodendrocyte lineage were identified in tissue processed for GLR by criteria defined for O4-labeled cells. We identified a population of early cells of the oligodendrocyte lineage that consistently showed intense cytoplasmic GLR staining (2.13±0.25 counts/mm2; Figure 3). Staining was completely absent from sections when the primary antibody was omitted from the staining protocol (Figures 3A and 3B), and when the glutamate antibody was pre-incubated with 100 μmol/L glutamate (see also Wilke et al, 2004). Both light, medium (Figure 3E), and dark (Figures 3A to 3F) oligodendroglial cells were found in the periventricular white matter, with a larger proportion of dark cells observed in these sections than was found in those described above. The similarities between the oligodendroglial cells and the O4(+) cells included numerous mitochondria, a narrow bore endoplasmic reticulum that was usually studded with ribosomes and the characteristic nature of the chromatin distribution in the medium and dark cells (compare Figures 2 and 3). Glial filaments, glycogen particles, and dark bodies were absent from the oligodendroglial cells and the morphologic differences between a dark oligodendroglial cell and an astrocyte can be seen in Figure 3F. In these GLR studies, very little reactivity was observed in the extracellular space and gold particles were only occasionally seen on the outer aspect of cell membranes, indicating that extracellular glutamate levels did not approach those found within cell.
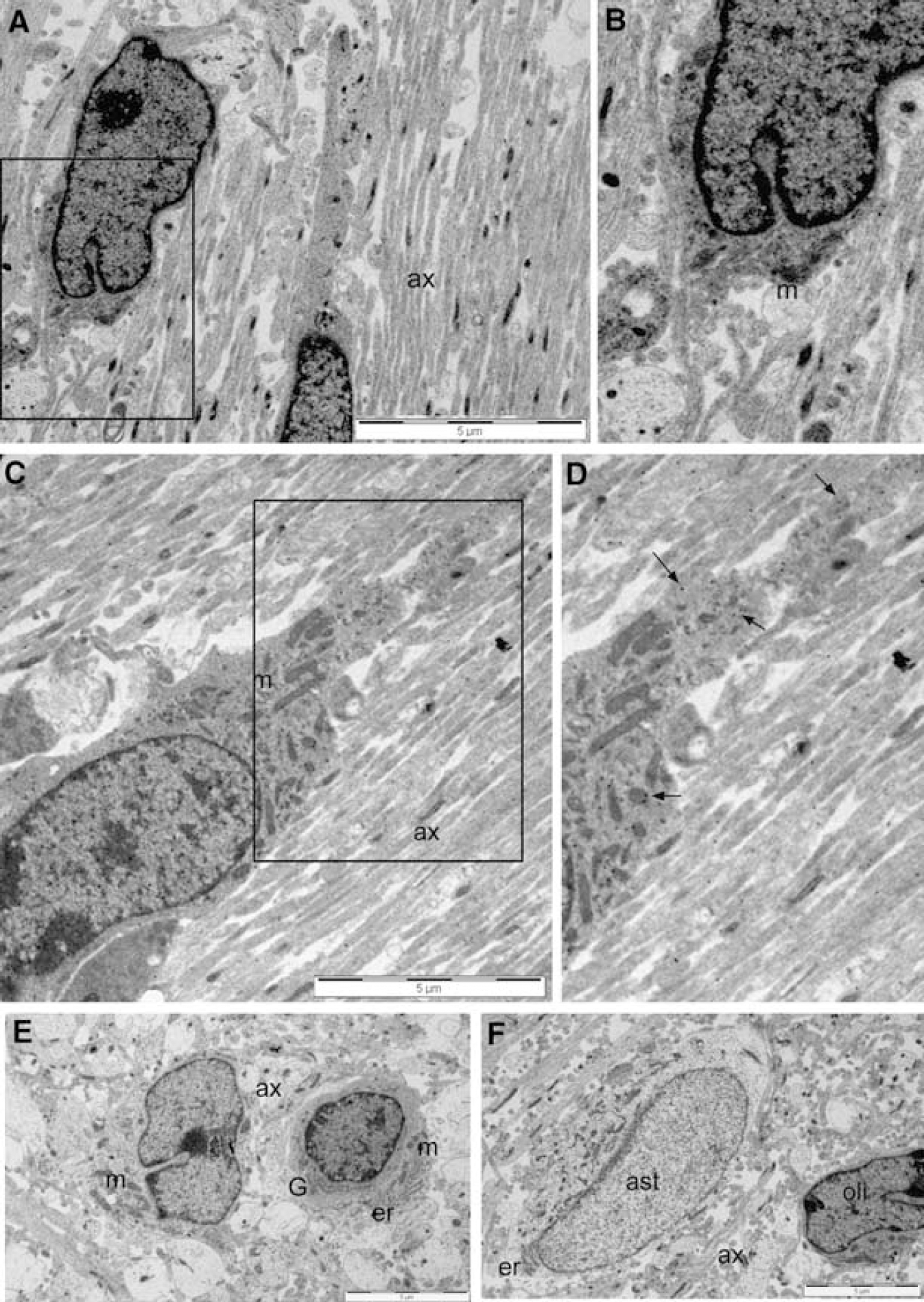
Oligodendroglial cells in control periventricular white matter. (
Immature oligodendroglial cells in post-ischemic periventricular white matter could be reliably identified on the basis of the morphologic features seen in the healthy cells, and tended to have low levels of GLR (0.93 + 0.15; P < 0.001 versus control; Figure 4). Dark oligodendroglial cells retained their typical clustered chromatin, dark cytoplasm, and numerous mitochondria, while the narrow-bore endoplasmic reticulum was often absent (Figure 4A). Occasionally, the processes of larger dark cells could be traced out to contact points with large axons (Figure 4A). The dark oligodendroglial cells contained numerous vacuoles (v) that presumably represent to some extent endoplasmic reticulum that has swollen beyond recognition. In cells that had formed immature processes, these structures were particularly affected by vacuoles and in many cases appeared to be disintegrating (Figure 4A). Intermediate (Figures 4B, 4C, and 4E) and light (Figure 4D) oligodendroglial cells showed a similar range of morphologic changes to those seen in the dark cells. Necrotic changes and loss of cell membrane integrity was not seen in any of the classes of oligodendroglial cell, including those with immature morphologic features. The mitochondria within the oligodendroglial cells in general appeared normal, although damaged mitochondria were occasionally observed (Figure 4C, arrow head). The extent of mitochondrial damage was significantly less than that seen in astrocytes (Figures 4 and 6).
