
Abstract
Alterations in cerebral autoregulation and cerebrovascular reactivity after traumatic brain injury (TBI) may increase the susceptibility of the brain to secondary insults, including arterial hypotension. The purpose of this study was to evaluate the consequences of mild hemorrhagic hypotension on hemodynamic and histopathologic outcome after TBI. Intubated, anesthetized male rats were subjected to moderate (1.94 to 2.18 atm) parasagittal fluid–percussion (FP) brain injury. After TBI, animals were exposed to either normotension (group 1: TBI alone, n = 6) or hypotension (group 2: TBI + hypotension, n = 6). Moderate hypotension (60 mm Hg/30 min) was induced 5 minutes after TBI or sham procedures by hemorrhage. Sham-operated controls (group 3, n = 7) underwent an induced hypotensive period, whereas normotensive controls (group 4, n = 4) did not. For measuring regional cerebral blood flow (rCBF), radiolabeled microspheres were injected before, 20 minutes after, and 60 minutes after TBI (n = 23). For quantitative histopathologic evaluation, separate groups of animals were perfusion-fixed 3 days after TBI (n = 22). At 20 minutes after TBI, rCBF was bilaterally reduced by 57% ± 6% and 48% ± 11% in cortical and subcortical brain regions, respectively, under normotensive conditions. Compared with normotensive TBI rats, hemodynamic depression was significantly greater with induced hypotension in the histopathologically vulnerable (P1) posterior parietal cortex (P < 0.01). Secondary hypotension also increased contusion area at specific bregma levels compared with normotensive TBI rats (P < 0.05), as well as overall contusion volume (0.96 ± 0.46 mm3 vs. 2.02 ± 0.51 mm3, mean ± SD, P < 0.05). These findings demonstrate that mild hemorrhagic hypotension after FP injury worsens local histopathologic outcome, possibly through vascular mechanisms.
Posttraumatic secondary events—including ischemia, hypoxia, hypotension, hyperthermia, and intracranial hypertension—are commonly observed in head-injured patients (Bouma and Muizelaar, 1992; Chesnut et al., 1993; Golding et al., 1999; Kelly et al., 1997; Marion et al., 1991; Muizelaar et al., 1984; Obrist et al., 1984). It is also well documented that secondary insults contribute to the mortality and morbidity associated with clinical (Chesnut et al., 1993; Muizelaar et al., 1984) and experimental (Bramlett et al., 1999; Cherian et al., 1996; Clark et al., 1997; Davis et al., 1998; DeWitt et al., 1992a, 1992b, 1995; Dietrich et al., 1996;Golding et al., 1999; Ishige et al., 1987; Jenkins et al., 1989; Kroppenstedt et al., 1999; Nawashiro et al., 1995; Schmoker et al., 1992) traumatic brain injury (TBI). Cerebrovascular perturbations, including structural (Cortez et al., 1989; Dietrich et al., 1994; Povlishock et al., 1978; Wei et al., 1980) and hemodynamic perturbations (Armstead and Kurth, 1994; Davis et al., 1998; DeWitt et al., 1986, 1992a, 1992b; Dietrich et al., 1996a, 1998; Yamakami and McIntosh, 1991), are a common consequence of experimental TBI and may underlie the increased sensitivity of the posttraumatic brain to secondary insults.
Alterations in global and regional cerebral blood flow (rCBF) have been documented in patients with head injuries (Bouma and Muizelaar, 1992; Marion et al., 1991; Martin et al., 1997; Obrist et al., 1984). In models of moderate fluid–percussion brain injury (FP), rCBF reductions are common in the mild to moderate range (Yamakami and McIntosh, 1991; DeWitt et al., 1986, 1992a, 1992b; Dietrich et al., 1996a), whereas severe FP injury leads to ischemic levels of rCBF (Dietrich et al., 1998). In contrast, models of controlled cortical impact injury are known to produce regional cerebral ischemia (Bryan et al., 1995; Dietrich et al., 1998; Kochanek et al., 1995; Nilsson et al., 1996). In human TBI, histopathologic damage in selectively vulnerable brain regions, including the hippocampus, is observed (Adams et al., 1982; Graham et al., 1978). Thus, under specific experimental and clinical conditions, posttraumatic ischemia is an acute response to TBI and may participate in the pathophysiology of this injury.
Dysfunction of cerebral autoregulation also has been reported in animal models of TBI (Golding et al., 1999). In the acute stages of injury, several studies have reported that TBI impairs and/or abolishes the CBF response to induced hypotension, increased CO2, or other vasoactive substances (Armstead, 1998; Engelborghse et al., 2000; Law et al., 1996; Lewelt et al., 1980; Newell et al., 1996; Wei et al., 1980). DeWitt and colleagues (1992b) demonstrated that global and regional autoregulation are absent in response to hemorrhagic hypotension after midline FP brain injury in cats. Recently, Giri and colleagues (2000) reported that after cortical impact injury in rats, rCBF also is further reduced at the impact site when the injured brain is confronted with a secondary ischemic insult. This impairment in rCBF maintenance with respect to changes in Pco2 and cerebral perfusion pressure also have been observed in the traumatically brain-injured patient (Muizelaar et al., 1984; Newell et al., 1996).
There are two prevailing mechanisms that have been proposed in mediating autoregulation. One is a metabolic component whereby neuronal activity induces the release of some neurochemicals (for example, nitric oxide and adenosine) that vasodilate arterioles (Golding et al., 1999). Acute disturbances in glucose metabolism or strong vasoconstrictors such as endothelin have been reported in experimental models of TBI and might participate in these abnormalities (Ginsberg et al., 1997; Yoshino et al., 1991). Consequently, experimental TBI models may have a propensity for autoregulatory dysfunction by depleting a vasodilator or eliciting an abnormal increase in a vasoconstrictor, resulting in the loss of vascular coupling.
