
Abstract
Disturbances in the nitric oxide (NO) vasodilatory pathway have been implicated in acute vasoconstriction and ischemia after subarachnoid hemorrhage (SAH). The authors hypothesize that blood released during SAH leads to vasoconstriction by scavenging NO and limiting its availability. This was tested by measuring the major NO metabolites nitrite and nitrate in five different brain regions before and after experimental SAH. The basal NO metabolites levels were as follows (mean ± SD, μmol/mg wet weight): brain stem, 0.14 ± 0.07; cerebellum, 0.12 ± 0.08; ventral convexity cortex, 0.22 ± 0.15; dorsal convexity cortex, 0.16 ± 0.11; and hippocampus, 0.26 ± 0.17. In sham-operated animals, no effect of the nitric oxide synthase (NOS) inhibitor
Keywords
Cerebral blood flow decreases acutely and ischemic injury occurs after subarachnoid hemorrhage (SAH) in both experimental and clinical studies. Cerebral arteries have been shown to respond to SAH with a biphasic constriction. An acute constriction begins minutes after the bleed, and delayed vasospasm begins 48 hours later (Delgado et al., 1985). Although the significance of delayed vasospasm is recognized (Sobey and Faraci, 1998) the contribution of acute vasoconstriction to ischemic brain damage after SAH is less clear (Bederson et al., 1998; Schwartz et al., 1999; Sehba et al., 1999). Different mechanisms may underlie the two processes, and numerous mechanisms have been proposed (Delgado et al., 1988; Edwards et al., 1992; Svendgaard et al., 1987; Svendgaard et al., 1985; Wellum et al., 1985; Willette et al., 1997).
Resting cerebrovascular tone and blood flow are maintained by a balance between opposing vasoconstrictive and vasodilatory forces. One of the most important of these is the resting vasodilatory influence of nitric oxide (NO). NO has been shown to induce endothelium-dependent vasodilation through a cGMP-mediated mechanism (Rapoport and Murad, 1983). Decreased cGMP production (Kim et al., 1989; Kim et al., 1992) and loss of endothelium-mediated vasodilation (Kim et al., 1988; Nakagomi et al. 1987) contributes to delayed vasospasm after SAH. Furthermore, NO also may cause hyperpolarization and vessel relaxation through a cGMP-independent activation of potassium channels (Bolotina et al., 1994).
Acute constriction of cerebral blood vessels after SAH may result from inhibition of the vasodilatory influence of NO. This is supported by our recent findings that administration of NO donor can reverse acute vasoconstriction and restore CBF after SAH (Sehba et al., 1999). Because both the vasodilatory mechanism and the NO synthesizing ability of cerebral blood vessels are preserved after SAH (Kajita et al., 1994; Kim et al., 1989; Sehba et al., 1999), acute vasoconstriction observed after SAH may indicate a decrease in cerebral NO levels that leads to unopposed vasoconstriction. Hemoglobin has a high binding affinity for NO (Macdonald and Weir, 1991). Blood released in the subarachnoid space may scavenge NO, decreasing its availability to blood vessels (Afshar et al., 1995; Kajita et al., 1994; Sehba et al., 1999; Watkins, 1995). However, no direct supporting evidence has been presented. If SAH causes decreased NO availability, this should be detectable by measuring NO or its metabolites.
Several techniques have been used to measure NO content in brain. The three most commonly used techniques include the Griess reaction, the oxyhemoglobin assay, and NO microelectrodes (Malinski et al., 1993; Williams et al., 1997). The Griess reaction is a spectrophotometric assay that measures nitrite and nitrate, the two major metabolites of NO, after converting them into a diazo compound (Privat et al., 1997). It has been previously used to demonstrate an increase in NO production during cerebral ischemia in rats (Kader et al., 1993; Kumura et al., 1994; Shibata et al., 1996).