Immature oligodendroglial cells in the periventricular white matter after 90 mins of hypoxia–ischemia. (
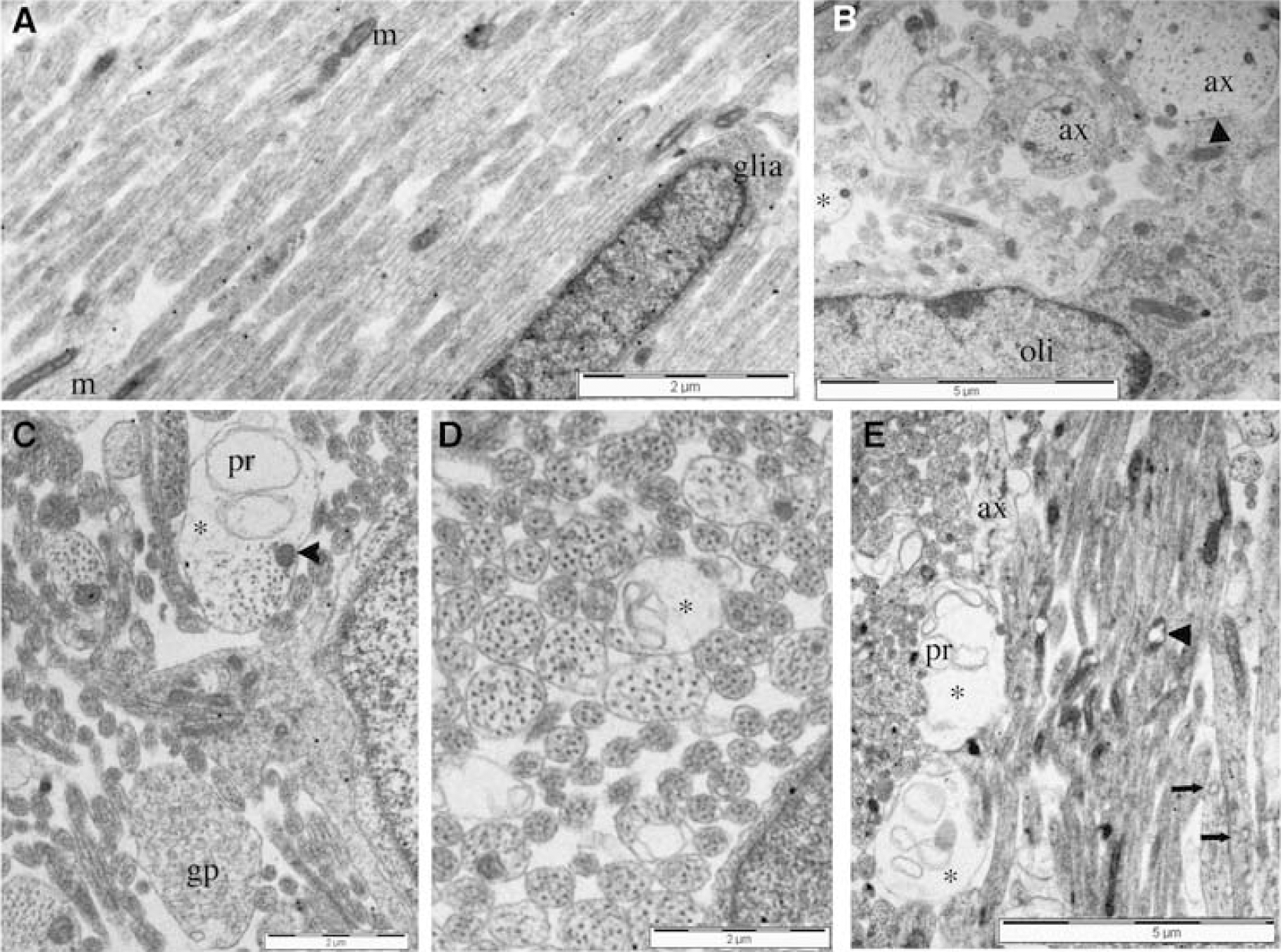
GLR in axonal regions of periventricular white matter. (
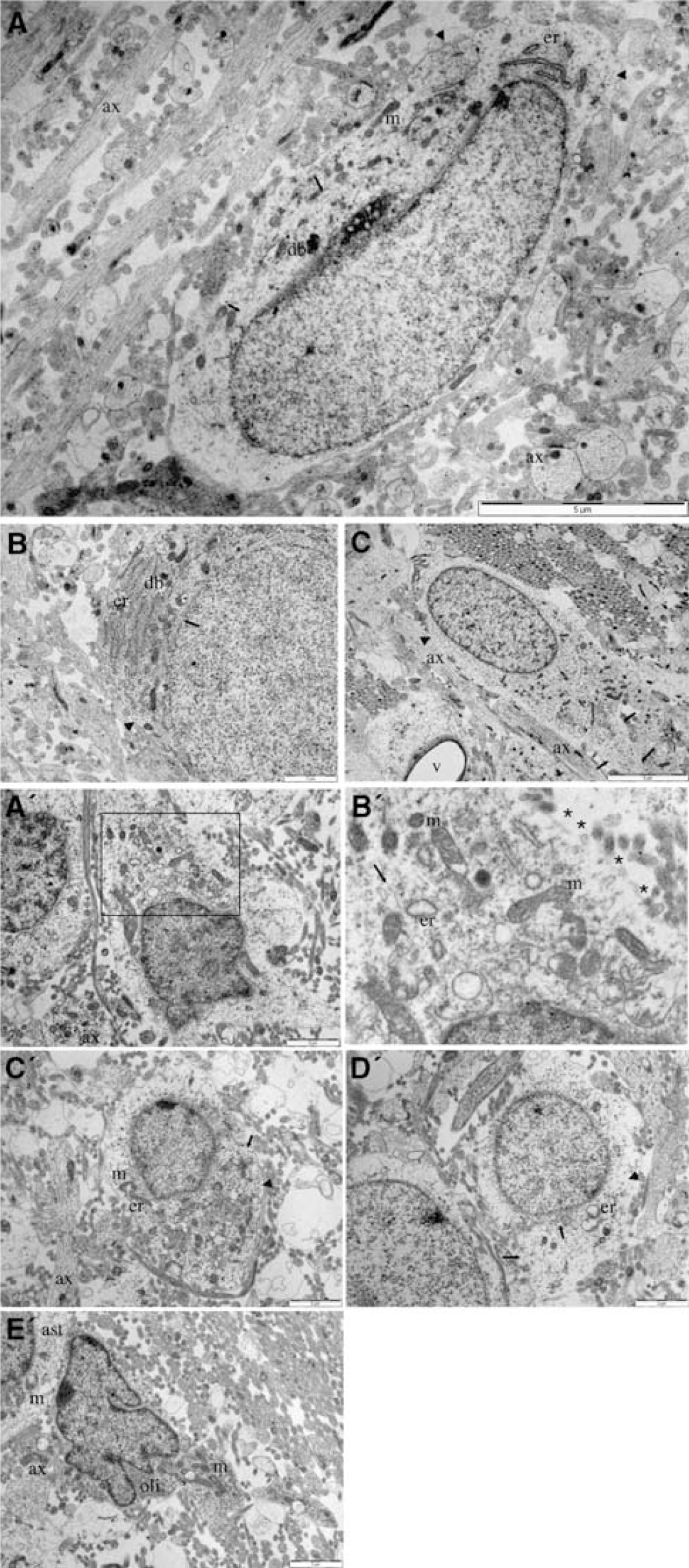
Astrocytes in control and post 90 mins hypoxiaischemia periventricular white matter. (
Probably the most striking feature of the postischemic oligodendroglial cells was the presence of large membrane-delineated profiles within the regions surrounding these cells (‘p’ in Figures 4A to 4E). These profiles were always approximately round, varied in diameter from ~ 1 to 5 μm and were either empty of electron-dense material or were filled with a grainy debris and sometimes contained smaller membrane-delineated profiles. In several instances in Figure 4 it appears that structures with the features of large axons are degenerating into forms similar to these profiles and more details of this feature are given when axonal changes are addressed below.
Localization of Glutamate-Like Reactivity to Axons
Axons within the white matter layer adjacent to the subventricular zone were generally small in diameter and were closely packed and aligned together (Figure 5A). In this region, glial cells were scarce and often appeared immature, while glial processes were virtually absent. Axons in the white matter adjacent to the deep cortex were more randomly aligned with larger axons (2 to 3 μm in diameter) interspersed among smaller axons (Figure 5B). In this region, glial cells were more developed and oligodendroglial cells extended processes to contact axons, but ensheathment and myelination were not apparent. Long, dark mitochondria were common in both large and small axons, which had characteristic microtubules and neurofilaments. In both of the regions described above, marked GLR was found within the axoplasm