The other proposed autoregulatory mechanism is myogenic. This cellular response is because of the interaction of smooth muscle cells to changes in vascular wall tension. Thus, contraction of the vessel wall will occur if perfusion pressure increases and the opposite occurs if pressure decreases. Golding and colleagues (1998) have demonstrated temporal changes in the myogenic response in the middle cerebral artery (MCA) after severe cortical impact injury. They found that although the mechanisms of contraction were present, changes existed in the interaction of myogenic tone or response indices to pressure at 24 hours after trauma. Recently, Mathew and colleagues (1999) reported that cerebral arterial vasodilatory responses to progressive reductions in intraluminal pressure also are affected after moderate central FP injury. In Mathew's study, MCA segments harvested from traumatized rats exhibited abnormal vasodilation during pressure reductions. When regional autoregulation is dysfunctional, secondary insults, such as hypotension, might lead to regional ischemia and worsen outcome (Bouma and Muizelaar, 1992).
Posttraumatic hypoxia has been reported to worsen outcome after TBI (Bramlett et al., 1999; Clark et al., 1997; Ishige et al., 1987; Nawashiro et al., 1995). In one study, 30-minute hypoxia after moderate FP brain injury significantly increased cortical contusion volume and the frequency of CA1 hippocampal neuronal damage compared with normoxic TBI rats (Bramlett et al., 1999). Because TBI hypoxic animals in that study also exhibited mild hypotension, the interpretation of which specific mechanisms were responsible for increased damage (that is, hypoxia vs. hypotension) was unclear. Hypotensive episodes occur in 15% to 35% of patients with severe head injuries and may increase mortality and morbidity associated with that injury (Chesnut et al., 1993). Therefore, the current study was conducted to determine whether posttraumatic moderate hypotension (50 to 60 mm Hg) alone would alter the histopathologic consequences of TBI. Although posttraumatic hypotension has been reported to alter the hemodynamic consequences of TBI, few studies have determined whether regional hemodynamic perturbations influence regional histopathologic outcome.
MATERIALS AND METHODS
Animal groups
Forty-five male Sprague-Dawley rats (254 to 342 g) were housed individually under controlled environmental conditions (12 hours light/12 hours dark cycle) with food and water available ad libitum before being used in the experiments. All animal procedures followed the National Institutes of Health Guide for the Care and Use of Laboratory Animals (National Institutes of Health Publication 86–23, revised 1985).
Fluid–percussion traumatic brain injury procedure
Animals were anesthetized with equithesin (a mixture of pentobarbital, propylene glycol, ethanol, MgSO4, and chloral hydrate) 24 hours before injury and were surgically prepared for FP injury as described previously (Bramlett et al., 1999). Briefly, a craniectomy (4.8 mm in diameter) was performed over the right parietal cortex (3.8 mm posterior to the bregma and 2.5 mm lateral to the midline), leaving the dura intact. The plastic injury tube (2.6-mm inner diameter, approximately 4.8-mm outer diameter) was positioned over the exposed dura and bonded by adhesive. The tube then was cemented to the skull with dental acrylic. The scalp was sutured closed and the animals were returned to their home cages and allowed to recover.
After fasting overnight, a FP device was used to produce experimental TBI through the injury tube (Dixon et al., 1987). Intubated anesthetized rats (0.5% halothane, 30% oxygen, and 70% nitrous oxide) were subjected to a pressure pulse of moderate (1.94 to 2.18 atm) intensity. Before FP injury, catheters (PE50) were inserted into both femoral arteries for blood sampling, measurement of blood pressure, and withdrawal of reference blood samples. A silastic catheter was inserted into the femoral vein and was used to induce hypotension by exsanguination into a syringe. The right axillary artery was cannulated and the catheter (PE10) was advanced into the left ventricle for injection of microspheres. Brain temperature was monitored indirectly through a thermocouple probe inserted into the temporal muscle and maintained at 37.0°C ± 0.5°C. Rectal temperature also was monitored and maintained at 37.0°C ± 0.5°C.
Secondary hypotension
After moderate FP injury, animals were maintained for 30 minutes at either a normotensive (group 1: TBI only, n = 6) or hypotensive (group 2: TBI + hypotension, n = 6) level to simulate secondary hypotension. Hypotension (mean arterial blood pressure = 60 mm Hg) was induced by the gradual withdrawal of blood into a heparinized syringe 5 minutes after TBI. After 30 minutes of hypotension, shed blood was reinfused to restore arterial blood pressure. Blood gas measurements were taken before TBI and 10, 25, and 55 minutes after injury. Sham animals underwent all surgical procedures, including a similar period of hypotension (group 3: sham + hypotension, n = 7), but were not subjected to the FP pulse. For hemodynamic studies, an additional normotensive control group (group 4: n = 4) was produced. These animals did not undergo sham procedures.
Regional cerebral blood flow measurement
Radiolabeled microspheres (15 ± 0.5 μm in diameter; New England Nuclear, Wilmington, DE, U.S.A.) were injected into the left ventricle for measurement of rCBF. Three of 5 possible isotopes—141Ce, 114In, 103Ru, 95Nb, and 46Sc—were randomly selected for injection into each animal at baseline, 20 minutes after TBI during hypotension, and 60 minutes after TBI after reinfusion of shed blood. The method for measurement of rCBF in rats has been previously described (Yamakami and McIntosh, 1989). Reference sample method for cardiac output and regional organ blood flow determinations in the rat also has been reported (Malik et al., 1976).
Approximately 125,000 microspheres were injected at each time point. Injecting this number of microspheres ensured that each brain tissue sample contained more than 400 microspheres. Beginning 10 seconds before microsphere injections, reference blood was withdrawn from the femoral artery at a rate of 0.5 mL/minute during normotension and 0.3 mL/minute during hypotension. Withdrawal continued for 45 seconds after each injection. Reference blood was replaced with 0.9% saline (1:3) after each injection. A postmortem examination was performed after each experiment to confirm position of the left ventricular catheter.
