
Abstract
Transient middle cerebral artery (MCA) occlusion results in substantially smaller cortical infarcts than permanent MCA occlusion if reperfusion is initiated within the first few hours. Only little information is available on the long-term functional outcome of the cortical regions “salvaged” by early reperfusion. To address this issue we examined basic electrophysiologic parameters in vitro using standard extracellular recording techniques at 7 and 28 days after transient MCA occlusion (1- and 2-hour ischemia) in rats. Both neocortical areas ipsi- and contralateral to MCA occlusion were systematically mapped to delineate the extent of periinfarct and remote alterations. In the periinfarct region we found a significant reduction of field potential amplitudes up to 3 mm when measuring from the infarct border at 7 days and up to 7 mm at 28 days. Paired-pulse inhibition, an indicator of GABAergic transmission, was only moderately impaired in this region at 7 days and not significantly different from control at 28 days. Remote effects were observed both ipsi- and contralaterally. Ipsilaterally they were restricted to a region close to the midline (presumably motor cortex) and were most likely attributable to the degeneration of corticostriatal connections. The extent of the contralateral excitability changes was clearly related to the size of the neocortical infarcts with large infarcts resulting in the widespread reduction of field potential amplitudes and an impairment of paired-pulse inhibition. The results show that there is a relatively large periinfarct region with decreased overall excitability after transient MCA occlusion which is likely to have a profound effect on perilesional processes involved in functional recovery. Remote excitability changes may contribute to the functional deficit and are probably related to deafferentation.
Keywords
Over the past few years thrombolysis with recombinant tissue-plasminogen activator (rt-PA) has become the treatment of choice for stroke patients presenting within 3 hours of symptom onset (Hacke et al., 1995; National Institute of Neurological Disorders and Stroke rt-PA Stroke Study Group, 1995; Lyden et al., 1997). The aim of thrombolysis is to induce reperfusion at an early stage of stroke evolution before infarct size has become definite. This strategy prevents the integration of critically ischemic (but still viable) brain regions, i.e., the penumbra (Astrup et al., 1981; Hossmann, 1994; Obrenovitch, 1995), into the irreversibly damaged ischemic core. The acute mechanisms involved in infarct growth (unless reperfusion occurs) such as periinfarct depolarizations (Hossmann, 1996), disturbances of calcium ion homeostasis and release of excitatory amino acids (Siesjö and Bengtsson, 1989), inhibition of protein synthesis (Mies et al., 1991) and apoptosis (Li et al., 1995; Charriaut-Marlangue et al., 1996), as well as the mechanisms involved in reperfusion injury (Mori et al., 1992; del Zoppo et al., 1998) have been investigated in detail.
Comparatively little, however, is known about the long-term functional outcome of the regions “salvaged” by early thrombolysis-induced or spontaneous reperfusion, i.e., those regions next to the infarcts which remain intact (or at least almost intact). Most studies investigating functional disturbances (in particular excitability changes) after different types of brain injury focused either on the acute and chronic changes after global ischemia or, when using focal ischemia models, on the alterations in tissue that was not ischemic at the time of the insult (for review see Luhmann, 1996; Witte and Stoll, 1997). Chronic excitability changes observed in focal ischemia models or lesion studies were generally associated with a reduced level of intracortical GABAergic inhibition and an increase in NMDA receptor-mediated responses (Schiene et al., 1996; Buchkremer-Ratzmann et al., 1998; Neumann-Haefelin et al., 1998; Qu et al., 1998b). Taken together these studies provide unequivocal evidence that excitability changes can occur in primarily unaffected tissue after different types of cortical injury, but the changes in the transiently ischemic periinfarct region have not been investigated in detail.
The aim of the present study was, therefore, (1) to delineate the extent of the functional changes in the periinfarct region using a model (transient middle cerebral artery (MCA) occlusion) with a large ischemic border zone and (2) to investigate the secondary effects in brain regions remote from the infarct. To this purpose we used standard extracellular in vitro recording techniques which are well suited to systematically map field potential amplitudes (as a measure of the general excitability) and paired-pulse inhibition (as an estimate of GABAergic intracortical inhibition).
MATERIAL AND METHODS
Experimental procedure
A total of 29 adult male Wistar SPF strain rats (weighing 270 to 330 g) were used for the experiments. Both the animals in the experimental group (n = 20) and the controls (n = 9) were kept on a 12-hour light cycle. All experimental procedures were conducted according to protocols approved by the Governmental Animal Care Committee.
