
Abstract
The purpose of this study was to investigate: 1) the temporal and regional profile of polymorphonuclear leukocyte (PMNL) infiltration after moderate traumatic brain injury using the parasagittal fluid percussion model and 2) the effects of posttraumatic hypothermia (30°C) and hyperthermia (39°C) on the acute and subacute inflammatory response. We hypothesized that posttraumatic hypothermia would reduce the degree of PMNL accumulation whereas hyperthermia would exacerbate this response to injury. In the first series of experiments we quantitated the temporal profile of altered myeloperoxidase activity under normothermic (37°C) conditions (n = 20). The rats were allowed to survive for 3 hours, 24 hours, 3 days, or 7 days after trauma, and brains were dissected into cortical and subcortical regions ipsilateral and contralateral to injury. Additional animals were perfused and fixed for the immunocytochemical visualization of myeloperoxidase (n = 15). In the second series of experiments, rats (n = 25) were killed 3 hours or 3 days after the 3-hour monitoring period of normothermia (36.5°C), hypothermia (30°C), or hyperthemia (39°C) (n = 4 to 5 per group), and myeloperoxidase activity was again quantitated. In normothermic rats, the enzymatic activity of myeloperoxidase was significantly increased (P < 0.05) at 3 hours within the anterior cortical segment (213.97 ± 56.2 versus control 65.5 ± 52.3 U/g of wet tissue; mean ± SD) and posterior (injured) cortical and subcortical segments compared to shamoperated rats (305.76 ± 27.8 and 258.67 ± 101.4 U/g of wet tissue versus control 62.8 ± 24.8 and 37.28 ± 35.6 U/g of wet tissue; P < 0.0001, P < 0.05, respectively). At 24 hours and 7-days after trauma only the posterior cortical region (P < 0.005, P < 0.05, respectively) exhibited increased myeloperoxidase activity. However, 3 days after trauma, myeloperoxidase activity was also significantly increased within the anterior cortical segment (P < 0.05) and in posterior cortical and subcortical regions compared to sham-operated cortex (P < 0.0001, P < 0.05, respectively). Immunocytochemical analysis of myeloperoxidase reactivity at 3 hours, 24 hours, 3- and 7-days demonstrated large numbers of immunoreactive leukocytes within and associated with blood vessels, damaged tissues, and subarachnoid spaces. Posttraumatic hypothermia and hyperthermia had significant effects on myeloperoxidase activity at both 3 hours and 3 days after traumatic brain injury. Posttraumatic hypothermia reduced myeloperoxidase activity in the injured and noninjured cortical and subcortical segments compared to normothermic values (P < 0.05). In contrast, posttraumatic hyperthermia significantly elevated myeloperoxidase activity in the posterior cortical region compared to normothermic values at both 3 hours and 3 days (473.5 ± 258.4 and 100.11 ± 27.58 U/g of wet tissue, respectively, P < 0.05 versus controls). These results indicate that posttraumatic hypothermia decreases early and more prolonged myeloperoxidase activation whereas hyperthermia increases myeloperoxidase activity. Temperature-dependent alterations in PMNL accumulation appear to be a potential mechanism by which posttraumatic temperature manipulations may influence traumatic outcome.
Ischemic and traumatic brain injury (TBI) lead to inflammatory events that are believed to contribute to outcome through secondary injury mechanisms (Hallenbeck et al., 1986; Giulian et al., 1989; Shoettlle et al. 1990; Kochanek et al., 1992; Biagas et al., 1992; Chopp et al., 1994; Clark et al., 1994; del Zoppo and Garcia, 1995; Tomina and Fukuuchi, 1996; Hartl et al., 1997; Jean et al., 1998; Grady et al., 1999). Among the known critical factors that are involved in the spread of secondary damage after TBI is the acute inflammatory response to injury involving the infiltration and accumulation of polymorphonuclear leukocytes (PMNLs) (Soares et al., 1995; Rothwell et al., 1997). Potential consequences of PMNL activation and accumulation include increased microvascular permeability (Petito, 1979; Wahl et al., 1988; Cortez et al., 1989; Beynon et al., 1993), edema formation (Schoettle et al., 1990; Bareyre et al., 1997), intracranial hypertension (Uhl et al., 1994), and secondary neuronal injury (Chopp et al., 1994).
In various models of cerebral ischemia (Barone et al., 1991; Chopp et al., 1994), the involvement of PMNL recruitment as contributors to the acute inflammatory response has been reported (for reviews see Kochanek and Hallenbeck, 1992; Jean et al., 1998). Neutrophils have been reported to be the first inflammatory cells to arrive at the ischemic injury site, followed by the infiltration of mononuclear phagocytes (Schoettle et al., 1990; Clark et al., 1994). In models of TBI including weight drop, control cortical impact, intracerebral stab injury, and fluid percussion (F-P), an influx of neutrophil accumulation has been shown as early as 4 to 8 hours after trauma with peak values occurring at 48 hours (Persson 1976; Biagas et al., 1992; Horner et al., 1992; Clark et al., 1994; Dietrich et al., 1996b; Toyoda et al., 1996; Carlos et al., 1997). The infiltration and accumulation of activated PMNLs into extravascular tissues depend on PMNLs-endothelial interactions between cell surface leukocyte integrins and adhesion molecules such as endothelial selectins (E- and P- selectin) (Mackay and Imhof 1993; Smith 1993; Stoolman 1993; Carlos and Harlan 1994; Shappell and Smith, 1994; Soares et al., 1995; Hamada et al., 1996; McIntosh et al., 1996). Increased expression of intercellular adhesion molecule (ICAM-1) and platelet endothelial adhesion molecule-1 (PECAM-1) after brain trauma injury account partly for neutrophil adhesion to endothelial cells and transendothelial migration (Zhang et al., 1994b; Carlos et al., 1997; Whalen et al., 1991a, b, 1999).
After injury, activated neutrophils, macrophages, and disrupted endothelial cells produce a variety of cytokines that may also contribute to the pathologic process (Zimmerman, 1986; Lindholm et al., 1987; Hama et al., 1989; David et al., 1990; Marion et al., 1993; Taupin et al., 1993; Ott et al., 1994; Fan et al., 1995, 1996; Goss et al., 1995; Shohami et al., 1996, 1997; Mantovani et al., 1997). Cytokines are capable of altering vascular permeability (Beynon et al., 1993) and exacerbate subsequent brain edema formation (Albelda et al., 1994; Matsuo et al., 1994a, b ). These events may lead to the influx of more inflammatory cells, with the resultant production of harmful reactive oxygen species (Hallenbeck et al., 1986; Kochanek et al., 1987; Saito et al., 1988; Ward et al., 1988; Grogaard et al., 1989; Barone et al., 1991; Clark et al., 1994; Matsuo 1995).