The brain was removed and fixed in 10% buffered formalin for 24 hours and then dissected into 100- to 200-g sections. Briefly, a 3-mm-thick coronal brain slice was first made at the bregma levels 1.2 mm and −1.8 mm and included cortical and subcortical areas anterior to the histopathologically vulnerable posterior cortical and subcortical regions (that is, −1.8 mm and −4.8 mm) using rodent brain matrices (Harvard Apparatus, MA, U.S.A.). The ipsilateral anterior cortical and subcortical regions adjacent to the injury were labeled as A1 and A2, respectively. The ipsilateral posterior cortical and subcortical sites that included histopathologically vulnerable regions (that is, posterior cortical and subcortical segments) were dissected and labeled as P1 and P2, respectively. Homotopic cortical and subcortical regions of the contralateral hemisphere also were dissected and labeled (that is, A3, A4, P3, P4). Radioactivity of each sample of blood and tissue was counted in a gamma counter (Auto-Gamma 5000; Packard Instruments, Meriden, CT, U.S.A.). Tissue blood flow was calculated by the standard simultaneous equation technique, and rCBF was converted to mL min−1 100 g−1. Blood flow data are expressed as percent of baseline (group 4, mean ± SD).
Histopathology
Animals were killed at 72 hours after TBI (group 5: TBI only, n = 8; group 6: TBI + hypotension, n = 7) or sham surgery (group 7: sham + hypotension, n = 7). Semiserial sections were processed and stained for hematoxylin and eosin. Animals were anesthetized and perfused transcardially with isotonic saline at a pressure of 100 to 120 mm Hg for 60 seconds. This was followed by fixative for 20 minutes (FAM, a mixture of 40% formaldehyde, glacial acetic acid, and methanol; 1:1:8 by volume). After the perfusions, the heads were immersed in FAM for 24 hours. Brains then were blocked and embedded in paraffin for tissue sectioning. Coronal sections at various levels (0.8, 1.8, 3.3, 4.3, 5.8, 6.8, 7.3 mm posterior to bregma) were used for contusion area measurements. Contusion boundaries were demarcated by swollen axons, necrotic neurons, reactive astrocytes, and inflammatory cells. The cortical contusion was first traced at a power of 100× using a camera lucida microscope attachment. Contusion areas were calculated by retracing these drawings onto a digitizing table that was interfaced with a computer. Contusion volumes then were computed by using numeric integration of successive areas.
Statistical analysis
Physiologic variables and histopathologic data are expressed as mean ± SD. Group differences in contusion volume measurements and rCBF values were assessed using one-way analysis of variance.
RESULTS
Physiologic changes
Physiologic changes are summarized in Tables 1 and 2. In groups 1 and 2, mean arterial blood pressure (MABP) transiently increased at the moment of the impact, but returned to baseline values within 1 minute. Values for Paco2, Pao2, and pH did not differ among the experimental groups. In groups 2 and 3, MABP was significantly decreased compared with group 1 and was maintained at 59.7 mm Hg (59.7 ± 2.9 and 60.0 ± 1.3 mm Hg, respectively) during the 30-minute hypotensive period.
Physiologic parameters for CBF studies
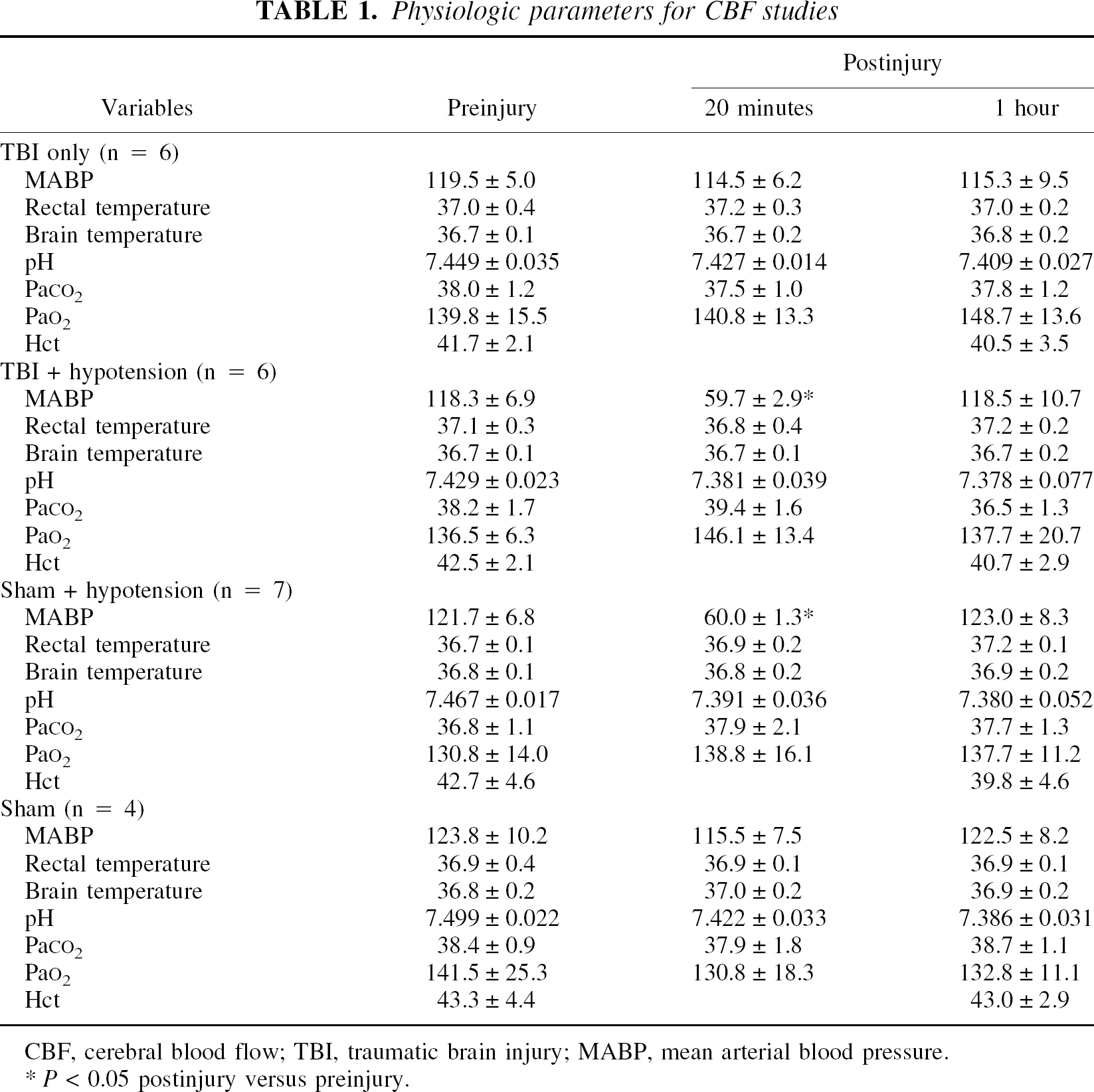
CBF, cerebral blood flow; TBI, traumatic brain injury; MABP, mean arterial blood pressure.