The current study examines the effect of SAH on basal NO levels by measuring nitrite and nitrate in five different regions of rat brain before and after SAH using the Griess reaction. Inhibition of NO synthesis by
MATERIALS AND METHODS
Animal model of subarachnoid hemorrhage
Male Sprague-Dawley rats (350 to 420 g) underwent experimental SAH using the endovascular suture model developed in this laboratory (Bederson et al., 1995; Bederson et al., 1998). Briefly, rats were anesthetized with chloral hydrate (35 mg/kg intraperitoneally), transorally intubated, ventilated, and maintained on inspired halothane (0.5% to 0.75% in oxygen-supplemented room air). Rats were positioned in a stereotaxic frame, and the femoral artery exposed and cannulated for blood gas and blood pressure (BP) monitoring. For measurement of intracranial pressure (ICP), the atlanto-occipital membrane was exposed and cannulated, and the cannula was affixed with methymethacrylate cement to a stainless steel screw implanted in the occipital bone (Barth et al., 1992). The CBF was measured by laser-Doppler flowmetry (0.8-mm diameter probes, model P-433, Vasamedics, Inc., St. Paul, MN, U.S.A.) advanced under stereotaxic control to the epidural surface exposed by small burr holes over the middle cerebral artery territory 5 mm lateral to the midline at the coronal suture.
Subarachnoid hemorrhage was induced by advancing a suture retrograde through the ligated right external carotid artery and distally through the internal carotid artery until the suture perforated the intracranial bifurcation of the internal carotid artery. This event was confirmed by a rapid rise in ICP and bilateral decrease in CBF (Bederson et al., 1995). Animals were monitored for 20 minutes before SAH and 10, 60, or 180 minutes after SAH. Sham-operated rats underwent a similar experimental procedure except for induction of SAH.
Brain preparation
After the experiment, animals were killed, and the brains were removed and placed on ice. Brains were divided in the midline. One hemisphere (the left side) was used for the assay. In preliminary experiments, no significant difference in NO metabolite content was found between right and left hemispheres. Five different brain regions (brain stem, cerebellum, ventral convexity cortex, dorsal convexity cortex, and hippocampus) were isolated. Tissues were quickly weighed and placed in boroscilicate glass tubes containing chilled phosphate-buffered saline, volume 10 times the tissue weight. Tissues were homogenized on ice using a tissue homogenizer (Tissumizer, Tekmar, Cincinnati, OH, U.S.A.). The homogenate was centrifuged at 10, 000 × g for 20 minutes. The supernatant was collected and centrifuged again at 75, 000 × g for 15 minutes. A total of 500 μL of supernatant was filtered through a micron filter system (Microcon YM-30, Amicon Inc. Beverly, MA, U.S.A.) at 14, 000 × g for 20 minutes to eliminate proteins above 30, 000-kDa molecular weight, which may interfere with the absorbance of nitrite. Filtrate was collected and refrigerated at 4°C until use. In preliminary experiments, refrigeration of the filtrate did not affect NO−2/NO−3 measurements, whereas freezing brain tissues or filtrate decreased the contents of NO metabolites.
Measurement of NO metabolite levels
The measurements were performed using a Nitrate/Nitrite Colorimetric assay kit (Cayman Chemicals Co., Ann Arbor, MI, U.S.A.). For NO metabolites determination, a 50-μL sample was incubated with nitrate reductase and cofactors at room temperature. After 90 minutes, Griess reagents No. 1 (sulfanilamide) and No. 2 (N-(1-naphthyl)ethylenediamine) were added, and absorbance at 540 nm was measured using an ultraviolet plate reader (Biorad, 2000 Alfred Nobel Drive, Hercules, CA, U.S.A.). Each experiment included a standard curve for nitrite and nitrate. Standard curves were used to determine NO metabolite concentration (μmol/L), which was divided by wet tissue weight and expressed as μmol/mg wet tissue weight. Nitrite contents of the sample (100 μL) were determined by reading absorbance after adding Griess reagents in the absence of nitrate reductase. Subtraction of nitrite values from the total NO metabolites allowed quantification of nitrate. All measurements were performed in duplicate.
Drug treatment
Animals were divided into six groups. Group 1 (n = 9) underwent sham operation and was observed for 15 minutes before killing and brain removal. Groups 2 (n = 6) and 3 (n = 5) underwent sham operation followed by intravenous injection of
Data acquisition and statistical analysis
Cerebral blood flow, ICP, and mean arterial blood pressure data were continuously recorded starting 20 minutes before and 10 to 60 minutes after SAH using customized analog-to-digital conversion hardware and software (Labview v 4.0, National Instruments, Austin, TX, U.S.A.) and stored at a rate of 0.25 Hz. The CBF data were normalized to an average (baseline) value obtained for 10 minutes before SAH and expressed as a percentage of baseline. The NO metabolites were read from standard curves and divided by the wet tissue weight and expressed as μmol/mg wet tissue weight. Statistical evaluation was performed using a one-way analysis of variance followed by Bonferroni post hoc test setting significance at P < 0.05.