Posttraumatic hypothermia has been shown in several models of TBI to improve histopathologic and behavioral outcome (Jiang et al., 1991; Arnould et al., 1993; Clifton et al., 1993; Lyeth et al., 1993; Palmer et al., 1993; Dietrich et al., 1994a, b , 1996c; Bramlett et al., 1995; Globus et al., 1995; Clark et al., 1996; Marion et al., 1997). In brain trauma models, posttraumatic hypothermia has been reported to preserve the integrity of the blood-brain barrier (BBB) (Jiang et al., 1992), reduce neuronal injury, decrease contusion volume (Dietrich et al., 1990b; Palmer et al., 1993), and reduce cerebral oxygen consumption (Rosomoff and Haladay, 1954). Cytoskeletal components including microtubule-associated protein have been reported to be protected from TBI by hypothermia (Taft et al., 1993). In addition, hypothermia applied after TBI attenuates the increase in extracellular levels of excitatory amino acids and oxygen-free radical production (Globus et al., 1995). Recent reports using the control cortical impact model of TBI demonstrated that PMNL accumulation in the injured cortex was significantly decreased in rats maintained at 32°C versus 39°C at 4 hours after TBI (Whalen et al., 1997a, b ).
In contrast to hypothermia, hyperthermia has been shown to aggravate outcome in models of brain injury (Dietrich et al., 1996b). After cerebral ischemia for example, hyperthermia has been reported to worsen neuronal and cerebrovascular outcome (Dietrich et al., 1990a, b ; Chen et al., 1991; Morikawa et al., 1992). In TBI, hyperthermia (39°C) applied 24 hours after F-P injury increased contusion volume, BBB breakdown, axonal injury, and mortality (Dietrich et al., 1996b). Despite extensive data indicating the beneficial effects of posttraumatic hypothermia on outcome and the detrimental effects of posttraumatic hyperthermia, the effect of posttraumatic temperature manipulations on the acute and subacute inflammatory response to F-P brain injury has not been investigated.
To quantitate the accumulation of PMNLs at the injury site, we conducted a regional determination of myeloperoxidase activity which has been reported to be a reliable marker of PMNL accumulation (Barone et al., 1991; Zhang and Chopp, 1997). Immunocytochemical studies were conducted in parallel to document the regional distribution of neutrophils in the traumatized brain. Our findings demonstrate that PMNLs accumulate within 3 hours after moderate F-P injury within the histopathologically vulnerable brain regions. In addition posttraumatic temperature modifications significantly influence the inflammatory response to TBI.
MATERIALS AND METHODS
Animal groups
The myeloperoxidase enzymatic activity was performed on 20 male Sprague-Dawley rats weighing between 300 and 400 g obtained from Charles River Breeders. They were kept at a constant temperature (∼25°C) in an air-conditioned room for at least 7 days before the study and exposed to a 12-hour light-dark cycle. Rats were allowed free access to food and water before and after the surgery. The studies were conducted in two phases. In the first phase, the temporal profile of myeloperoxidase activity was determined: group 1, the normothermic group, time = 3 hours, 24 hours, 3 days, or 7 days and sham (n = 3 in each group). The same temporal profile of immunocytochemical detection of myeloperoxidase was performed on 12 male Sprague-Dawley rats weighing between 300 and 400 g (n = 4 in each group). Sham-operated control rats underwent all surgical procedures but were not traumatized and killed 24 hours later. In the second phase, the effects of posttraumatic hypothermia or hyperthermia on myeloperoxidase activity at 3 hours and 3 days were assessed: group 2, normothermic (37°C) group 3-hour survival (n = 4); group 3, hypothermic (30°C) group 3-hour survival (n = 4); group 4, hyperthermic (39°C) group 3-hour survival (n = 5); group 5, 3 hours of posttraumatic normothermia and 3-days survival (n = 4); group 6, 3 hours of posttraumatic hypothermia and 3 days survival (n = 4); group 7, 3-hour posttraumatic hyperthermic group and 3-days survival (n = 4). After the 3-hour temperature monitoring period the animals were allowed to recover and had free access to food and water. The 3-hour period was chosen because this represented the earliest time after the temperature monitoring period that could be assessed. The 3-day period represented the maximal myeloperoxidase response after TBI.
Surgical preparation
All animal use procedures were in strict accordance with the NIH Guide for the Care and Use of Laboratory Animals. The basic surgical preparation for the F-P brain injury was performed according to methods previously described (Dietrich et al., 1994a). Briefly, 1 day before injury, the rats were anesthetized with Equitensin (a mixture of nembutal, propylene glycol, ethanol, MgSO4, and chloral hydrate). Rats were then placed in a stereotaxic frame, and a 4.8 mm craniectomy was made overlying the right parietal cortex (3.8 mm posterior to bregma and 2.5 mm lateral to the midline) (Zilles, 1985). A plastic injury tube was then placed over the exposed dura, was bound by adhesive, and then dental acrylic was poured around the injury tube. After the acrylic had hardened, the scalp was closed, and the animal was returned to its cage and allowed to recover overnight before TBI.
The next day, a fluid percussion (F-P) device was used to produce experimental TBI (Dixon et al., 1987; McIntosh et al., 1989). The rats were reanesthetized with 3.0% halothane in a gas mixture of 70% nitrous oxide and 30% oxygen and maintained at 0.5% halothane. An endotracheal tube was inserted orally, and rats were mechanically ventilated. During ventilation, the animals were paralyzed with pancronium bromide (1 mg/kg per hour, intravenously). Moderate head injury ranging from 1.8 to 2.2 atm was next produced. The F-P device consists of a saline-filled Plexiglass cylinder that is fitted with a transducer housing and injury screw adapted for the rat's skull. The metal screw is firmly connected to the plastic injury tube of the anesthetized rat, and the injury is induced by the descent of a pendulum that strikes the piston. In the first phase of experiments where the temporal profile of myeloperoxidase was assessed, temporalis muscle temperature was monitored as an indirect measure of brain temperature throughout the experiment and maintained at a normothermic (37.0°C) level using a feedback heating lamp above the animal's head (Jiang et al., 1991). Rectal temperature was also monitored and maintained at normothermic levels throughout the study using a feedback heating lamp above the animal's body. Arterial blood pressure was monitored by means of the right femoral artery and recorded for up to 60 minutes after TBI. Blood gases were obtained from arterial samples taken from the right femoral artery just before TBI and 60 minutes later. Blood glucose levels were also monitored. The animals in the sham groups for myeloperoxidase activity measurements were subjected to all of the same surgical procedures except for the actual insult, including anesthesia and the placement of a plastic injury tube.