P< 0.05 postinjury versus preinjury.
Physiologic parameters for histopathologic studies
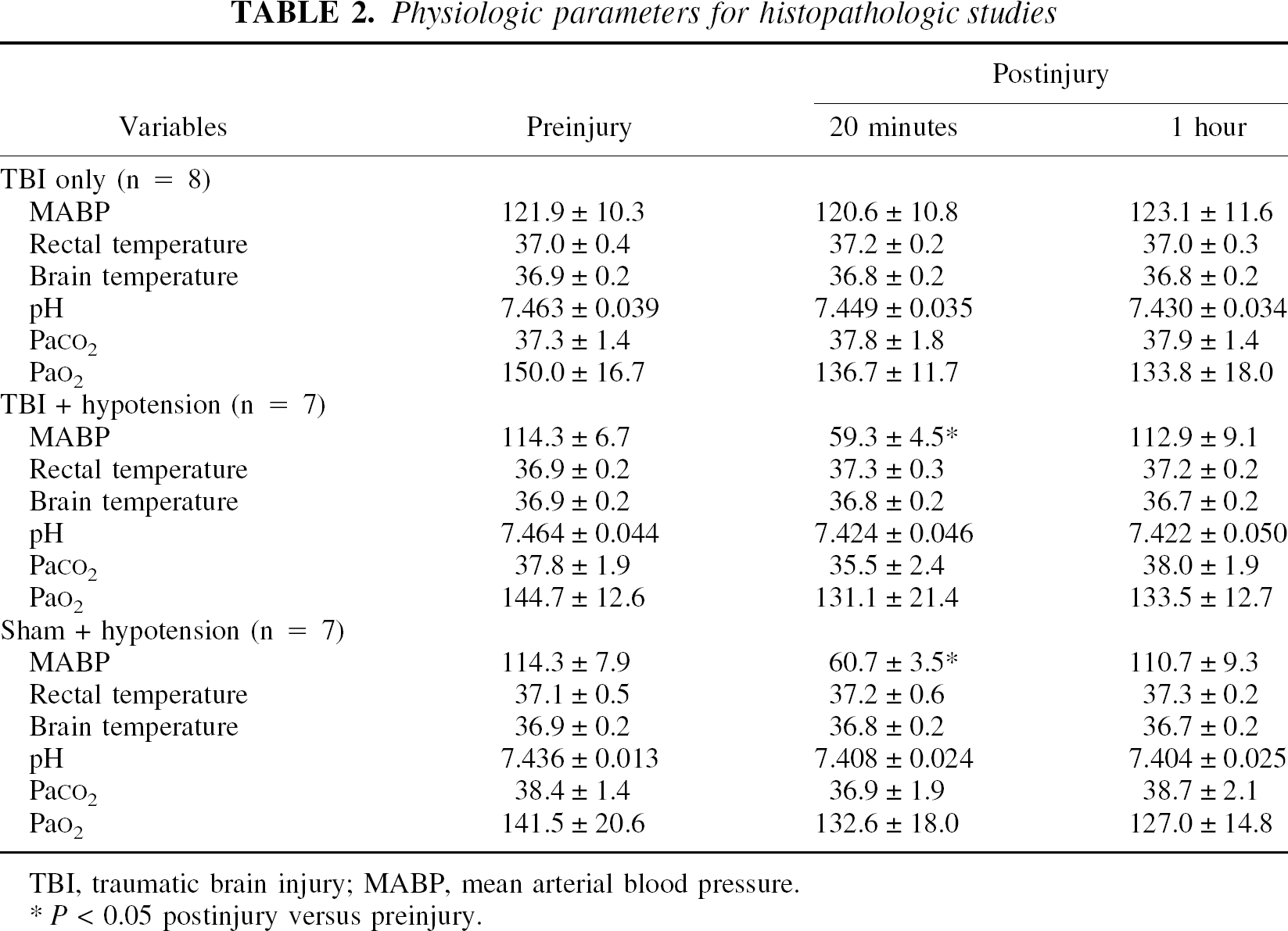
TBI, traumatic brain injury; MABP, mean arterial blood pressure.
P< 0.05 postinjury versus preinjury.
Regional cerebral blood flow
There were no significant differences in rCBF among groups at baseline and no significant changes over time in normotensive controls. Twenty minutes after normotensive TBI, blood flow was significantly reduced in total forebrain (58% ± 12% of baseline, P < 0.01) and ipsilateral (50% ± 15%, P < 0.01) and contralateral cerebral hemispheres (65% ± 17%, P < 0.05; Fig. 1A to 1C). Hypotension further reduced blood flow 20 minutes after TBI in total forebrain (36% ± 17%, P < 0.03) and contralateral (39% ± 22%, P < 0.05), but not ipsilateral, cerebral hemispheres (33% ± 15%, P < 0.07), although there was a trend for the latter also to decrease with hypotension. Sixty minutes after TBI, blood flow was not significantly different from control in total forebrain and the contralateral cerebral hemisphere; however, in the ipsilateral hemisphere, blood flow remained less than control in both TBI groups (Fig. 1).