RESULTS
Standardization of the Griess reaction for cerebral NO metabolites detection and quantification
The efficiency of the Griess reaction to detect changes in NO metabolites levels would be limited if the quantity of metabolite is outside of the minimum or maximum sensitive range of the assay. The absorbance-to-volume ratio using increasing volumes of brain tissue filtrate (20, 40, 60, and 80 (μL) was therefore studied. A linear increase in absorbance at 540 nm was found with increasing quantity of brain tissue homogenate for all the regions assayed (Fig. 1A). A volume of 50 μL was used for the subsequent experiments, since absorbance by this volume would fall in the middle of linear portion of the graph.
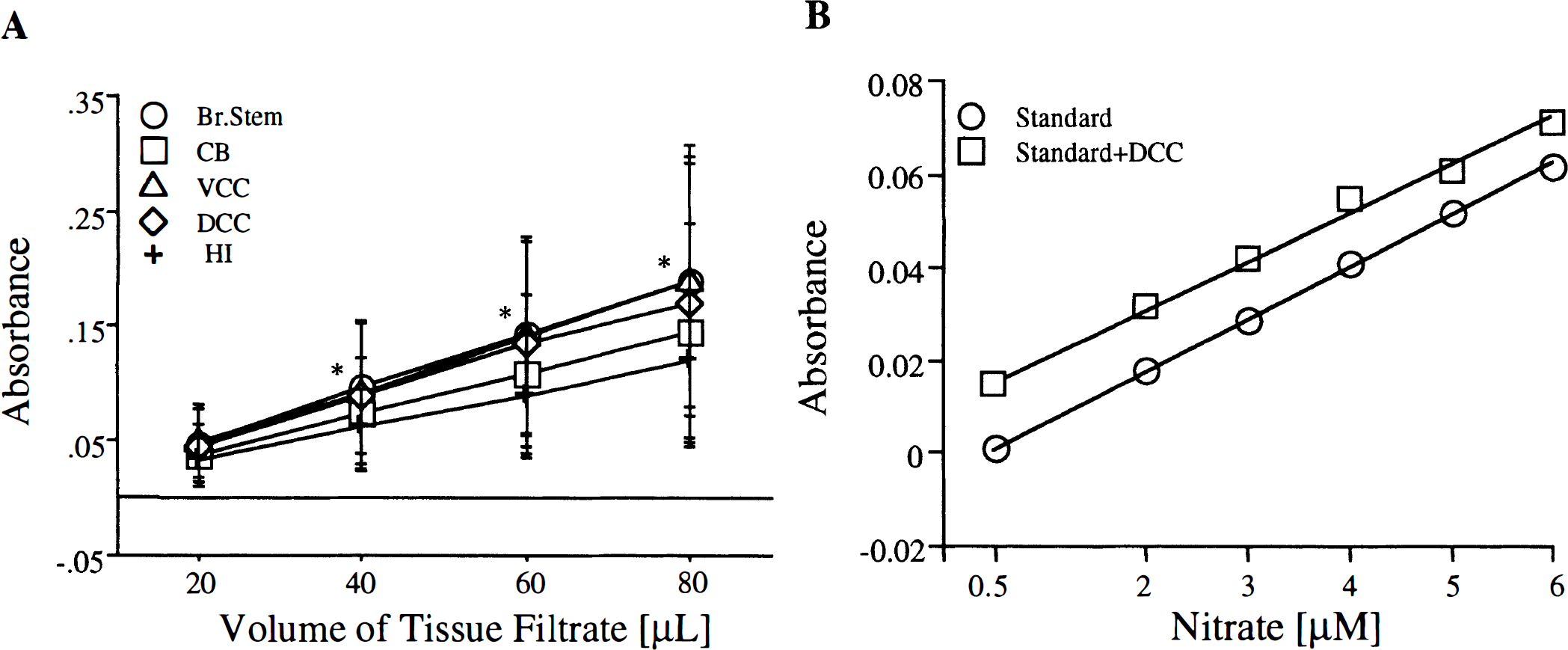
Characterization of the Griess reaction. (
Analytic recovery of NO metabolites theoretically could be affected by the presence of substances in brain filtrates that interfere with the activity of nitrate reductase or have absorbance near 540 nm. This was investigated by creating a standard curve of nitrate in presence or absence of a fixed amount of brain tissue filtrate. Because it was assumed that filtrate from all six regions of brain would act in a similar manner, dorsal convexity cortex from a control animal was used for this experiment. A linear increase in absorbance was found with increasing concentration of nitrate in the presence or absence of cerebral cortex filtrate, indicating the analytical capability of the assay to accurately measure nitrate content (Fig. 1B).