In the second phase of experiments the effects of posttraumatic hypothermia and hyperthermia on myeloperoxidase activity were assessed. Posttraumatic rats were brought to the target temporalis muscle temperature within 5 minutes and maintained under normothermic (36.62 ± 0.086°C), hypothermic (30.18 ± 0.14°C), or hyperthermic (39.68 ± 0.217°C) conditions for 3 hours after TBI. In animals in which brain temperature was lowered, cold air was blown directly onto the head.
Myeloperoxidase assay
Myeloperoxidase, a heme lysosomal enzyme present in azurophilic granules of neutrophils and in smaller quantities in monocytes, has been commonly used as a marker of PMN accumulation (Barone et al., 1991). The assay method used in this study was a modification of a previously described method by Barone et al., (1991). To eliminate intravascular blood, rats were intracardially perfused with 250 mL of ice-cold 0.9% NaCl (at a pressure of 100 mm Hg). A 3-mm thick coronal brain slice was first made at the bregma levels 1.2 mm and −1.8 mm and included cortical and subcortical areas anterior to the histopathologically vulnerable posterior cortical and subcortical regions (i.e., −1.8 mm and −4.8 mm) using Rodent Brain Matrices (Harvard Apparatus, Inc. MA, U.S.A.) (Fig. 1). The anterior cortical and subcortical regions adjacent to the injury were labeled as ACips and ASCips, respectively. The posterior cortical and subcortical sites that included histopathologically vulnerable regions (i.e., posterior cortical and subcortical segments) were dissected and labeled as PCips and PSCips. The homotopic cortical and subcortical regions of the contralateral hemisphere were also dissected and labeled as ACcont, ASCcont, PCcont, and PSCcont. Brain samples were frozen in liquid nitrogen then stored at −80°C until use. A 10% (w/v) tissue homogenate was mixed with 20 mmol/L potassium phosphate buffer (pH 6.0) containing 0.5% hexadecyltrimethyl ammonium bromide (Sigma), sonicated for 30 seconds, subjected to three freeze/thaw cycles, and then sonicated again for 30 seconds. The samples were centrifuged at 12, 000 rpm, at 4°C for 20 minutes. The supernatant was assayed for myeloperoxidase activity.
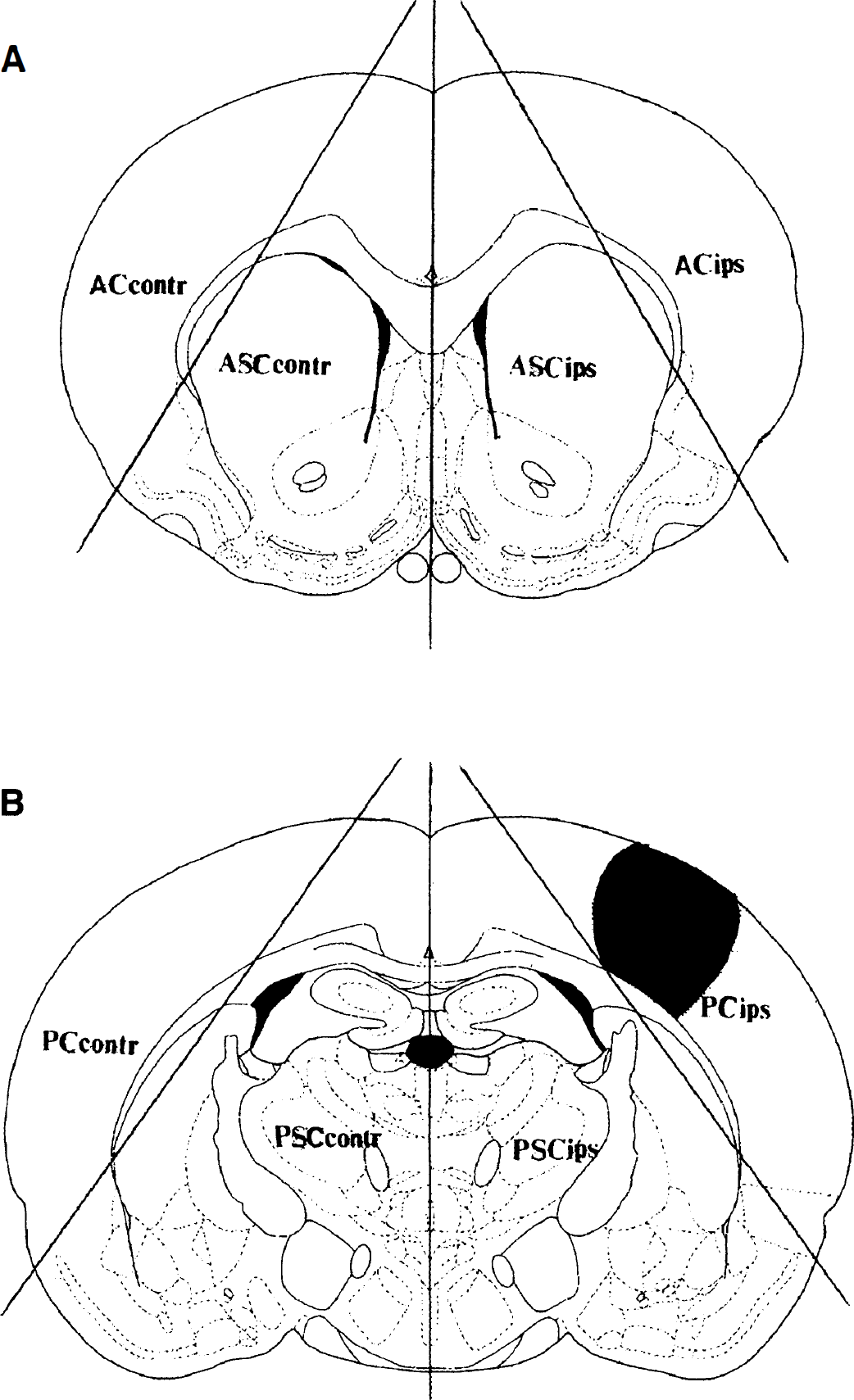
Diagrammatic illustration of forebrain dissection into eight segments. (
MPO activity in supernatant was determined in 0.3 mL of 0.1 mol/L potassium phosphate buffer (pH 6.0) containing 1.25 mg/mL o-dianisidine and 0.05% H2O2. The assay was started by adding 30 to 40 μL of sample. A change in absorbance at 460 nm after 5 minutes was measured in an Elisa Plate Reader and the myeloperoxidase activity was calculated using a standard curve prepared for purified myeloperoxidase (Sigma). The linear portion of the absorbance at 460 nm was used to determine myeloperoxidase activity. One unit of myeloperoxidase activity was defined as that degrading 1 μmol of H2O2 in 1 minute. The myeloperoxidase enzymatic activity was proportional to the amount of sample assayed (20, 40, 80, and 100 μL) and was abolished by boiling the extract for 3 minutes. Data were expressed as units per gram of wet tissue, and an increase or decrease in enzymatic activity of ipsilateral or contralateral structures was compared with activities from sham-operated animals.