of large and small axons and mean GLR in axonal regions was 1.84±0.20 counts/μm2. Axoplasmic GLR in post-ischemic white matter was greatly reduced in both the shallow and deep white matter regions (Figures 5C to 5E), and GLR in axonal regions fell to 0.72±0.20 counts/μm2 (P < 0.001).
While apparent axons surrounding oligodendrocytes had frequently degenerated into swollen and empty membrane profiles (Figure 4), axons that were not in close proximity to oligodendrocytes were largely intact after hypoxia–ischemia. In these regions some axon damage was apparent in the form of occasional swollen axons containing lacunae that were membrane bound with one or more membrane profiles present, in particular within larger axons (Figures 5C to 5D). In regions where axons run in close apposition and that did not contain glial somata, the proportion of axons with evident pathologic changes such as loss of microtubules, axolemma distortion, or mitochondrial swelling increased from 1.9%±0.08% (n =19) in control white matter to 7.7%± 1.9% (n = 10) in Hypoxia–Ischemi white matter (P < 0.001). These changes did not generally include frank loss of axolemma integrity, even in the apparently grossly swollen axons neighboring oligodendroglia (Figures 4 and 5). Potential sources of the membrane profiles within axonal lacunae include mitochondria. Indeed, axons caught in longitudinal section sometimes had vacuolated mitochondria (Figure 5E), although normal mitochondrial profiles were also seen within evolving lacunae (Figure 5C, arrowhead). Alternatively these profiles might be derived from endoplasmic reticulum, since small vacuoles of apparent distended endoplasmic reticulum were sometimes seen (Figure 5E, arrows). Despite the presence of these axon vacuoles, the majority of axons retained a normal appearance with the usual compliment of microtubules.