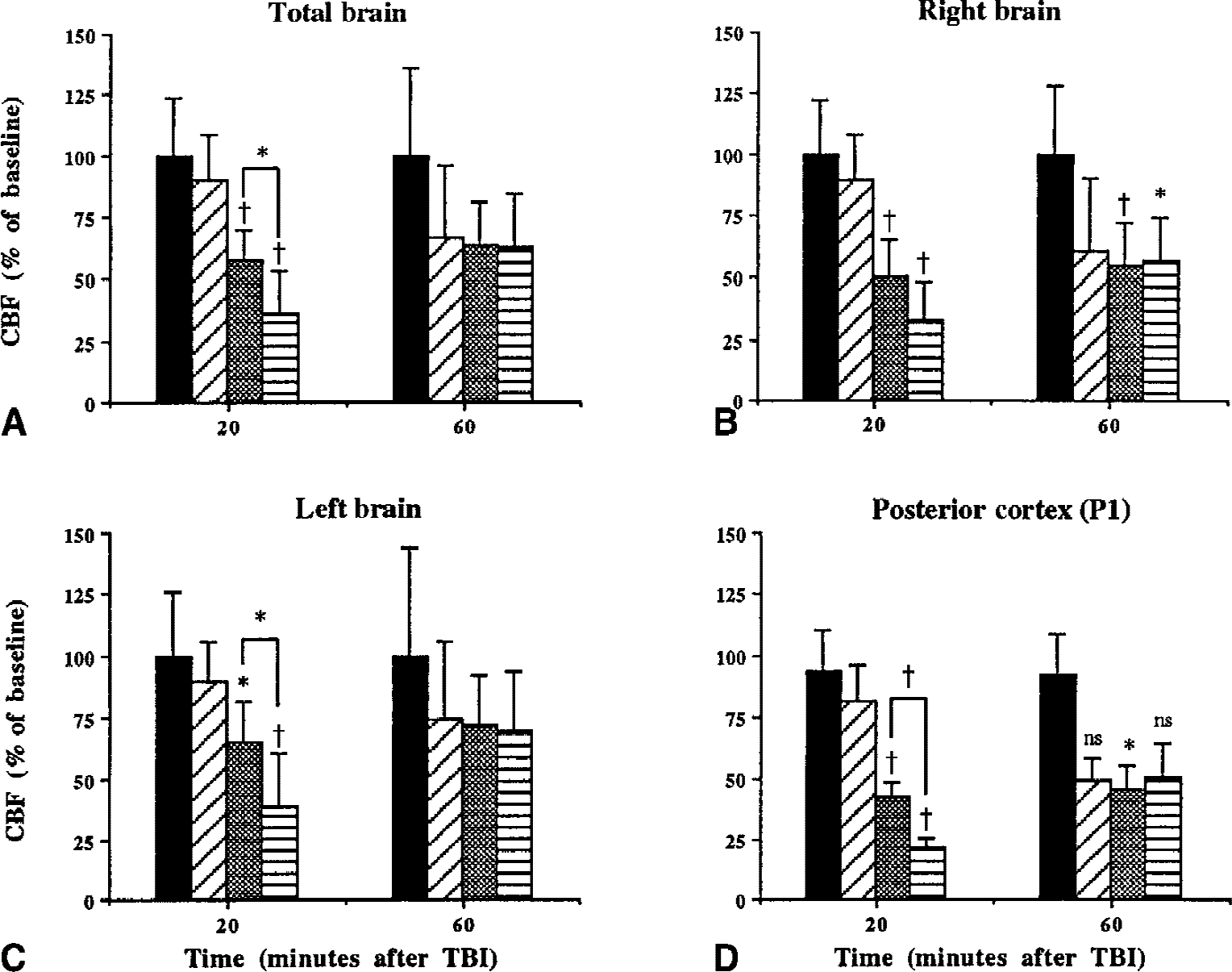
Values for cerebral blood flow (CBF) of total brain
Within the histopathologically vulnerable parietal cortex (P1), induced hypotension significantly reduced rCBF 20 minutes after TBI compared with normotensive TBI values (21% ± 10% vs. 43% ± 15% of control, P < 0.01; Fig. 1D). Sixty minutes after TBI, rCBF remained less than control in normotensive TBI rats (46 ± 23, P < 0.05) but only tended to be less in hypotensive TBI rats (51% ± 34%, P = 0.09). Analysis of rCBF in other regions revealed that 20 minutes after TBI, the hypotensive group had 7 of 8 regions with rCBF less than control, whereas in the normotensive group, only 5 of 8 regions were significantly reduced (Table 3). Sixty minutes after normotensive TBI, rCBF remained less than control primarily in ipsilateral regions, whereas after reinfusion of shed blood after hypotensive TBI, rCBF was less than control in only the parietal cortex anterior to the impact (Table 3).
Regional CBF values as % of baseline (mean ± SD) 20 and 60 minutes after TBI
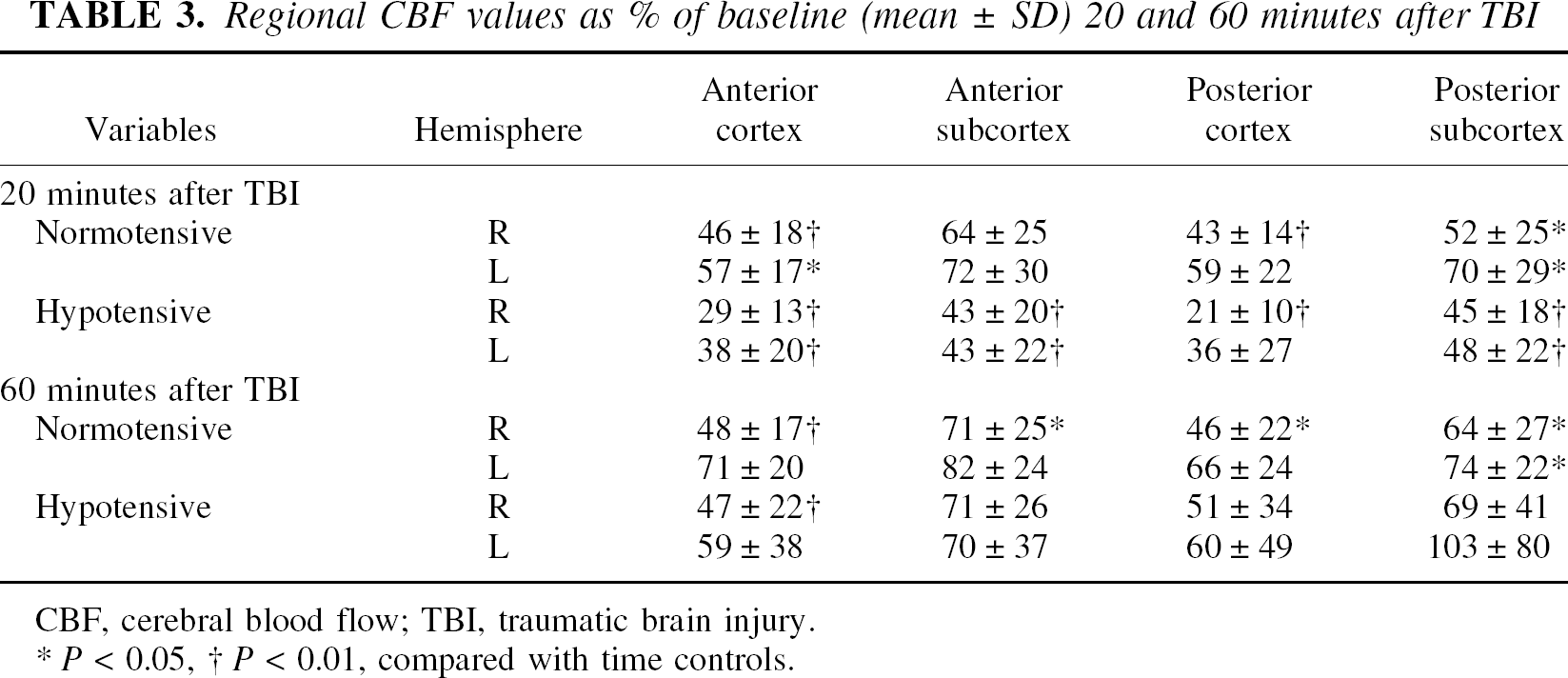
CBF, cerebral blood flow; TBI, traumatic brain injury.