Immunohistochemical studies
At 3 hours, 1 day, 3 days, or 7 days after TBI or sham operation, a subset of rats under normothermic conditions were deeply anesthetized with halothane and perfused transcardially with 250 mL of cold physiologic saline for 1 minute and then FAM (a mixture of 40% formaldehyde, glacial acetic acid, and methanol; 1:1:8 by volume) for 20 minutes (25°C at a pressure of 100 mm Hg). Brains were extracted and stored in chilled fixative overnight. The next day, brains with the dura attached were embedded in paraffin and semi serial 10-μm sections were taken through the neuraxis. Sections were mounted on slides, rehydrated, and placed in 6% H2O2 to block endogenous peroxidase activity. The tissue was rinsed and steamed with a solution of citrate buffer for 20 minutes. Then the tissue was rinsed with 0.05 mol/L phosphate-buffered saline. Nonspecific activity was blocked with normal horse serum. Subsequently, brain sections were incubated at room temperature for 1 hour in a 1:1500 dilution of rabbit antihuman myeloperoxidase (DAKO Corporation, Carpitaria, CA, U.S.A.). To test for nonspecific staining, negative controls were conducted where the primary antibody was omitted during tissue processing. Further rinsing was done with phosphate-buffered saline and secondary antibody applied. The slides were processed using the LSAB staining kit (DAKO Corp., Carpitaria, CA, U.S.A.) per the manufacturer's instructions. myeloperoxidase staining was visualized with peroxidase, and diaminobenzidine was used as a chromogen to yield a brown reaction product. Slides were washed in 0.5% Triton X-100 followed by 1% cupric sulfate to further intensify staining. Counterstaining was done with hematoxylin, and tissue was then dehydrated and coverslipped.
Statistical analysis
Data were expressed as mean values ± SD. Two-way analysis of variance (ANOVA) for repeated measures was used to compare physiologic variables measured over time. Betweengroup comparisons of physiologic data and myeloperoxidase data sets were made using ANOVA one-way analysis of variance. Differences were considered significant if the P value was < 0.05.
RESULTS
Physiologic Parameters
Group 1. The physiologic variables obtained 5 minutes before and 15 minutes after TBI are presented in Table 1. No significant difference was observed between groups.
Physiologic variables in rats for MPO measurements

Values are mean ± SD. MABP, mean arterial blood pressure; TMT, temporalis muscle temperature.
Groups 2 to 4 (3-hour survival). The physiologic variables measured 5 minutes before and 3 hours after TBI are presented in Table 2. During the 3 hour experimental period, brain temperatures differed significantly between normothermic (36.62 ± 0.172°C), hypothermic (30.18 ± 0.28°C), and hyperthermic (39.68 ± 0.44°C) groups (P < 0.05). P
Physiologic variables in rats for MPO measurements at 3 hours

Values are mean ± SD. Brain temperature differed between groups 5 minutes posttrauma (P < 0.05, two-way analysis of variance [ANOVA] for repeated measures). MABP, mean arterial blood pressure; TMT, temporalis muscle temperature.
dR Significantly different compared to normothermic values (P<0.05 by one-way ANOVA).
Groups 4 to 6 (3-day survival). The physiologic variables measured 5 minutes before and 15 minutes and 3 days after TBI are presented in Table 3. At 5, 15, minutes, and 3 hours after TBI brain temperatures differed significantly between the normothermic, hypothermic, and hyperthermic groups (P < 0.05). Baseline P
Physiologic variables in rats for MPO measurements at 3 days

Values are mean ± SD. Brain temperature differed between groups 5 minutes after trauma (P<0.05, two-way analysis of variance [ANOVA] for repeated measures). MABP, mean arterial blood pressure; TMT, temporalis muscle temperature.
Significantly different compared to normothermic values (P< 0.05 by one-way ANOVA).
MPO activity after normothermic TBI
First phase of experiments. The time course of myeloperoxidase activity in the injured and noninjured cortex is illustrated in Fig. 2. In normothermic rats, the enzymatic activity of myeloperoxidase was significantly increased (P < 0.05) at 3 hours within ipsilateral brain regions including the anterior (1.2 to −1.8 mm bregma) cortical segment (213.97 ± 56.2 versus control 65.5 ± 52.3 U/g of wet tissue; mean ± SD) and the posterior (−1.8 to −4.8 mm bregma, injured) cortical and subcortical segments compared to sham-operated rats (305.76 ± 27.8 and 258.67 ± 101.4 U/g of wet tissue versus control 62.8 ± 24.8 and 37.28 ± 35.6 U/g of wet tissue; mean ± SD, P < 0.0001, P < 0.05, respectively). At 24 hours after trauma only the injured cortical region (200.89 ± 57.7 versus control 62.8 ± 24.94 U/g of wet tissue; mean ± SD, P < 0.005) exhibited increased myeloperoxidase activity. However, 3 days after trauma, myeloperoxidase activity was significantly increased within the ipsilateral anterior cortical segment (183.3 ± 101.1 versus control 65.5 ± 52.3 U/g of wet tissue; mean ± SD, P < 0.05) and in posterior cortical and subcortical regions compared to sham-operated cortex (619.68 ± 53.2 and 133.86 ± 57.22 U/ g of wet tissue versus control 62.88 ± 24.94 and 37.28 ± 34.76 U/g of wet tissue, respectively; mean ± SD, P < 0.0001, P < 0.05). At 7 days there was a significant increase in myeloperoxidase activity only at the posterior cortical region compared to control (124.13 ± 48.02 versus 48.64 ± 24.8 U/g of wet tissue, respectively; mean ± SD, P < 0.05). No significant increase in myeloperoxidase activity was documented in the 24-hour sham-operated control brains.