The basal NO metabolites levels per wet tissue weight were as follows (μmol/mg wet tissue weight, mean ± SD): brain stem, 0.14 ± 0.07; cerebellum, 0.12 ± 0.08; ventral convexity cortex, 0.22 ± 0.15; dorsal convexity cortex, 0.16 ± 0.11; and hippocampus, 0.26 ± 0.15. Basal nitrite and nitrate levels also were determined. The highest nitrite and nitrate levels were found in the hippocampus (0.04 ± 0.01, and 0.23 ± 0.17 μmol/mg wet tissue weight, respectively) and lowest in the cerebellum (0.01 ± 0.008, and 0.11 ± 0.08 μmol/mg wet tissue weight, respectively, Fig. 2).
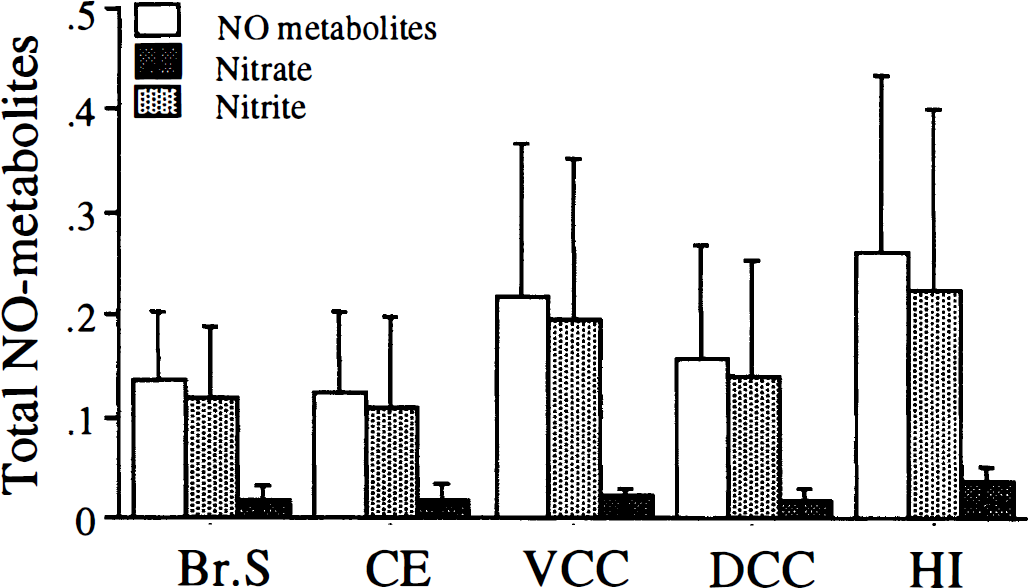
Measurement of nitric oxide (NO) metabolites in five different regions of rat brain. Tissue filtrates from brain stem (Br. Stem), cerebellum (CE), ventral convexity cortex (VCC), dorsal convexity cortex (DCC), and hippocampus (HI) were assayed for total NO metabolites or nitrite. Nitrate was calculated by subtracting nitrite values from NO metabolite values (see Materials and Methods). Data are expressed as μmol/mg wet tissue weight and are mean ± SD, n = 9. Asterisk indicates a significant difference from hippocampus at P < 0.05.