The animals in the experimental group (n = 20) were subjected to transient MCA occlusion using the intraluminal thread model originally described by Koizumi et al. (1986) with only minor modifications. Briefly, animals were spontaneously breathing, and anesthesia was induced with enflurane (1.5%) in a mixture of N2O:O2 (70%:30%). Body temperature was kept constant at 36.5 ± 0.5°C using a heating pad. The right cervical carotid bifurcation was exposed, and both the proximal common carotid artery (CCA) as well as the external carotid artery (before the origin of the occipital artery) were ligated. A 4-0 silicone-coated filament was introduced into the distal common carotid artery and advanced into the internal carotid artery between 16 and 17.5 mm (as measured from the carotid bifurcation) until a faint resistance was felt. The filament was secured in this position using a tight ligature around the CCA. Anesthesia was then discontinued, and animals were allowed to recover. The sham procedure consisted of placing sutures around the right CCA and external carotid artery without tightening them.
After occlusion times of 1 (n = 17) or 2 (n = 3) hours the intraluminal thread was removed, and the CCA was permanently occluded distal to the insertion site. Reperfusion was not directly assessed, but arterial backflow through the internal carotid artery (as evident just before tightening the ligature around the distal CCA) occurred in all animals, indicating that no major clots had formed at the tip of the filament which might have resulted in permanent MCA occlusion. Immediately before removal of the intraluminal thread the neurologic status of the animals was assessed using the score introduced by Bederson (Bederson et al., 1986).
In vitro electrophysiology
The electrophysiologic recordings were performed using coronal neocortical slices (400 μm) obtained from experimental animals after survival times of 7 days (n = 13; number of slices = 37) and 28 days (n = 3; number of slices = 8) as well as from controls (n = 9; number of slices = 16). For the preparation of the slices, the brains were removed under deep enflurane anesthesia (≈2 minutes) and rapidly cooled in artificial cerebrospinal fluid (composition in mmol/L: NaCl 124, NaHCO3 26, KCl 3, CaCl2 2, MgSO4 2, NaHPO4 1.25, and glucose 10 equilibrated with carbogen to pH 7.4; temperature ≈4°C) for 90 to 120 seconds before sectioning on a vibratome (Leica VT 1000). The slices were collected at positions roughly corresponding to Bregma 0 mm to Bregma −2.8 mm and were maintained in an interface recording chamber at 33°C superfused with artificial cerebrospinal fluid equilibrated with carbogen to pH 7.4. In total, preparation of the slices required 10 to 15 minutes, but only the first ≈2 minutes were under normothermic conditions (see above). Slices were left undisturbed for at least 1 hour before recording.
Field excitatory postsynaptic potentials (fEPSP) were recorded using glass micropipettes (filled with artificial cerebrospinal fluid; tip size, ≈1 μm; impedance, ≈2 to 3.5 MΩ) positioned in layers II/III. The bipolar stimulation electrode was placed in layer VI underneath the recording electrode, on the same radial trajectory perpendicular to the cortical surface as the recording electrode. A double-pulse stimulation protocol (pulses of 50-microsecond duration; interval: 20 milliseconds; stimulation strength: 10 to 60 V) was used to investigate both the general excitability (amplitude of the first fEPSP) and paired-pulse-inhibition (ratio: fEPSP2 to fEPSP1) (Domann et al., 1993; Buchkremer-Ratzmann et al., 1998). To map the responses across the slices, both the stimulation and extracellular recording electrodes, positioned in layers II/III and VI, respectively, were moved in parallel, and recordings were serially obtained between 2 and 10 mm lateral to the midline at intervals of 1 mm.
Assessment of infarct size
Both cortical and striatal infarcts were generally easily recognizable when looking at the slices in the recording chamber. After the electrophysiologic recordings all experimental slices were Nissl-stained for documentation purposes after sectioning the slices on a cryostat (thickness of sections: 20 μm). Areas judged as being macroscopically infarcted during the electrophysiologic recordings were consistently found to correspond to necrotic regions histologically.
Data analysis
For each recording position fEPSP1 amplitudes and fEPSP2 to fEPSP1 ratios were determined for all stimulation strengths (10 to 60 V). For statistical analysis we used the largest fEPSP1 amplitudes and the smallest of the fEPSP2 to fEPSP1 ratios obtained at supramaximal stimulation strengths, i.e., those stimulation conditions yielding maximal fEPSP1 amplitudes (amplitudes of at least 95% of the maximal amplitude were considered maximal).