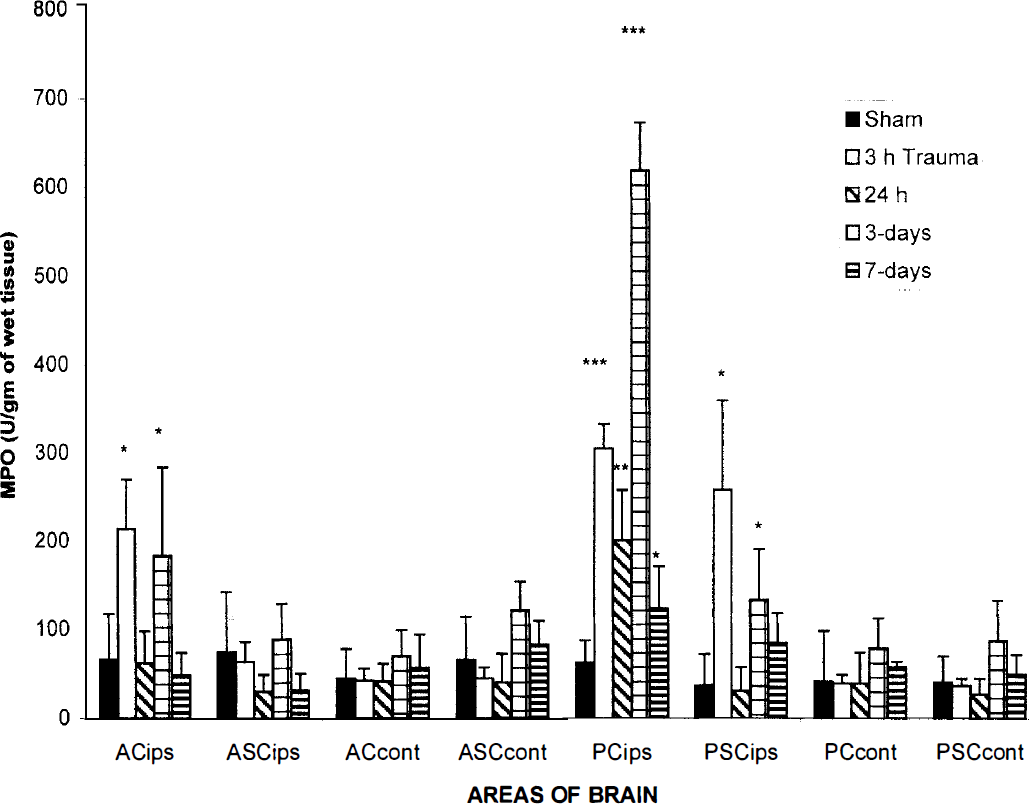
Time course of neutrophil accumulation (myeloperoxidase activity) in injured cortical and subcortical brain regions and in respective contralateral regions or sham-operated animals. Within the ipsilateral cerebral cortex, a significant increase of myeloperoxidase activity was observed at 3 hours, 24 hours, 3 days, and 7 days after TBI (significantly different compared to sham-operated rats, *P < 0.05, **P < 0.005, P < 0.0001 versus control by one-way ANOVA). Values are the mean ± SD of n = 4 in each group.
Second phase of experiments. Figures 3 and 4 depict the effects of posttraumatic hypothermia and hyperthermia on myeloperoxidase activity at 3 hours and 3 days after TBI. At both survival periods, posttraumatic hypothermia significantly reduced myeloperoxidase activity throughout the ipsilateral hemisphere including anterior and posterior cortical and subcortical structures compared to normothermic levels (P < 0.05). For example posttraumatic hypothermia significantly reduced myeloperoxidase activity in the injured posterior cortical region at 3 hours after TBI (47 ± 23.08 versus normothermic control 160.2 ± 44.6 U/g of wet tissue, n = 4). In contrast, posttraumatic hyperthermia significantly elevated myeloperoxidase activity in the posterior (injured) cortical region compared to normothermic values; 3 hours after TBI (473.5 ± 258.4 versus control 160.2 ± 44.66 U/g of wet tissue, n = 4, n = 5 respectively, P < 0.05) and 3 days after TBI (100.11 ± 27.58 versus 40.52 ± 21.8 U/g of wet tissue, mean ± SEM, n = 4, respectively, P < 0.05). In preliminary experiments we found that in normothermic TBI rats, extending the period of physiologic monitoring to 3 hours after TBI significantly reduced overall myeloperoxidase activity (Figs. 3 and 4). For this reason a separate set of normothermic rats was used in studies where 3 hours of posttraumatic monitoring was conducted. Although the mechanism for this consequence in myeloperoxidase activity was not investigated, the potential effects of prolonged anesthesia on myeloperoxidase activity should be considered.