Effects of nitric oxide synthase inhibition on physiology and NO metabolites
At 40 or 120 minutes before killing and brain removal,
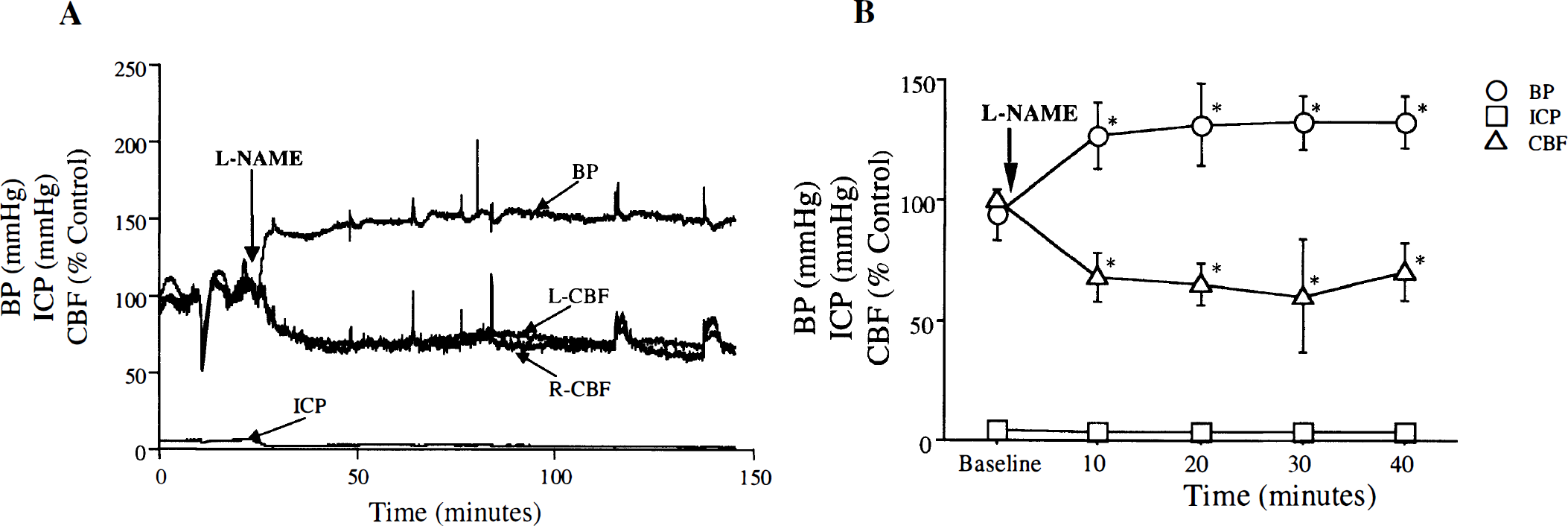
Effect of
Administration of
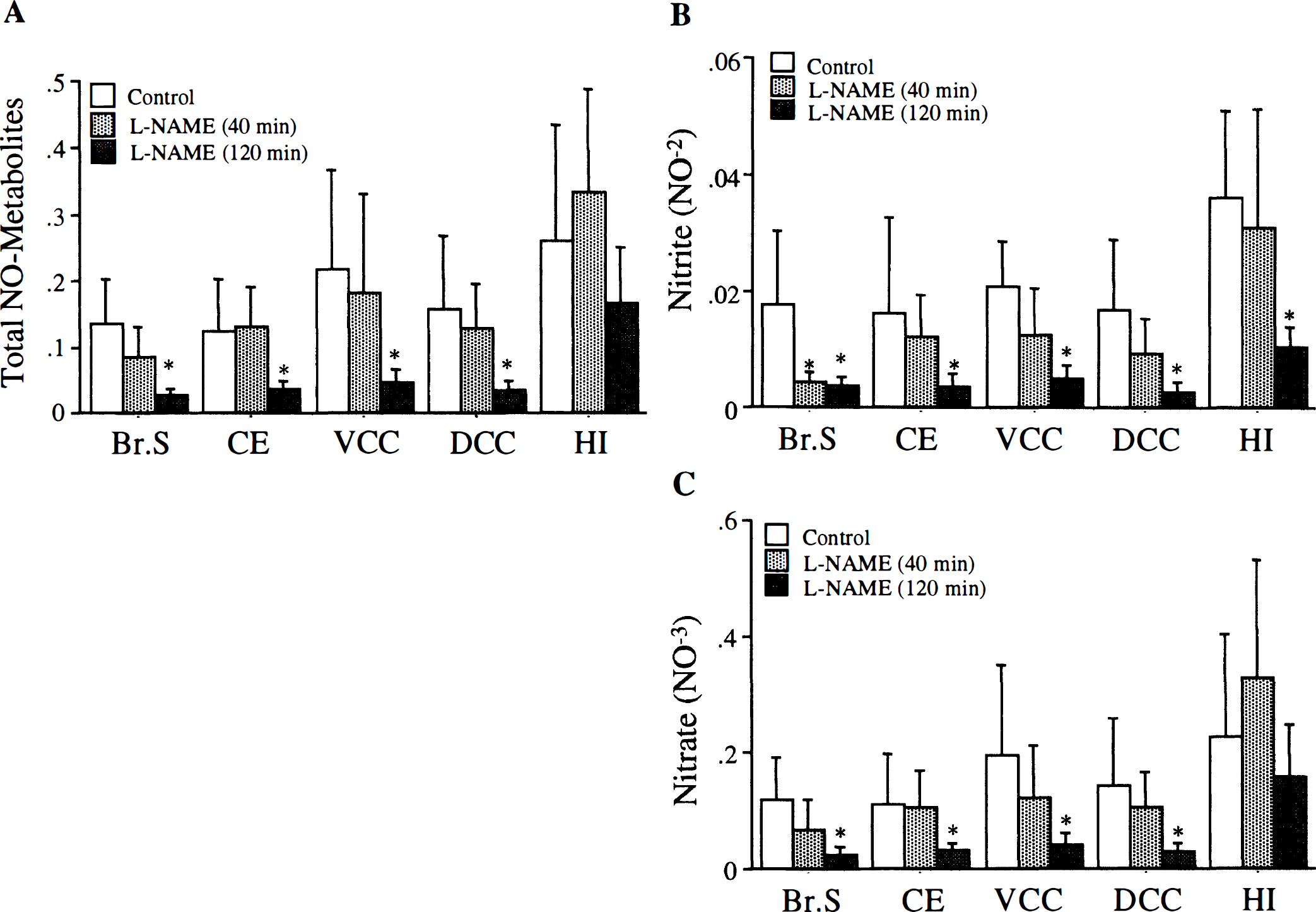
Effect of
Effect of subarachnoid hemorrhage on cerebral levels of NO metabolites
Animals were killed 10 minutes (group 4, n = 6), 60 minutes (group 5, n = 5), or 180 minutes (group 6, n = 6) after SAH induction. At the time of SAH induction, arterial blood P
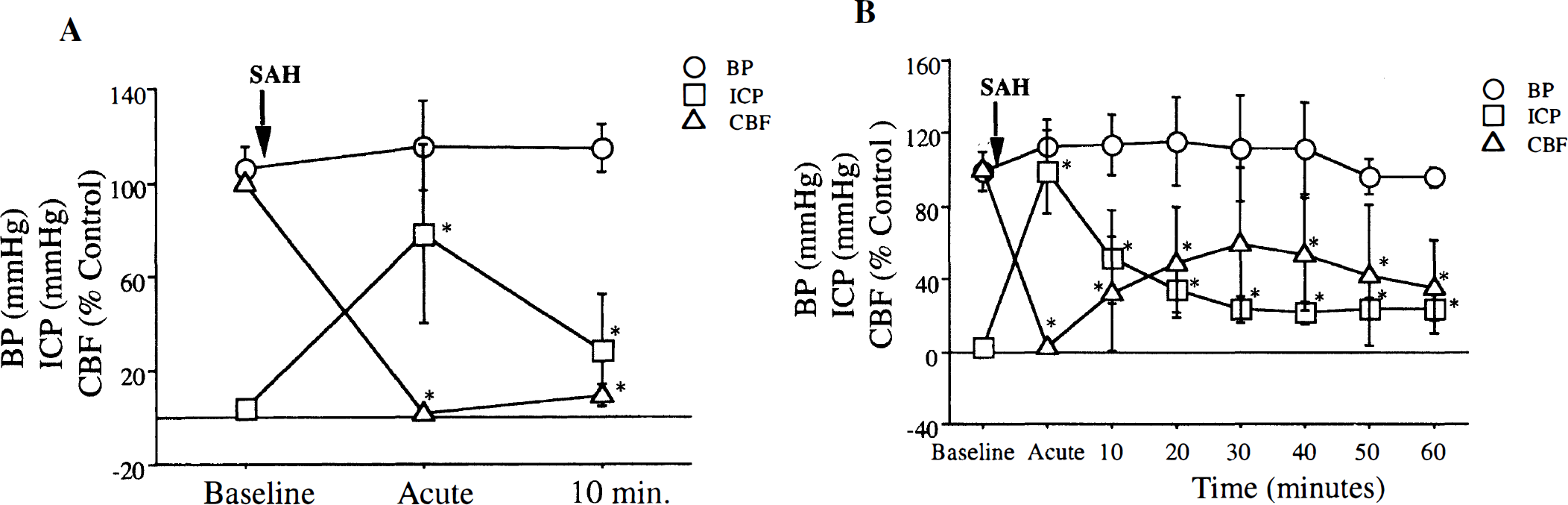