P < 0.05
P< 0.01, compared with time controls.
Histopathologic outcome
Group 7 (sham + hypotension) did not exhibit any type of cortical or subcortical damage. As seen in Fig. 2, both group 5 and 6 animals contained a well-demarcated intracerebral contusion overlying the right external capsule (Fig. 2A and 2B). However, group 6 animals showed more widespread cortical damage and a larger contusion compared with group 5. Overlying the contusion, a high frequency of necrotic cortical neurons were seen in the lateral cerebral cortex (Fig. 2C to 2F). Also, FP hypotensive rats showed more extensive cortical neuronal damage compared with normotensive rats. With hypotension, TBI rats demonstrated large numbers of shrunken eosinophilic cortical neurons in an edematous-appearing neuropil (Fig. 2F).
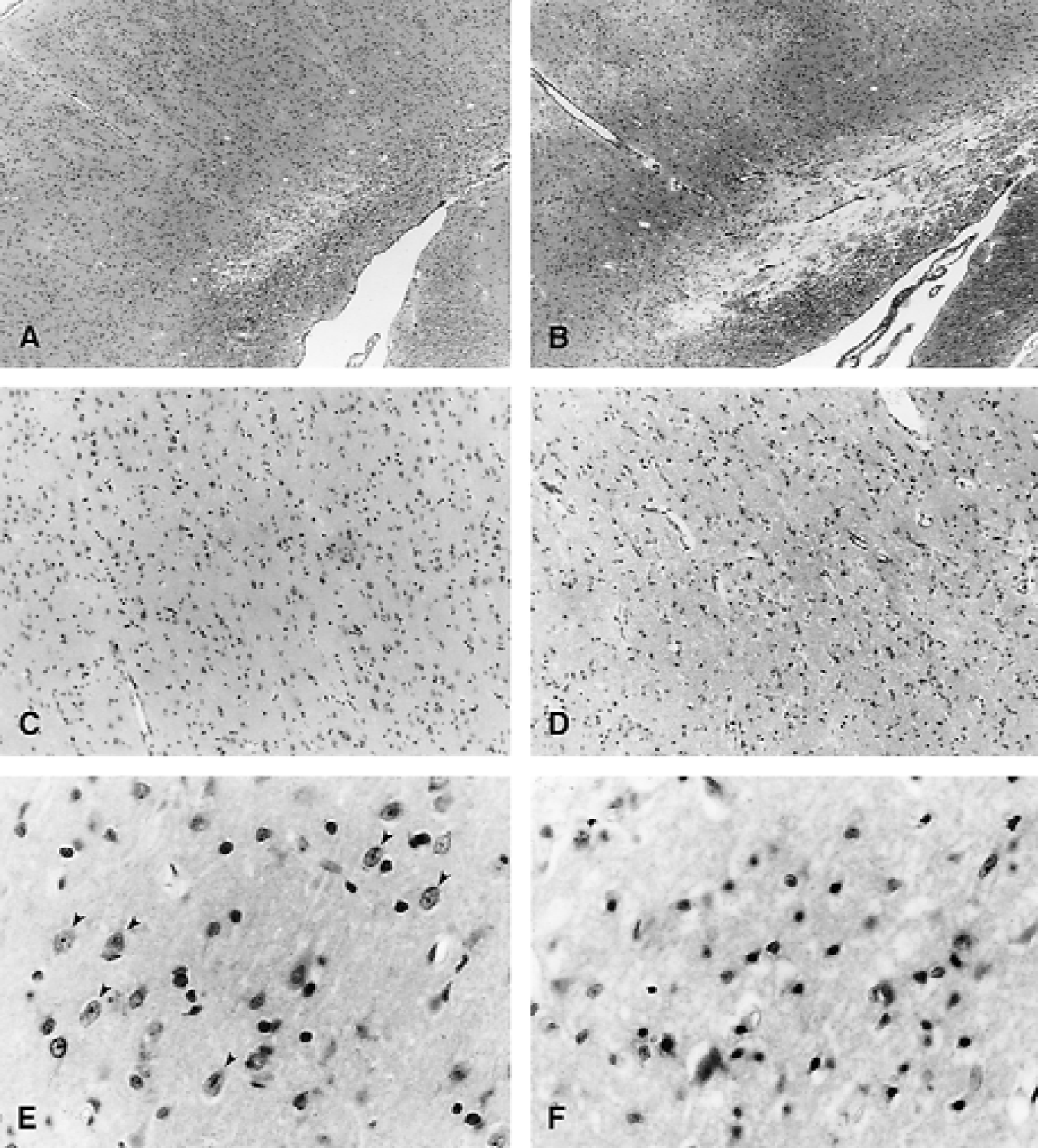
Photomicrographs displaying paraffin-embedded sections of rat brains three days after fluid-percussion (FP) injury. Hematoxylin and eosin staining.