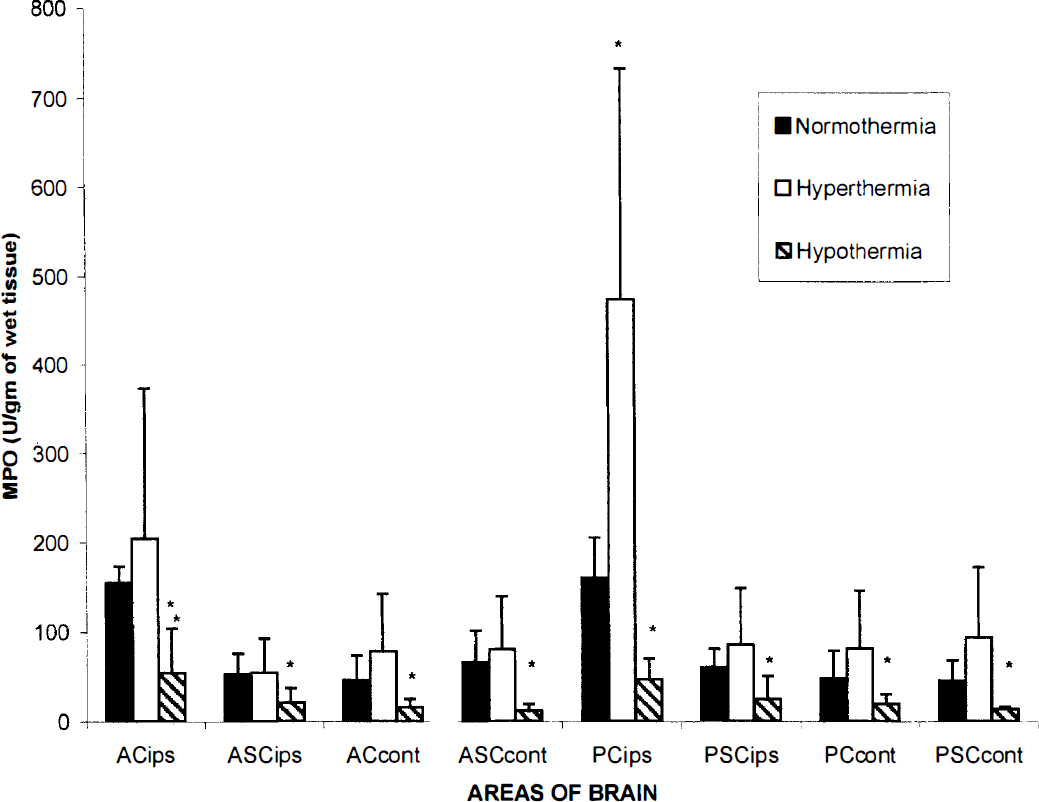
Graph depicting myeloperoxidase activity at 3 hours in cortical and subcortical areas anterior and posterior to the histopathologically vulnerable region. *P < 0.05 for 30°C versus 37°C and 39°C versus 37°C. Brain temperature was controlled for 3 hours at 30°C, 37°C, or 39°C after TBI (*P < 0.05 versus normothermic control by one-way ANOVA). Values are the mean ± SD of n = 4 to 5 in each group.
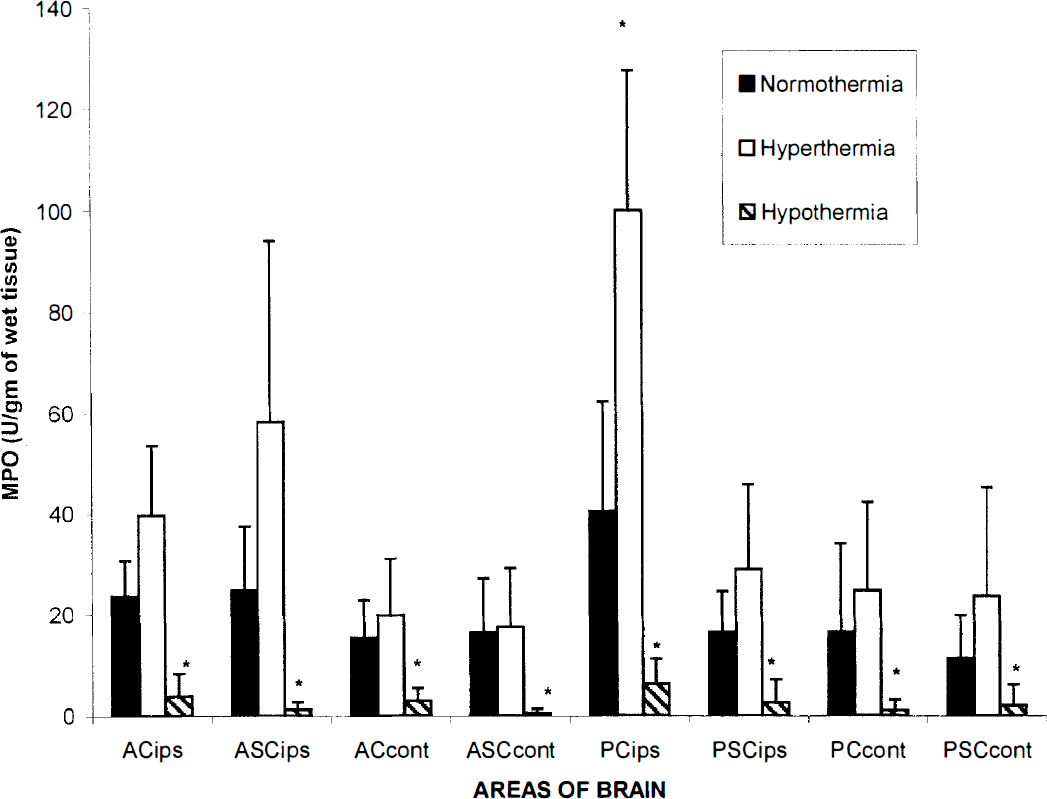
Graph depicting myeloperoxidase activity at 3 days in cortical and subcortical areas anterior and posterior to the histopathologically vulnerable region. *P < 0.05 for 30°C versus 37°C and 39°C versus 37°C. Brain temperature was controlled for 3 hours at 30°C, 37°C, or 39°C after TBI. The rats were kept alive for 3 days and then killed after saline perfusion (*P < 0.05 versus normothermic control by one-way ANOVA). Values are the mean ± SD of n = 4 to 5 in each group.
Immunocytochemical findings
Immunocytochemical analysis at 3 hours, 24 hours, 3 days, and 7 days demonstrated large numbers of immunoreactive vascular and parenchymal leukocytes associated with normal appearing and damaged tissue (Fig. 5). At 24 hours after TBI, myeloperoxidase immunoreactive PMNLs were seen within subarachnoid spaces overlying the histopathologically vulnerable cortical region (Figs. 5A and 5B). In addition, PMNLs were associated with blood vessels within and adjacent to the contusion site. High-magnification analysis demonstrated both luminal and perivascular leukocytes (Figs. 5C and 5D). Scattered immunoreactive cells were observed within the damaged cortical tissue (Fig. 5E) and within the subarachnoid space (Fig. 5F). PMNLs were also identified within pial vessels, the interhemispheric fissure, hippocampus, and dentate gyrus. At 3 days immunoreactive leukocytes remained present within the histopathologically damaged cerebral cortex and contusion area. At 7 days PMNLs were observed within the injured cortical tissue and associated with newly formed vessels. In contrast, sham-operated control brain sections did not demonstrate abnormal neutrophil accumulation.