Statistical analysis was performed separately for the ipsi- and contralateral hemispheres. Ipsilaterally, we grouped the data (both fEPSP1 amplitudes and fEPSP2 to fEPSP1 ratios) by distance from lesion border rather than distance from midline as for the contralateral data. We chose this approach because the size of the neocortical infarcts was quite variable even in groups with identical MCA occlusion times; it allowed us to analyze the excitability changes with respect to the distance from the lesion border despite the variability in infarct sizes. In addition, we subdivided the experimental group into three broad subgroups based on the size of the infarcts (no cortical infarct, moderate, and large infarcts; see first paragraph of Results section for details) because an initial data exploration suggested differences between animals with different infarct sizes. To test for differences between the groups we first performed a one-way analysis of variance (ANOVA) for each recording position followed by Dunnett's procedure for multiple post hoc comparisons against a single control group (if ANOVA resulted in a significant F test). Data are given as mean ± standard deviation unless stated otherwise. A P value of 0.05 was considered significant.
RESULTS
Infarct assessment and experimental subgroups
Most animals in the experimental subgroup with 1-hour MCA occlusion and 7-day survival (n = 13) had neocortical infarcts of moderate size (n = 7). Infarct sizes were arbitrarily defined as moderate if there was neocortical infarction sparing more than 4 mm of tissue when measuring from the midline. A more sophisticated analysis of infarct sizes was unfortunately not possible because basal brain structures had to be removed to facilitate slice preparation thereby excluding all methods using a complete hemisphere as reference. In three