Localization of Glutamate-Like Reactivity to Astroglia
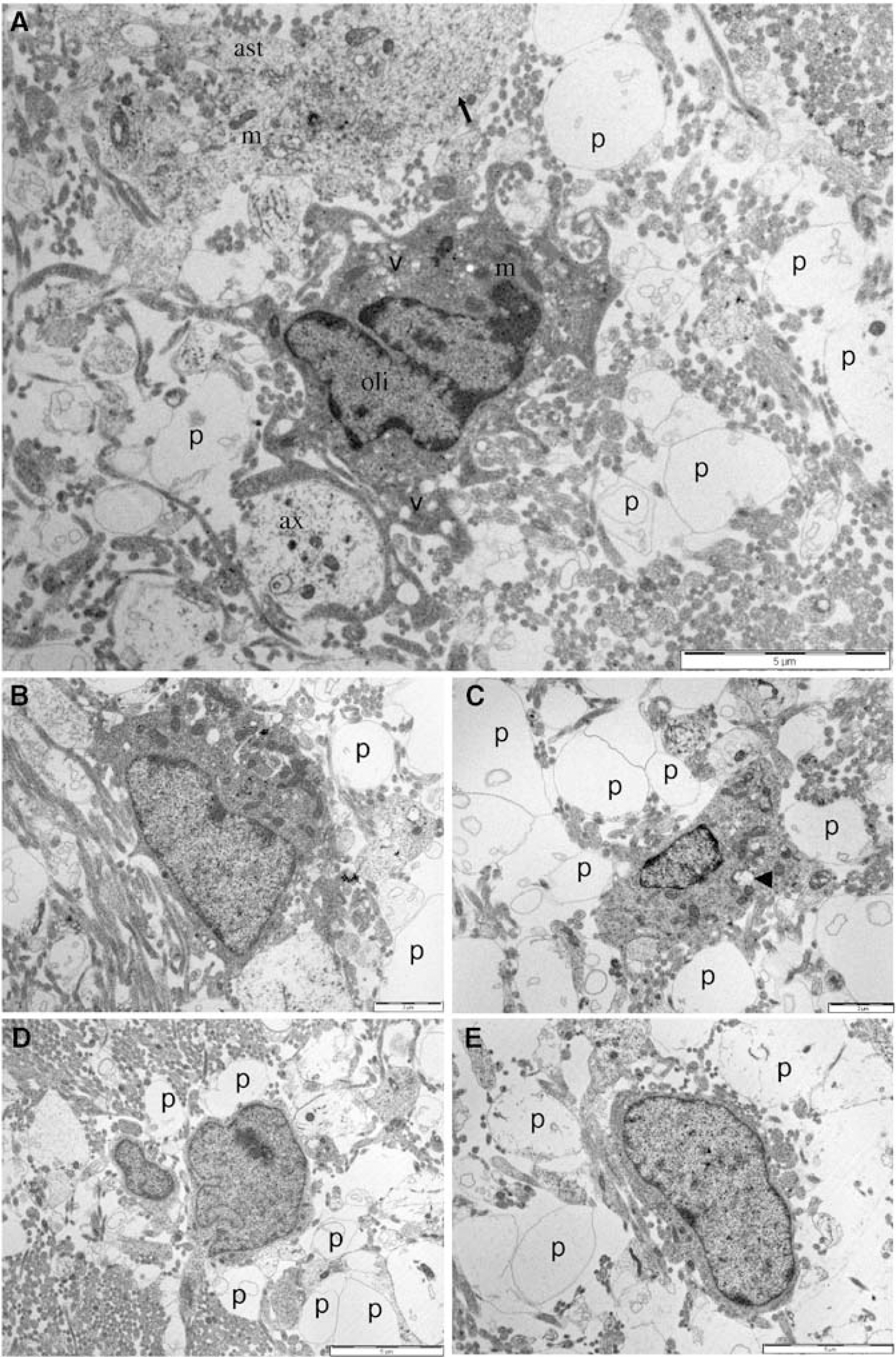
Astrocytes could be clearly identified in the deeper periventricular white matter (Figure 6). Features of these cells included an electron-lucent cytoplasm containing glial filaments and many small particles previously described as glycogen beta particles, dark bodies in the cytoplasm, a characteristic wide-bore endoplasmic reticulum that was sometimes studded with ribosomes, and a large nucleus of dispersed hetero-chromatin with no nucleolus (Mori and Leblond, 1969; Vaughn, 1969). Astrocytes generally had an immature morphology without processes but occasionally a single large process was observed (Figure 6C). As can be seen in Figure 6, GLR levels in astrocytes in non-ischemic white matter were generally lower than in oligodendroglial cells or axons (0.7±0.07 counts/mm2).