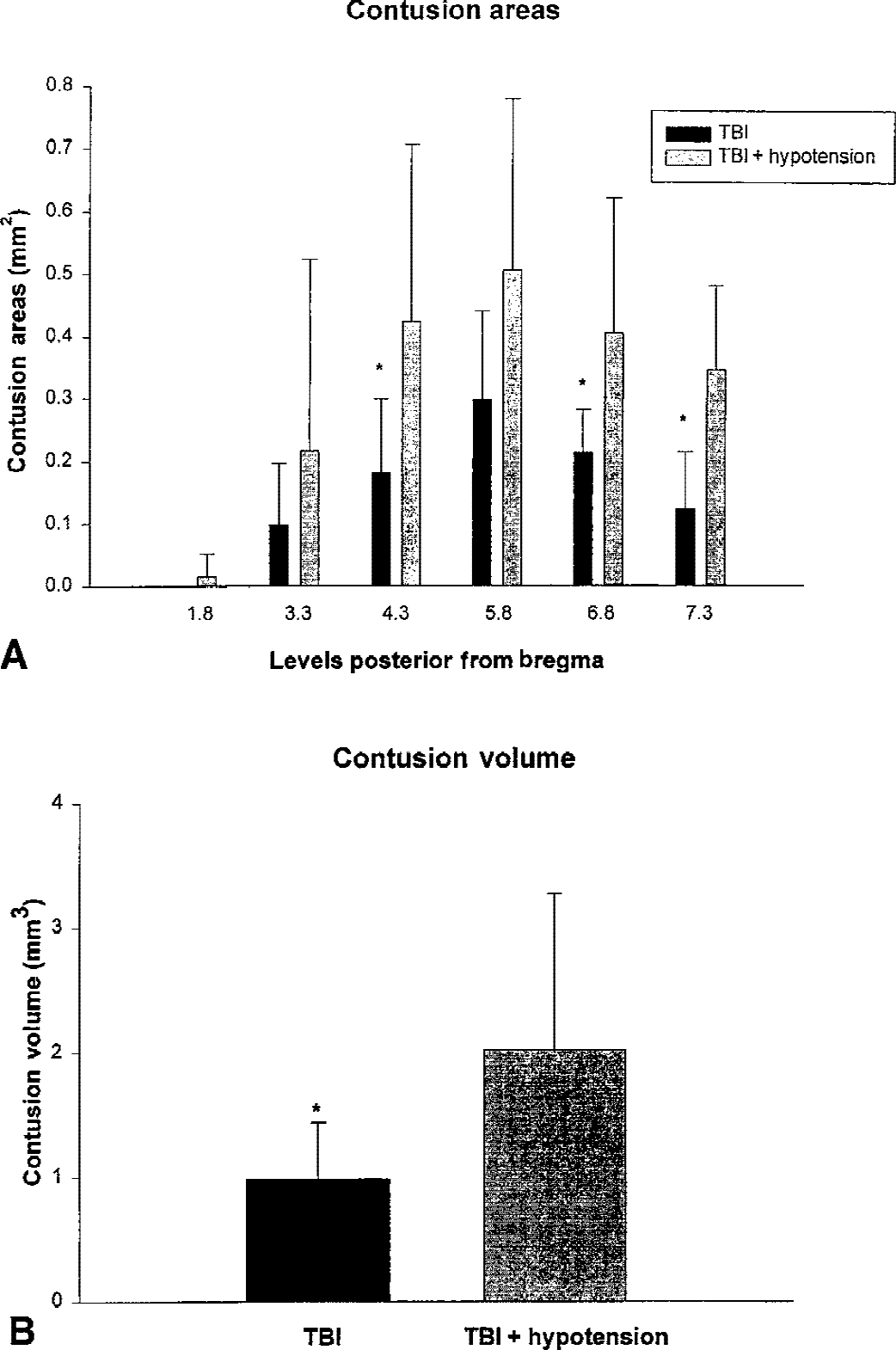
Figure 3A shows the contusion areas at the different bregma levels in normotensive and hypotensive rats. Significant differences in contusion areas were seen at several bregma levels. Figure 3B shows overall contusion volumes. There was a significant difference in contusion volume between group 5 and 6 (0.96 ± 0.46 mm3 vs. 2.02 ± 1.25 mm3, P < 0.05).
DISCUSSION
The current study demonstrated that a mild hypotensive event after moderate FP injury significantly affects the hemodynamic and histopathologic consequences of this traumatic insult. Although previous studies have reported the effects of mild hypotension on rCBF after TBI (DeWitt et al., 1992a, 1992b; Armstead, 1999; Kroppeenstedt et al., 1999; Lewelt et al., 1980; Schmoker et al., 1992), limited data exists regarding histopathologic correlates of posttraumatic hypotension. In the current study, contusion areas and volumes were significantly increased in hypotensive FP rats compared with normothermic animals. Increased structural damage was associated with more severe hypotension-induced rCBF changes that reached ischemic levels. The current data support previous findings and extend this investigative field by showing aggravated histopathologic outcome with induced hypotension. Posttraumatic blood pressure remains an important physiologic variable in experimental and clinical TBI studies.
Measurement of rCBF revealed that normotensive TBI caused widespread reductions in rCBF in the cerebral cortex and subcortex bilaterally 20 minutes after injury. During induced hypotension, the number of regions with significant reductions in rCBF increased, and in the cerebral cortex, a further decrease in rCBF occurred within the histopathologically vulnerable PI region. These findings demonstrate the well-known loss of autoregulation after TBI and suggest that secondary ischemic injury may have caused the enhancement of the histopathologic injury seen in the hypotensive TBI group (Armstead, 1999; DeWitt et al., 1992b; Golding et al., 1999). The return of rCBF to values not different from control after reinfusion of shed blood and restoration of normal blood pressure proves that with greater perfusion pressure rCBF can be increased in the traumatized brain, and that persistent depression of rCBF did not occur after a brief period of hypotension.