animals (n = 3) there was no macroscopic evidence of neocortical infarction, but striatal infarcts were clearly visible both in the electrophysiologic recording chamber and on Nissl stains. One animal had a large infarct (less than 4 mm of intact tissue); this animal was lumped together with the animals of the experimental subgroup with 2-hour MCA occlusion and 7-day survival (n = 3), all of which had large infarcts. All animals in the last subgroup (1-hour MCA occlusion, 28-day survival; n = 3) had neocortical infarcts of moderate size. Three animals died before day 7. All animals had a Bederson score of ≥2 before reperfusion, i.e., decreased resistance to lateral push (grade 2) or circling (grade 3).
Excitability changes ipsilateral to middle cerebral artery occlusion
Figure 1 depicts the changes ipsilateral to MCA occlusion in animals with neocortical infarcts of moderate size at the 7th day of survival, and Table 1 summarizes the results obtained for all subgroups. At 7 days after 1 hour of MCA occlusion fEPSP1 amplitudes were significantly reduced in the periinfarct region (up to 3 mm when measuring from the lesion border). In the remaining ipsilateral cortex as well as at all positions in the subgroup without cortical infarcts (group A in Table 1) fEPSP1 amplitudes were not significantly changed. This periinfarct reduction of fEPSP1 amplitudes was still present at 28 days and extended even further from the lesion border.
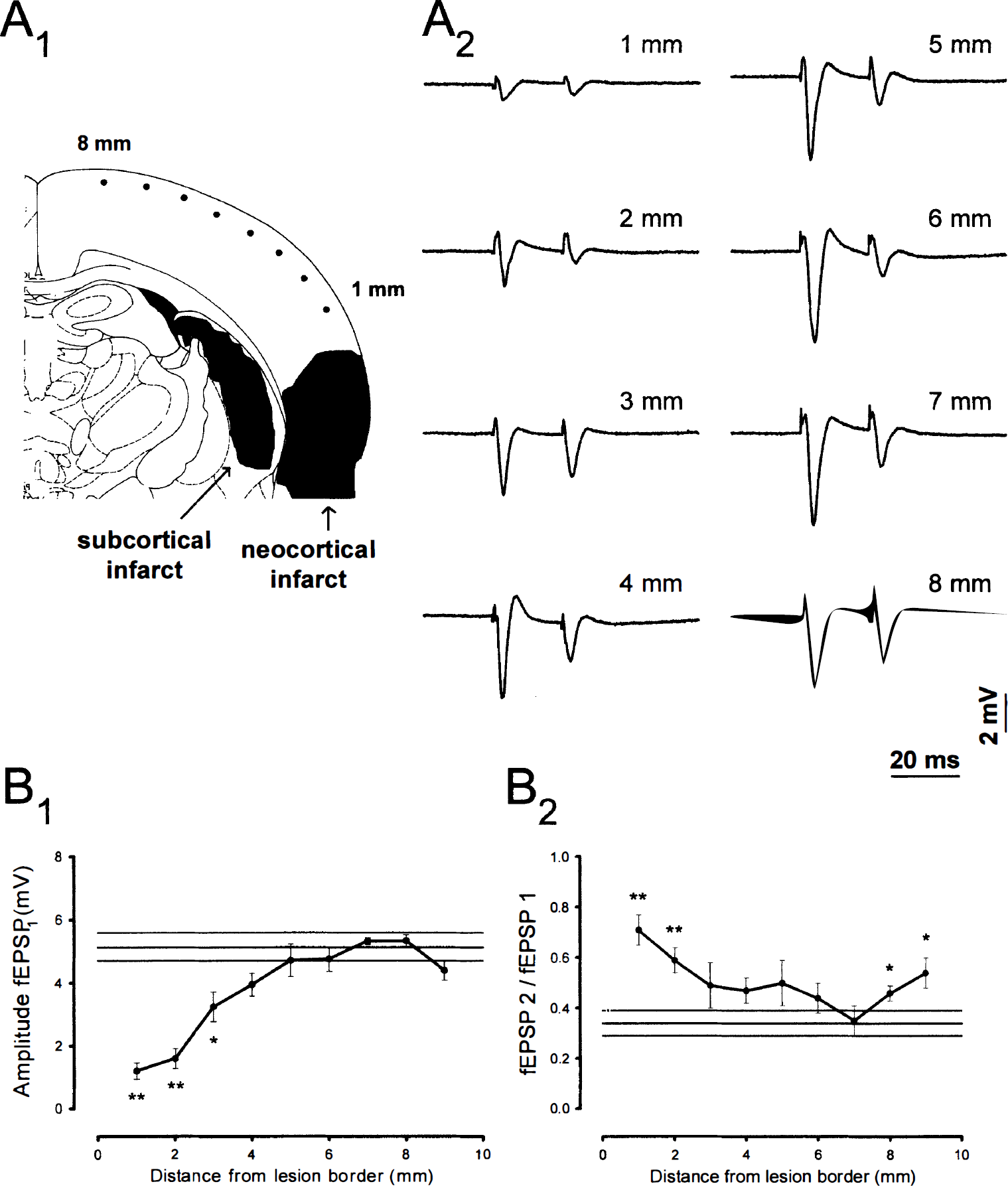
Ipsilateral excitability changes 7 days after 1-hour MCA occlusion.
Ipsilateral excitability changes
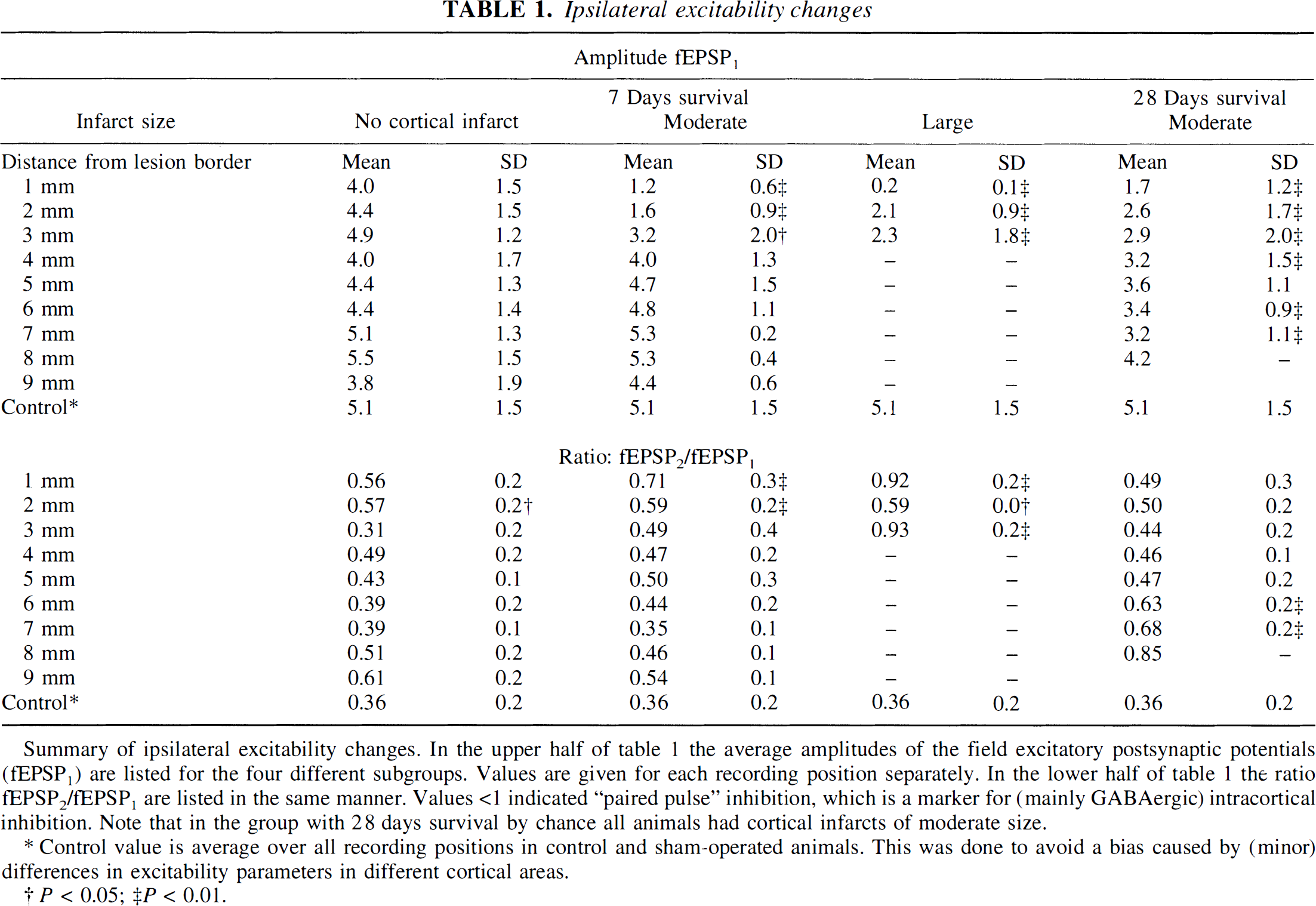