In periventricular white matter subject to hypoxia–ischemia, astrocytes retained an electron-lucent cytoplasm and the presence of glial filaments, allowing the cells to be reliably identified (Figure 6). Wide-bore endoplasmic reticulum was also common, although the cisternae were often severely distended (Figure 6B‘), as previously reported for astrocytes in post-ischemic immature white matter (Thomas et al, 2004). Glycogen particles were either greatly depleted in the cytoplasm (Figure 6C' and 6D') or absent (Figure 6A' and 6B'). Mitochondria were swollen and showed disruption of the internal architecture. Some astrocytes that had lost cell membrane integrity were present (Figure 6A' and 6B'), and in general astrocytes showed more ischemic damage than was apparent in oligodendroglial cells (Figure 4). As can be seen in Figure 6A' to 6D', the GLR level in the post-ischemic astrocytes was low (mean 0.57±0.15 counts/mm2).
Localization of Glutamate-Like Reactivity to Ependymal and Choroid Epithelial Cells
Glutamate-like reactivity labeling was present in both choroid epithelial and ependymal cells (Figures 7A and 7B). In nonischemic brain, the choroid epithelial cells extended a dense layer of microvilli, which intermingled with cilia and sparse microvilli projecting from ependymal cells (see (Gabrion et al, 1998). The ependymal cells formed a single cell layer covering the small diameter !QJ;axons that course in parallel next to the ventricular surface, or they formed a multi-cell layer (not shown). Both the ependymal and choroid epithelial cells contained ER and large numbers of mitochondria, and both were highly invaginated, particularly at intercellular junctions. Choroid epithelial cells were frequently observed to contain areas of electron-lucent material, which appeared to be a physiologic characteristic since nearby organelles were normal.
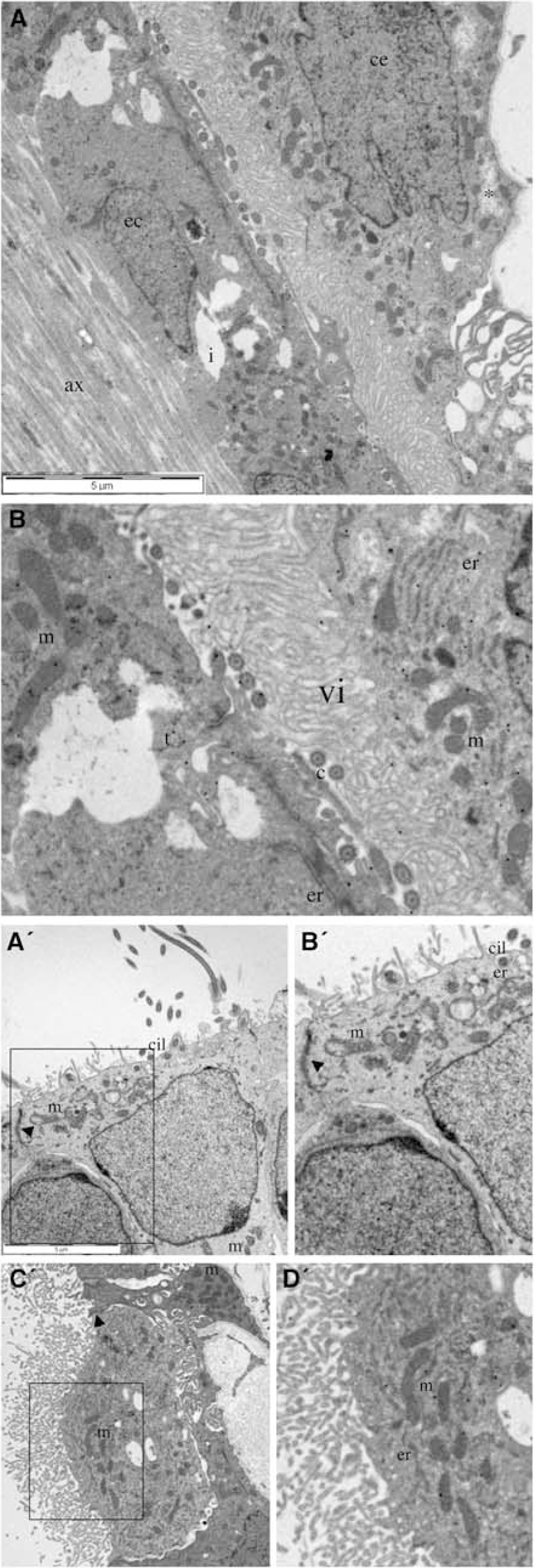
Choroid epithelial cells (ce) and ependymal cells (ec) in control brain and after 90 mins of hypoxia–ischemia. In this region the choroid plexus runs closely apposed to the ependymal cell lining of the subcortical ventricular surface, allowing both cell types to be seen. (
Ependymal cells in ischemic brain contained grossly swollen mitochondria and ER that had ballooned into vacuoles (Figure 7A' and 7B'). Despite these pathologic changes, the cell membrane was never disrupted and cell degeneration was not seen. Microvilli and cilia appeared normal and the tight junctions between cells were maintained. The organelles of choroid epithelial cells appeared normal in all cases, tight junctions were maintained, microvilli had a normal appearance and degenerating cells were not seen (Figure 7C' and 7D'). Control GLR levels were high in both choroid epithelial (1.90±0.20 counts/mm2), and ependymal cells (2.20±0.28 counts/μm2), and hypoxia–ischemia produced a significant fall in ependymal glutamate (0.97±0.08 counts/μm2: P>0.001).