Physiologic changes after SAH. An acute rise in intracranial pressure (ICP) and fall in CBF was seen after SAH induction. The CBF recovered to 9.5 ± 4.4% of baseline 10 minutes (n = 6) after SAH (
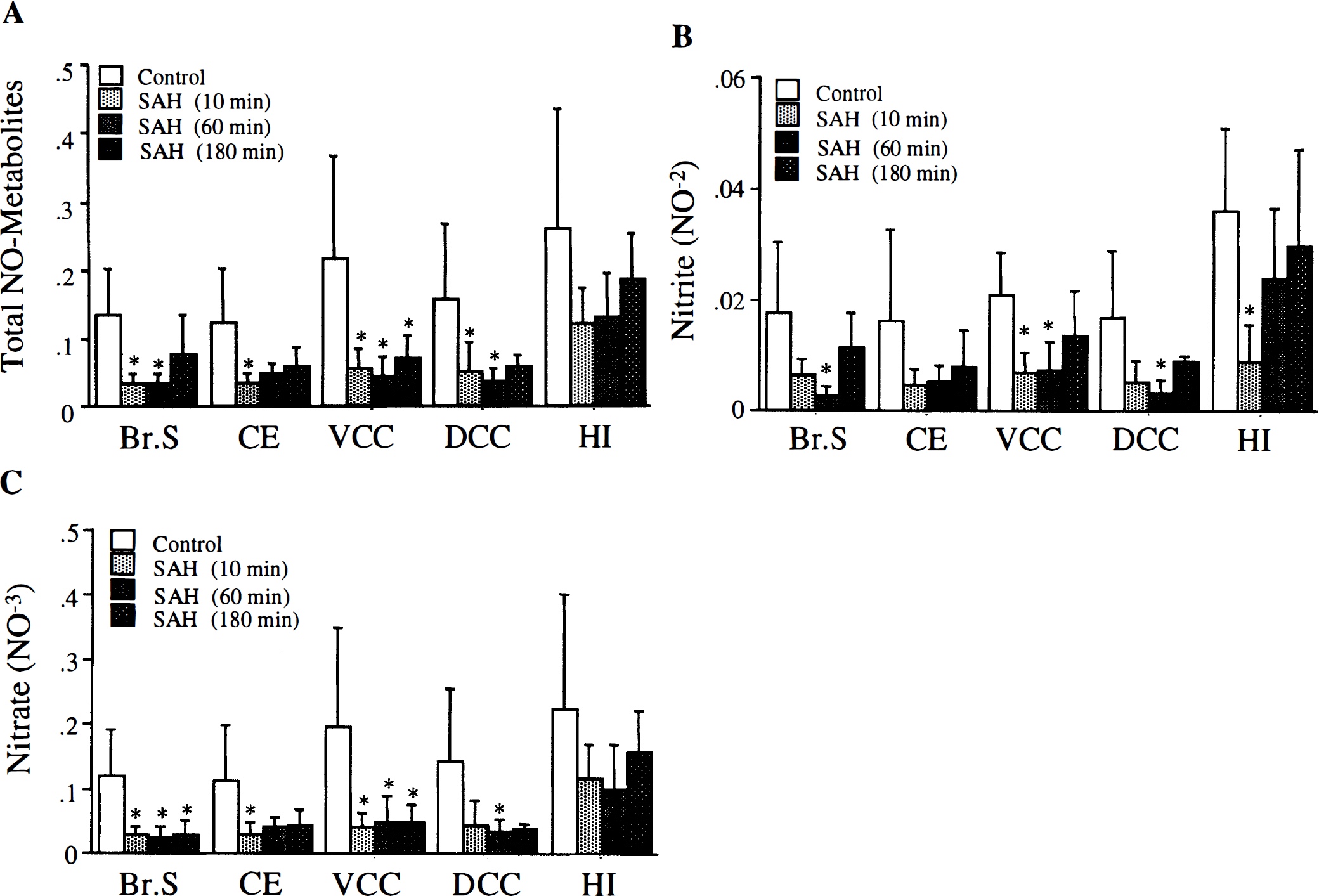
Effect of SAH on NO metabolites in rat brain. The SAH was associated with significant decrease in total NO metabolites at 10 minutes (n = 6) in all brain regions except hippocampus. (
DISCUSSION
Pathologic alteration of the
Both global and focal cerebral ischemia cause an increase in cerebral NO levels that can be attenuated by nitric oxide synthase (NOS) inhibitors, indicating that increased NOS activity is the source of ischemic NO overproduction (Kader et al., 1993; Kumura et al., 1994; Malinski et al., 1993; Shibata et al., 1996). Decreased NO levels observed in the current study makes SAH unique among the various forms of ischemic insults (Kader et al., 1993; Kumura et al., 1994; Malinski et al., 1993). An initial decrease in NO availability may contribute to SAH-induced brain injury by initiating a series of events including unopposed vasoconstriction, decreased CBF, ischemia, and glutamate release (Bederson et al., 1995; Bederson et al., 1998; Schwartz et al., 1999; Sehba et al., 1999) as well as Ca+2 influx, activation of NOS, and subsequently increased NO production.