Summary of ipsilateral excitability changes. In the upper half of Table 1 the average amplitudes of the field excitatory postsynaptic potentials (fEPSP1) are listed for the four different subgroups. Values are given for each recording position separately. In the lower half of Table 1 the ratio fEPSP2/fEPSP1 are listed in the same manner. Values <1 indicated “paired pulse” inhibition, which is a marker for (mainly GABAergic) intracortical inhibition. Note that in the group with 28 days survival by chance all animals had cortical infarcts of moderate size.
Control value is average over all recording positions in control and sham-operated animals. This was done to avoid a bias caused by (minor) differences in excitability parameters in different cortical areas.
P < 0.05; ‡P < 0.01.
The fEPSP2 to fEPSP1 ratio was significantly greater in the periinfarct region (up to 2 mm from the lesion border) than in controls at 7 days (group B and C in Table 1), but this effect was only observed after 7 days of survival. At 28 days the periinfarct increase of the fEPSP2 to fEPSP1 ratio was no longer significant, but now a remote area (close to the midline) was significantly different from controls.
Excitability changes contralateral to middle cerebral artery occlusion
The contralateral data are summarized in Table 2. Contralateral to MCA occlusion fEPSP1 amplitudes were statistically different from those of controls at only one position in the group with neocortical infarcts of moderate size (8 mm from midline) and completely normal in the group without neocortical infarcts at 7 days. Amplitudes of fEPSP1 were, however, significantly reduced at 6 of 9 positions in animals with large neocortical infarcts (Fig. 2). The fEPSP2 to fEPSP1 ratio was not significantly changed contralateral to MCA occlusion in the group without neocortical infarcts, was significantly changed at 9 of 10 positions in the group with large neocortical infarcts, and was changed at 3 of 9 positions (6, 7, and 8 mm from midline) in the group with neocortical infarcts of moderate size.