Cell-Type Dependent Changes in Glutamate-Like Reactivity Triggered by Hypoxia–Hschemia
A quantitative analysis of the relative density of GLR labeling in axonal regions, astrocytes, immature oligodendroglia, and the cells of the choroid plexus are shown in Figure 8. As noted above, control labeling showed a heterogeneous distribution of GLR in the immature brain. Astrocytes had the lowest density of GLR in the white matter, whereas immature oligodendroglia and axons had the highest (Figure 8A). Choroid epithelium and ependymal cells also had a high density of GLR. The degree of staining in axon regions was likely to underestimate the density of axoplasmic GLR, since these areas included a significant proportion of extracellular space (Figure 5). The pattern of GLR was altered in ischemic white matter (Figures 8A and 8B). Ependymal cells, axon regions, and immature oligodendroglial cells all lost a similar percentage of their normal GLR (~60%). Choroid epithelial cells did not show a significant loss of GLR, correlating with their apparently normal ultrastructure. Astrocytes also showed a degree of GLR loss.
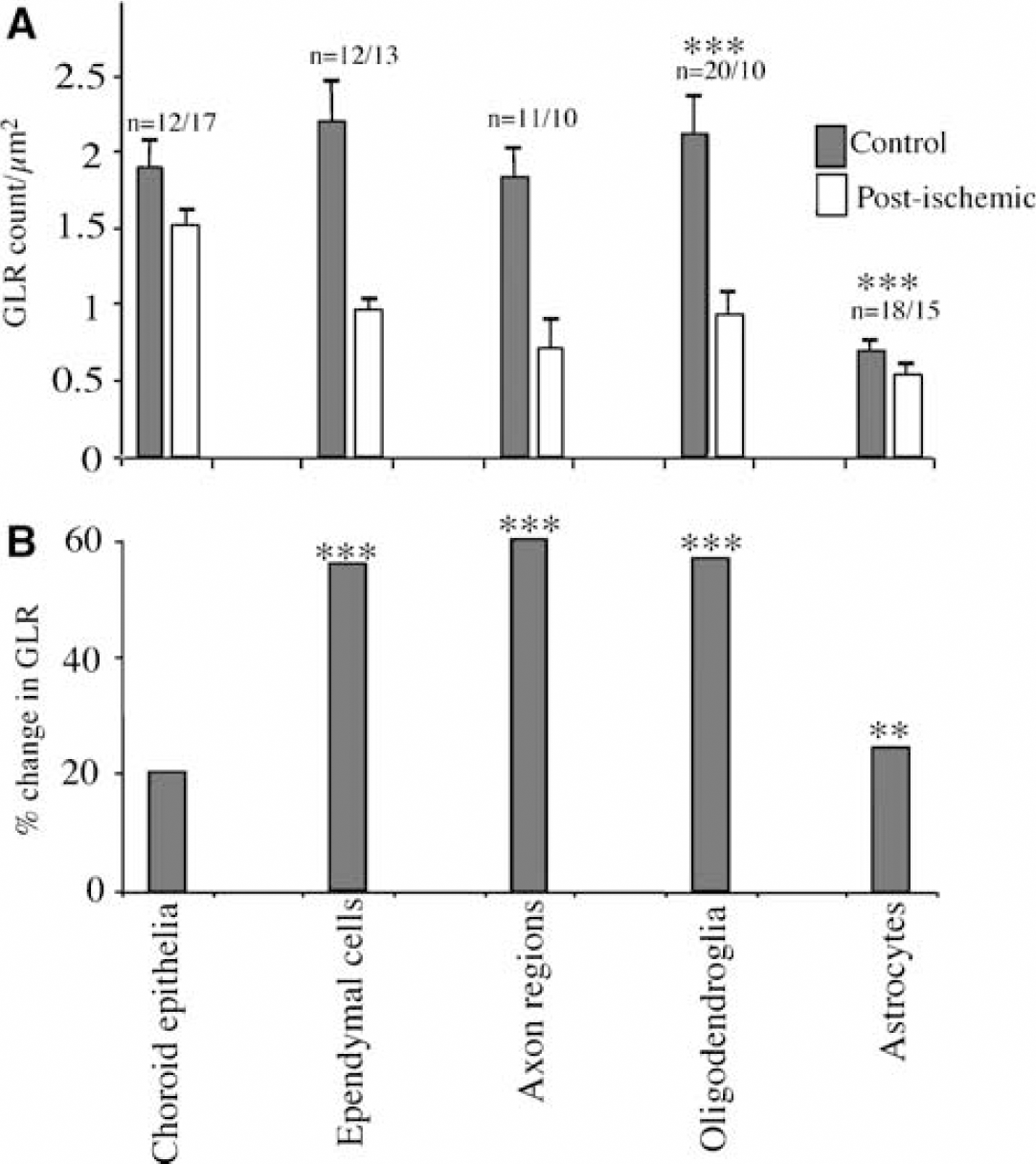
Glutamate-like reactivity (GLR) in immature white matter. (
Discussion
The pathogenesis of human PWMI remains a controversial but important topic, since it is the leading cause of chronic neurologic morbidity in survivors of premature birth (Back and Rivkees, 2004). Early cells of the human oligodendroglial lineage are a primary target of injury in PWMI (Back et al, 2005; Haynes et al, 2003). Major features of the early degeneration of these cells have been replicated in perinatal rodent models of acute hypoxia–ischemia (Back et al, 2002a; Follett et al, 2000; Lin et al, 2004; Liu et al, 2002; Ness et al, 2004; Ness and Wood, 2002). The evidence linking glutamate-mediated excitotoxicity to degeneration of early cells of the oligodendrocyte lineage is strong, and we have now defined the potential cellular pools of glutamate that are available for release in perinatal rat periventricular white matter, and have assayed how these sources of glutamate are affected by a relatively brief period of hypoxia–ischemia.