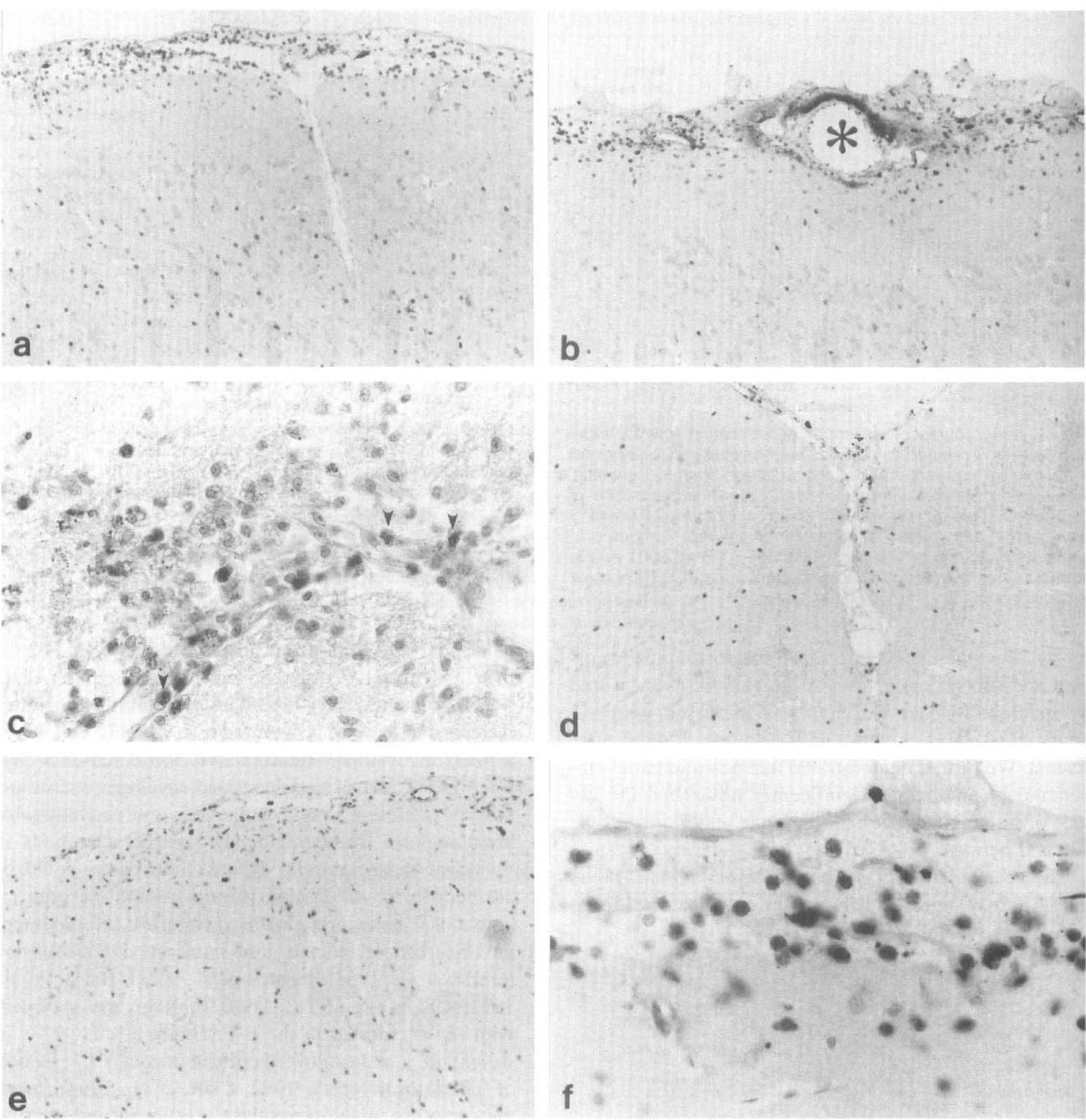
Immunocytochemical visualization of neutrophils that exhibit myeloperoxidase immunoreactivity in ipsilateral brain regions after TBI. PMNLs are present in the subarachnoid space overlying the histopathologically vulnerable cortical region (
DISCUSSION
The results of the present study demonstrate that in this F-P model of brain injury neutrophils accumulate as part of the acute and subacute inflammatory response to injury. The time-course data show that posttraumatic cortical and subcortical PMNL accumulation, as measured by myeloperoxidase activity, was increased within 3 hours, peaked at 3 days, and resolved 7 days after trauma. We further demonstrated that posttraumatic temperature manipulations significantly influenced the degree of inflammation at both the acute and more chronic survival periods.
The accumulation of neutrophils observed at 3 hours after F-P may represent hemorrhage associated with vascular injury resulting from shear forces generated by the primary impact (Cortez et al., 1989; Dietrich et al., 1990(3; Schoettle et al., 1990). We further demonstrated increased neutrophil accumulation at 24 to 72 hours with a peak of myeloperoxidase activity observed at 3 days after trauma. At 3 days increased myeloperoxidase activity was present in both vulnerable and nonvulnerable brain regions. These regionally specific alterations in myeloperoxidase activity at later time points could be attributable to widespread pathophysiologic events including subacute inflammatory responses, cerebrovascular disturbances, edema, and intracranial hypertension (Obrist et al., 1984; Shoettle et al., 1990; Biagas et al., 1992; Uhl et al., 1994). These findings are consistent with those reported in the F-P (Grady et al., 1999), in weight drop, and controlled cortical impact (CCI) models of TBI (Horner et al., 1992; Clark et al., 1994). In all these models, myeloperoxidase activity increased at 24 and 48 hours and resolved by 7 days after TBI. In addition, myeloperoxidase activity in the CCI model was reported to increase as a function of injury severity (Clark et al., 1996).
The time course of myeloperoxidase activity after TBI parallels traumatic events including alterations in local cerebral blood flow, cerebral edema, BBB damage, intracranial hypertension, and free radical formation (Chan et al., 1984; Obrist et al., 1984; Schoettle et al., 1990; Biagas et al., 1992; Uhl et al., 1994; Globus et al., 1995; Soares et al., 1995; Dietrich et al., 1996a). Previous studies by Bareyre et al. (1997) have reported the development of vasogenic edema resulting from BBB breakdown caused by cortical vascular endothelial injury as early as 1 hour and as late as 5 days after TBI. Focal ischemia studies also demonstrated the myeloperoxidase activity to be significantly increased at 24 hours after MCAO (Bareyre et al., 1997). In addition, Clark and colleagues (1996) in a CCI model have shown that neutrophil accumulation occurs rapidly in ischemic regions. Thus, the degree of abnormal perfusion after injury may be an important factor for PMNL accumulation (Zhang et al., 1994a). The possibility that the alterations in myeloperoxidase activity are attributable to vasodilation/capillary recruitment or neovascularity at later time points should also be considered.