Subarachnoid hemorrhage induces an acute global ischemic insult (Bederson et al., 1998), which is the primary cause of morbidity and mortality in humans (Broderick et al., 1994; Kassell et al., 1985). We previously demonstrated that acute cerebral vasoconstriction contributes to this ischemia (Bederson et al., 1998) and that decreased NO availability plays important role in this process (Schwartz et al., 1999; Sehba et al., 1999).
Although our sample collection technique excluded blood vessels of the Circle of Willis, it is presumed that measured NO was produced by both neuronal and endothelial NOS. Other investigators have found both NOS isoforms in neuronal tissues (Dinerman et al., 1994). Selection of brain regions for the current study was based on their anatomic location in relation to blood distribution in the subarachnoid space (Bederson et al., 1995; Sehba et al., 1999). Nitric oxide metabolite levels were reduced more in areas exposed to the most blood (ventral convexity cortex and brain stem) and less in the hippocampus, an area exposed only to blood in the lateral ventricle, which is minimal in this model (Bederson et al., 1995; Sehba et al., 1999). In the absence of hemoglobin, NO can diffuse across a distance of more than 100 μm to reach target cells (Kelm, 1999). These observations are consistent with the hypothesis that NO derived from both NOS isoforms is scavenged by hemoglobin immediately after SAH. A role of NO produced in perivascular nerves in neurogenic control of cerebrovascular tone has been observed (Iadecola et al., 1993; Toda and Okamura, 1996) and also could be altered by SAH.
Because of its highly reactive nature, the diffusion and availability of NO at its target depends on microenvironmental conditions such as the type and amount of oxygen-derived radicals, the pH, and concentration of transition metals and thiols. Furthermore, the nature of the final product of NO metabolism depends on local oxygen content at the time of its release and metabolism. Aerobic conditions favor production of methemoglobin and nitrate, whereas anaerobic conditions favor production of slowly dissociating nitrosylhemoglobin (Kelm, 1999; Wennmalm et al., 1992).
Administration of
In preliminary experiments, we found that, unlike nitrite, nitrate does not bind hemoglobin (data not shown). Because nitrate is an oxidation product of nitrite, measurable changes in its levels are preceded by changes in nitrite. These measurements are influenced by the time required for oxidation and accumulation of enough product to observe it through the Griess reaction. This may have contributed to the observed recovery of nitrite 180 minutes after SAH, a time when nitrate levels still are reduced in brain stem and ventral convexity cortex. Nevertheless, a significant decrease in nitrate levels in brain stem and cerebellum 10 minutes after SAH without a subsequent decrease in nitrite levels is surprising.
An additional explanation of the decrease in NO metabolites seen after SAH could be inhibition of NOS after SAH by an unknown mechanism. However, previous studies show that
The recovery of NO metabolites to baseline levels 180 minutes after SAH could indicate both increased NO production and saturation of hemoglobin. However, hemoglobin binding would influence measured NO levels, making an increase difficult to detect. The NOS activity could be increased by at least three mechanisms: (1) decreased product inhibition of NO on NOS, resulting from scavenging of NO (Vickroy and Malphurs, 1995); (2) activation of glutamate-activated N-methyl-
The decrease in CBF after
CONCLUSION
We have measured cerebral levels of NO metabolites after SAH and demonstrated significant decreases 10 minutes after the hemorrhage. We hypothesize that this decrease is a reflection of decreased NO availability that causes acute vasoconstriction after SAH and explains why NO donors are effective in reversing acute SAH-induced ischemia. Decreased NO levels after SAH may result from scavenging by hemoglobin and anaerobic metabolism into nitrosylhemoglobin. These mechanisms need to be investigated further, directly by measuring the levels of nitrosylhemoglobin, and indirectly by measuring methemoglobin after SAH.
Footnotes
Acknowledgment
The authors thank Dr. E. Fisher for his assistance.