Glutamate was depleted from oligodendroglial cells, astrocytes and axons in periventricular white matter at the termination of a 90 mins period of hypoxia–ischemia. This relatively short insult was sufficient to produce some glial and axonal pathology, but did not result in the widespread degeneration of early cells of the oligodendrocyte lineage that is observed after longer periods of hypoxia–ischemia and in human PWMI. The glutamate changes we observed in this study therefore reflect relatively early events that are likely to have significance for cellular injury. Mechanisms of glutamate release from axons (Kriegler and Chiu, 1993), and astrocytes (Anderson and Swanson, 2000; Bezzi et al, 2004; Ye et al, 2003), include reversal of Na-dependent uptake in both cases. Oligodendroglia express Na-dependent glutamate transporters and a Xc− transporter which are both potential sources of glutamate release during hypoxia–ischemia (Deng et al, 2003; Domercq et al, 1999; Fern and Moller, 2000; Oka et al, 1993). Unexpectedly, a significant depletion of glutamate occurred in the immature oligodendroglial cells, suggesting that PWMI may be related to an enhanced early release of glutamate in these particularly oligodendroglial–rich regions.
We have recently shown that 40 mins of oxygen–glucose deprivation in the isolated P10 rat optic nerve is associated with an accumulation rather than a depletion of glutamate in oligodendroglial cells (Wilke et al, 2004), the diametrical opposite to that found in P7 cortical white matter subject to 90 mins of hypoxia–ischemia in the current study. The oligodendroglial population of P7 periventricular white matter is less mature than that of the P10 optic nerve, which initiates myelination at ~P6 (Hildebrand and Waxman, 1984). Indeed, the cells studied using immuno-EM by Wilke et al (2004) were identified as oligodendrocytes partly on the basis of having compact myelin profiles within their processes, a phenomenon not seen in the P7 cortical oligodendrocytes studied here. Since different developmental stages of the oligodendroglial lineage exhibit rather different sensitivities to ischemic-type injury (Fern and Moller, 2000; Follett et al, 2000), this developmental factor may explain the difference in glutamate handling in the two populations of oligodendroglial cells. This would be consistent with the findings of Fern and Moller (2000) who found evidence for significant glutamate release during 40 mins of oxygen–glucose deprivation in cultured O4(+) oligodendroglial cells, which correspond developmentally to those examined in the current study. This indicates that developmental factors that affect glutamate handling in cells of the oligodendrocyte lineage may be of key importance to the pathogenesis of PWMI.
Ultrastructural Identification of Perinatal Oligodendrocytes
Although the ultrastructural features of mature myelinating oligodendrocytes have been extensively studied, definition of the features of earlier stages in the oligodendrocyte lineage has been limited because of the technical limitations of preserving perinatal brain for immuno-EM studies. To optimize the identification of O4(+) cells, we focused on white matter development at P7, when rat pups are sufficiently large to optimize preservation of cerebral ultrastructure. At P7, the white matter is populated mostly by immature oligodendrocytes that label with both the O4 and O1 antibodies (Craig et al, 2003). The enzyme CNPase is also specifically expressed on immature oligodendrocytes, and immuno-EM localization of rat CNPase at P6 identified a population of oligodendrocytes that was similar to that defined with the O4 antibody (Braun et al, 1988). Taken together, these findings indicate that immature oligodendrocytes can be reliably distinguished from other glia in premyelinating white matter.
We distinguished three types of O4(+) cells in P7 rat corpus callosum based on the density and distribution of nuclear chromatin. Our findings are consistent with those from adolescent rats (60 to 80 g) in which three types of oligodendrocytes, light, medium, and dark, were defined in corpus callosum according to their nuclear density (Mori and Leblond, 1970). The oligodendrocyte lineage stages corresponding to these three types of oligodendrocytes were not defined, but may be retrospectively predicted on the basis of their tritiated thymidine studies. Light oligodendrocytes were markedly more proliferative than medium oligodendrocytes and dark oligodendrocytes were post-mitotic. Since medium density cells were the most common in our preparation, these are likely to be a mixed population of pre-myelinating and early myelinating immature oligodendrocytes that predominate in the corpus callosum at P7 (Craig et al, 2003). Interestingly, we also infrequently detected light oligodendrocytes, consistent with the notion that they are mitotically active pre-oligodendrocytes, which are present at low density at P7 (Craig et al, 2003). To confirm that the light oligodendrocytes are preoligodendrocytes, future immuno-EM studies are needed to determine whether they immuno-label with the O4, but not the O1 antibody.
Differential Susceptibility of Perinatal Neural Cells to Hypoxia–Ischemia
Perinatal rat astrocytes are susceptible to even brief exposure to oxygen–glucose deprivation, as previously observed (Fern, 1998; Thomas et al, 2004). Interestingly, degenerating human astrocytes were not commonly seen in association with degenerating oligodendroglial cells in preterm human white matter lesions (Back et al, 2005). This suggests either that astroglial degeneration is less pronounced in human white matter, or that it is an early event that does not persist in later stages of ischemic injury. Indeed, the delayed reactive astrocytosis that follows CNS injury is likely to hide any early astrocyte injury (Fern, 2001), and early damage of astrocytes was found in the current study. In contrast to astrocytes, early cells of the oligodendroglial lineage typically displayed limited changes that included cytoplasmic vacuolization apparently of endoplasmic reticulum origin and occasionally mitochondrial swelling.