Trauma-induced alterations of neutrophils and macrophages/microglia may alter pathologic outcome through the secretion of proinflammatory cytokines including tumor necrosis factor-α, IL-1, IL-6, leukotrienes (Ellis et al, 1981; Goodman, 1986, McClain et al., 1991; Taupin et al., 1993; Shohami et al., 1994, 1996; Fan et al., 1995, 1996), complement, integrin (Kaczorowski et al., 1995), platelet activating factor (Patel et al., 1992), and platelet activating factor-induced active oxygen species (H2O2 and superoxide anion)(Arnould et al., 1993). When stimulated the activated PMNs consume oxygen (Nauseef et al., 1983) known as respiratory burst, phagocytose debris (Persson, 1976; Giulian et al., 1989) and release potent oxygen species that are antibacterial agents (Ward et al., 1988). The most important of these oxygen species are hydrogen peroxide (H2O2), the superoxide anion (O2−), hypochlorous acid, hydroxyl radicals, and nitric oxide (NO). However, a number of enzymes are also secreted by activated neutrophils, including superoxide dismutase that converts superoxide anion to H2O2 and myeloperoxidase which converts H2O2 to hypochlorous acid and oxygen-free radical (O−) (Zhang and Chopp, 1997). These reactive oxygen species can damage surrounding healthy nervous tissue by lipid peroxidation (Shappell and Smith, 1994; Anderson, 1995). Secreted factors may also lead to the upregulation of adhesion molecule expression by vascular endothelial cells which in turn attract circulating neutrophils. In this regard, selective therapeutic strategies targeted toward neutrophil depletion or blockers of PMNL adherence have been investigated in a number of cerebral ischemia studies (Sekiya et al., 1989; Biagas et al., 1992; Chen et al., 1994; Chopp et al., 1994; Uhl et al., 1994; Zhang et al., 1994b; McIntosh et al., 1996) and after F-P injury (Grady et al., 1999).
Previous studies have discussed the beneficial effects of mild and moderate hypothermia on secondary damage after TBI (Clifton et al., 1993; Marion et al., 1993; Palmer et al., 1993; Dietrich et al., 1994b; Chatzipanteli et al., 1999). The level of moderate hypothermia (30°C) used in our studies was similar to that used in previous experimental studies (Jiang et al., 1992; Lyeth et al., 1993; Dietrich et al., 1994b; Bramlett et al., 1995; Chatzipanteli et al., 1999). Other experimental studies have used more clinically relevant levels of mild hypothermia (Clark et al., 1996; Whalen et al., 1997a, b ). In clinical studies, mild hypothermia has also been used (Clifton et al., 1993; Marion et al., 1993; Clifton 1995; Marion and White, 1996). Possible hypothermic mechanisms underlying the beneficial effects include consequences on excitatory amino acids, metabolic changes, edema formation, BBB damage, local cerebral blood flow, marked reduction of IL-1 mRNA, and free-radical formation (Busto et al., 1989; Jiang et al., 1992; Clifton et al., 1993; Goss et al., 1995; Dietrich et al., 1996a; Toyoda et al., 1996). Previous studies have reported that posttraumatic hypothermia leads to reductions in cerebral blood flow (Marion et al., 1993; Marion and White, 1996; Zhao et al., 1999). The present findings emphasize the importance of posttraumatic brain temperature on inflammatory processes.
A study by Rosomoff et al. (1965) was the first to show the effects of hypothermia on the acute inflammatory response to experimental brain injury, where hypothermia (25°C) applied for 1 hour after injury delayed neutrophil accumulation up to 6 hours. Further studies have demonstrated that PMNL migration in vivo and in vitro was markedly reduced by profound hypothermia (29°C) (Biggar et al., 1984; Akriotis and Biggar, 1985). In a focal ischemia-reperfusion model, hypothermia (30°C) reduced the cell-mediated inflammatory response (quantified by myeloperoxidase measurements) in the pericore region (Toyoda et al., 1996). Recent myeloperoxidase activity data and histologic outcome measurements have also demonstrated that mild hypothermia (32°C) reduced PMNL accumulation after CCI injury compared to hyperthermia (39°C) by fourfold using immunohistochemistry and by eightfold using the myeloperoxidase assay (Whalen et al., 1997a). Interestingly, E-selectin activation 4 hours after CCI was decreased in that study only modestly by hypothermia (32°C). It was concluded that neutrophil accumulation is dependent on posttraumatic brain temperature and that this effect is independent of the changes in the expression of E-selectin, ICAM-1, and absolute neutrophil count (Whalen et al., 1997a, b ).
In contrast to hypothermia, posttraumatic hyperthermia appears to aggravate the inflammatory consequences of TBI. Previous TBI studies reported that delayed posttraumatic hyperthermia (39°C) increased mortality rate, contusion volume, white matter pathology, and PMNL accumulation in the injured brain during the initial 4 hours after TBI (Dietrich et al., 1994b; Whalen et al., 1997a, b ). Furthermore, hyperthermia had no effect in the expression of E-selectin, ICAM-1, and absolute neutrophil count at this early time point after TBI (Whalen et al., 1997a, b ). The present findings therefore support previous observations and provide quantitative data indicating that hyperthermia increases the acute and more chronic inflammatory response to TBI. These temperature findings do not prove a cause-and-effect relationship between posttraumatic hyperthermia and injury outcome. Thus, it will be important in future studies to determine specific pathomechanisms underlying hyperthermia-induced aggravated PMNL accumulation.
In conclusion, these results indicate that posttraumatic inflammation plays an important role in the pathophysiology of moderate parasagittal F-P brain injury. We demonstrated a temporal response of myeloperoxidase activity in histopathologic vulnerable and nonvulnerable brain regions. In addition, treatment with therapeutic hypothermia significantly decreased myeloperoxidase activity whereas hyperthemia increased myeloperoxidase activity. Thus, a possible mechanism by which posttraumatic temperature modifications influence traumatic outcome is by altering the acute and more chronic inflammatory response to injury. A major objective in TBI research is to develop novel treatments that will prevent secondary tissue damage and promote neuronal survival. It is possible that combination therapy including mild hypothermia with pharmacologic agents that block PMNL accumulation may prove useful in the treatment of TBI.