The authors have previously reported that a 30-minute period of posttraumatic hypoxia exacerbates cortical and hippocampal damage after moderate FP brain injury (Bramlett et al., 1999). However, when animals were made hypoxic after TBI, mild hypotension occurred during the hypoxic period. Although hypotension is often associated with an hypoxic insult (Chesnut et al., 1993), the question of whether decreased O2 or moderate hypotension was primarily responsible for the exacerbation of brain damage in that study remains unclear. The current results indicate that the hypotensive component of the hypoxic insult most likely had a significant impact on tissue injury. However, because the histopathologic consequences were more severe with secondary hypoxia compared with hypotension alone (Bramlett et al., 1999), the authors conclude that a transient period of reduced Po2 alone has a detrimental effect on traumatic outcome.
Kroppenstedt and colleagues (1999) reported that a reduction of MABP to 70 mm Hg significantly increased contusion volumes after cortical impact injury. Interestingly, the range of mean arterial pressure with no enlargement of contusion volume was between 70 and 105 mm Hg. Furthermore, a brief period of 30-minute hypotension was enough to cause a significant increase in contusion volume. Other studies have reported the effects of hypotension after TBI in a weight drop model, cryogenic model, and FP model (DeWitt et al., 1992a, 1992b; Giri et al., 2000; Golding et al., 1999; Lewelt et al., 1980). In each case, hypotension appeared to adversely affect the hemodynamic, electrophysiologic, or metabolic state of the traumatized brain. Because MABP of 60 mm Hg is apparently an important threshold for secondary injuries in rats, it may be important in future studies directed at mechanistic issues to also investigate more severe degrees of hypotension.
As previously reported, moderate parasagittal FP injury produces a well-demarcated contusion, primarily overlying the lateral external capsule, with damaged cortical neurons within the overlying parietal cortex (Bramlett et al., 1999; Dietrich et al, 1994, 1996b). This relatively focal lesion is a consequence of mechanically disrupted blood vessels because of shearing stress caused by FP injury (Cortez et al., 1989; Dietrich et al., 1994). Blood–brain barrier breakdown and subsequent reductions in local CBF (lCBF) occur in the damaged region (Cortez et al., 1989; Dietrich et al., 1994, 1996a; Yamakami and McIntosh, 1991). In the current study, induced hypotension increased overall contusion volume in the lateral parietal cortex. Because posttraumatic hypotension also aggravated the hemodynamic consequences of this specific brain region, vascular mechanisms most likely participated in the observed outcome.
In this regard, Barzo et al. (1996) reported that posttraumatic secondary insults after closed head injury prolonged the time of blood–brain barrier breakdown. Perturbations in blood–brain barrier function possibly leading to increased brain edema (Baldwin et al., 1996), coupled with induced hypotension, might lead to more severe hemodynamic disturbances. Such a consequence would be expected to cause additional brain damage, possibly by ischemic mechanisms. In this study, induced hypotension led to rCBF values less than 18 mL 100 g−1 min−1, which are considered ischemic in rodents (Bolander et al., 1989; Tyson et al., 1984). However, because of the complex metabolic consequences of posttraumatic brain, ischemic thresholds in normal brain may not directly apply to the posttraumatic brain (Giri et al., 2000). The regional measurement of adenosine triphosphate levels and other energy metabolites would also provide important information regarding the metabolic status of the posttraumatic brain after secondary hypotensive insult (Ishige et al., 1988).
Previous studies have demonstrated that metabolic rates are initially increased after TBI, with decreased rates observed at later study periods (Yoshino et al., 1991). Clinical and experimental data also have indicated that flow–metabolic uncoupling can occur in vulnerable brain regions after TBI and may complicate the interpretation of the significance of posttraumatic hemodynamic alterations (Ginsberg et al., 1997; Jaggi et al., 1990). In this regard, studies in which measurements of cerebral oxygenation have been undertaken have provided important data about the relative adequacy of CBF after brain injury. Cryogenic injury combined with reduction of mean arterial blood pressure of 50 mm Hg/45 minutes was reported to significantly decrease cerebral oxygen delivery and cerebral metabolic rate of oxygen (Lewelt et al., 1980). In a recent study, Giri and colleagues (2000) reported, using a cortical impact injury model, that brain tissue Po2 and rCBF decreased to low levels in the posttraumatic brain during a secondary carotid occlusion insult; thus, reduced CBF in that study was inadequate to prevent oxygen depletion. Similar studies are required in the current FP model to determine whether metabolic requirements are significantly influenced under induced hypotensive periods.
Mechanisms underlying the autoregulatory dysfunction after TBI require clarification (Golding et al., 1999). Traumatic brain injury is known to produce a marked increase in vasoactive substances, including prostaglandins (Armstead, 1998; DeWitt et al., 1997; Ellis et al., 1981) and oxygen free radicals (Kontos and Wei, 1986), that may affect rCBF and vascular reactivity. Several studies have demonstrated that pial arteries and arterioles do not exhibit normal vasodilatory responses to acetylcholine and serotonin after TBI (Kontos and Wei, 1992). Armstead (1999) measured marked elevations in cerebrospinal fluid concentrations of endothelin-1 after TBI in piglets and reported that pretreatment with an endothelin-1 antagonist partially restored pial artery dilation during hypotension. DeWitt and colleagues (1997) reported that treatment with superoxide dismutase and L-arginine prevented or reversed posttraumatic hypoperfusion after midline FP in rats. Thus, early treatment with agents that target free radical generation, nitric oxide production, or vasoactive substances may promote protection from secondary insults.
In summary, posttraumatic hypotension after FP injury significantly worsens histopathologic outcome in an established FP model of TBI. It is concluded that hypoxia-induced hypotension may have contributed to previous results regarding the detrimental consequences of posttraumatic hypoxia after TBI. However, because hypotension appears to have a milder histopathologic effect on outcome compared with hypoxia, critical levels of O2 delivery after TBI also appear to be a critical factor in secondary injury mechanisms. Strategies to prevent or reverse autoregulatory abnormalities and enhance normal O2 delivery to posttraumatic tissues offers a reasonable approach to preventing acute as well as progressive damage after TBI.
Footnotes
Acknowledgments:
The authors gratefully acknowledge the superb technical assistance of Susan Kraydieh and Charlaine Rowlette.