Axonal injury was focused in the regions adjacent to immature oligodendroglial cells, zones that will presumably accumulate the glutamate released from the oligodendroglial cell during hypoxia–ischemia. The mechanisms of axonal injury in immature white matter are not understood, but a role for glutamate receptor activation in injury has been shown in mature central axons (e.g., Tekkok and Goldberg, 2001). We have recently demonstrated that the processes of immature oligodendrocytes are damaged during ischemic conditions after activation of NMDA-type glutamate receptors that are clustered along processes (Salter and Fern, 2005). The P7 cortical white matter oligodendroglia examined in the current study are at a less mature stage than the P10 mouse optic nerve cells studied by Salter and Fern (2005), and in general did not have extensive processes. However, vacuolization and partial degeneration of processes was observed, and may be associated with the release of factors such as reactive oxygen species. Such damaging elements may also be released by the oligodendroglial somata, and a mechanism of this type has been suggested to mediate axonal injury in central white matter in the adult (Tekkok and Goldberg, 2001).
Axon damage was most commonly seen in the larger diameter axons. Axons undergo a rapid increase in diameter just before the onset of myelination (Hildebrand and Waxman, 1984), and these larger axons will be more advanced in forming a relationship with the myelinating processes of oligodendrocytes. Indeed in the P7 rat we have identified apparent pre-myelin sheaths that appear before the immunohistochemical detection of myelin basic protein (Craig et al, 2003). The immature nature of these glial–axonal units is shown by the presence of O4-labeled partial segments of single membranous sheaths that were closely apposed to the axonal outer membrane. We have previously identified a developmental window in early human myelinogenesis when the periventricular white matter also contains mostly immature oligodendrocytes and pre-myelin sheaths that preceded the generation of mature myelin (Back et al, 2002b).
Clinico-Pathologic Correlations
Emerging evidence indicates that the timing of glutamate receptor expression during human and rat white matter development coincides with the window of cerebral white matter vulnerability. Until recently, evidence suggested that non-NMDA iono-tropic glutamate receptors were central to ischemic injury of cells of the oligodendrocyte lineage. Increased expression of the GluR4 subunit on O4(+) cells occurs between 23 and 32 weeks gestation in human parietal white matter and P7 rat corpus callosum (Follett et al, 2004). GluR4 is expressed on pre-oligodendrocytes and immature oligodendrocytes, and O4(+) cells also express calcium permeable AMPA receptors (Fern and Moller, 2000; Follett et al, 2004; Ong et al, 1996; Sanchez-Gomez and Matute, 1999). Hence, functional glutamate receptors localize to oligodendrocyte lineage cells during a developmental window that coincides with the propensity for glutamate release from immature oligodendrocytes during acute hypoxia–ischemia. Recent studies have shown the focal expression of NMDA receptors on immature oligodendrocyte processes. These receptors mediate the rapid, Ca2+ dependent, disintegration of the oligodendrocyte processes under ischemic conditions (Salter and Fern, 2005; Karadottir et al, 2005). Taken together, these findings suggest a mechanism whereby glutamate release from cells of the oligodendrocyte lineage and axons in response to hypoxia–ischemia can activate receptors on adjacent oligodendrocytes, leading to injury. The observation that astrocytes show only a small loss of glutamate during hypoxia–ischemia may indicate that these cells are neither a significant source nor sink for glutamate in developing white matter. Caution is required however when correlating changes in cell glutamate levels with changes in glutamate uptake, in particular in astrocytes which are believed to retain to some degree an ability to convert glutamate to glutamine during ischemic conditions (Pascual et al, 1998).
There is currently little information regarding the potential mechanisms of glutamate re-uptake after release into ischemic perinatal cerebral white matter. The X−c transporter exchanges glutamate for cystine across the plasma membrane (Sato et al, 1999, 2002), and in the presence of high extracellular glutamate this antiporter mediates the intracellular depletion of glutathione in both preoligodendrocytes and mature oligodendrocytes in vitro (Back et al, 1998; Oka et al, 1993). The relative resistance of mature oligodendrocytes to this form of oxidative stress and to hypoxia–ischemia, in general, suggests that other mechanisms may compensate for the downstream sequelae of extracellular glutamate release. These include the maturation of anti-oxidant enzyme systems (Baud et al, 2004a,b; Folkerth et al, 2004) and potentially glutamate reuptake mechanisms. The pronounced depletion of glutamate that we observed in oligodendroglial cells and astrocytes suggests that re-uptake mechanisms may be immature in the perinatal brain or dysfunctional during perinatal hypoxia–ischemia. Indeed, glutamate transport inhibition in the adult optic nerve in vivo resulted in excitotoxic degeneration of both oligodendrocytes and axons mediated by AMPA and kainate receptor overactivation (Domercq et al, 2005).
The timing of hypoxia–ischemia during development appears to determine the extent of white matter damage and the cell types that degenerate. The cerebral white matter of normal term infants is generally more resistant to injury than that of preterm infants (Volpe, 2001). As the perinatal rat periventricular white matter matures during the first post-natal week, it more closely resembles the term infant and oligodendrocyte lineage cells display increasing resistance to hypoxia–ischemia (Back et al, 2002a; Craig et al, 2003). Our data suggest that the extent of white matter damage in the term cerebrum may be influenced by cellular-maturational factors that reflect increasing resistance of oligodendroglia but increasing vulnerability of axons.
Footnotes
Acknowledgements
We are grateful to Dr Steven Levison for generous advice regarding the optimization of conditions for electron microscopy studies in perinatal rodents.