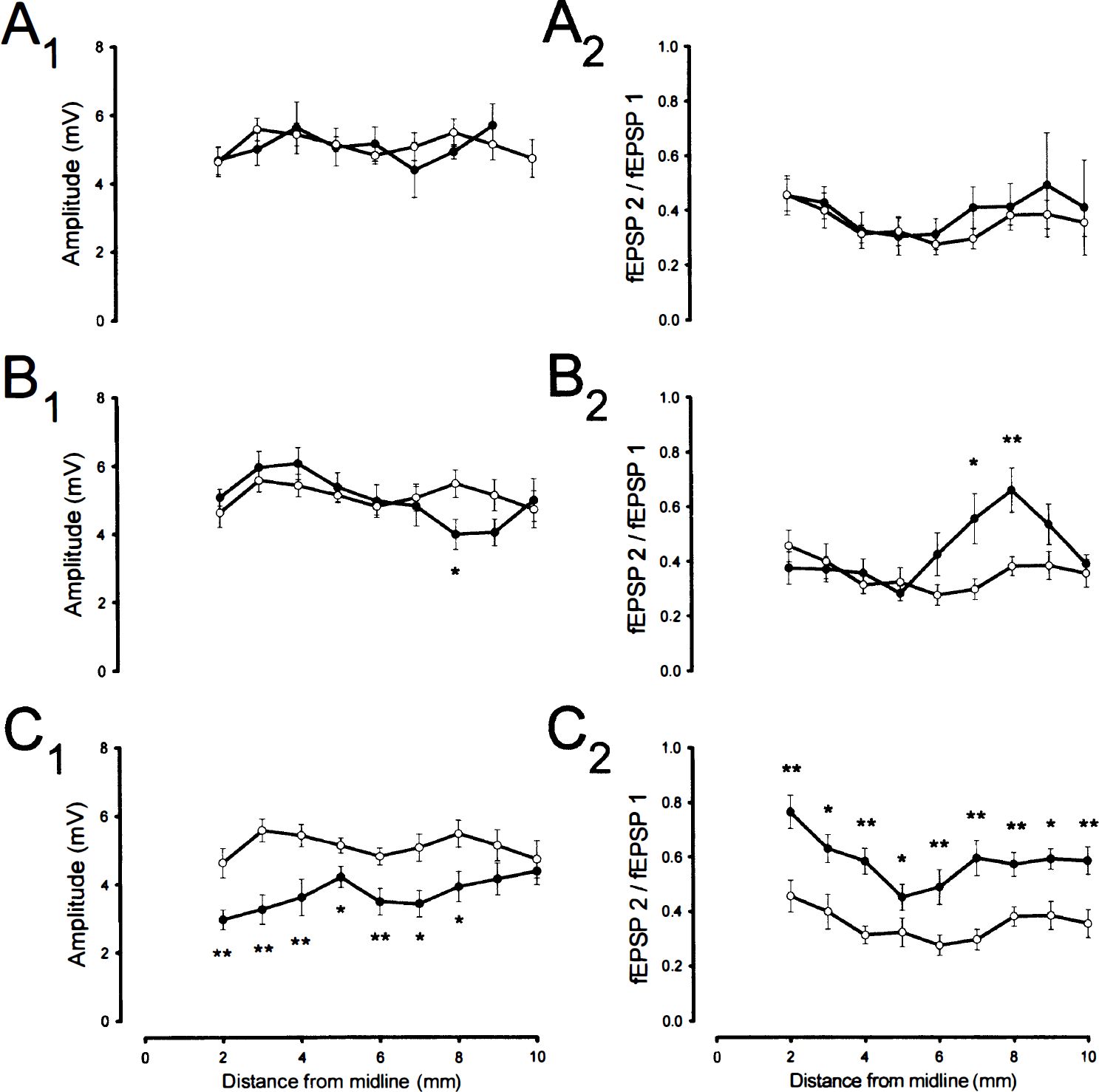
Contralateral excitability changes 7 days after 1-hour MCA occlusion in animals with no cortical infarcts
Contralateral excitability changes
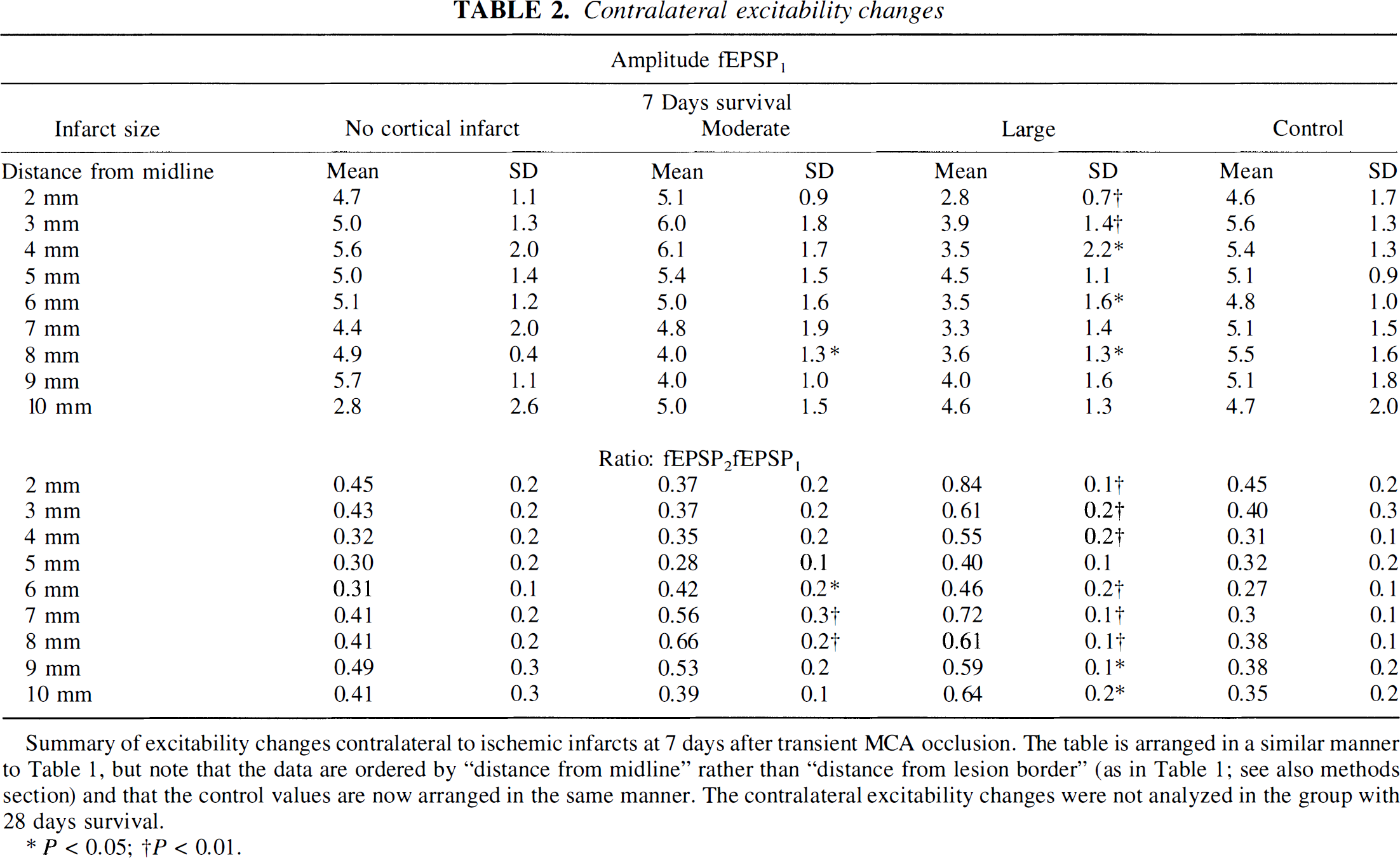
Summary of excitability changes contralateral to ischemic infarcts at 7 days after transient MCA occlusion. The table is arranged in a similar manner to Table 1, but note that the data are ordered by “distance from midline” rather than “distance from lesion border” (as in Table 1; see also methods section) and that the control values are now arranged in the same manner. The contralateral excitability changes were not analyzed in the group with 28 days survival.
P < 0.05;
P < 0.01.
DISCUSSION
With our in vitro investigation we provide evidence for two types of excitability changes in macroscopically intact tissue after transient MCA occlusion: (1) changes in the periinfarct region and (2) remote changes both ipsi- and contralateral to the side of the infarct. These changes were observed 7 days after MCA occlusion and persisted (at least partially) until day 28.
In the periinfarct region we found a very marked reduction of field potential amplitudes indicating a reduction of overall excitability in this region. In addition, there was a relatively moderate increase in the ratio of fEPSP2 to fEPSP1 amplitudes, i.e., a decrease of paired-pulse inhibition compared to that of controls. Impairment of cortical paired-pulse inhibition is a sensitive (indirect) marker of impaired GABAA receptor-mediated inhibitory mechanisms (Domann et al., 1993) and has been shown to correlate well with GABAA receptor autoradiography (Qu et al., 1998a). These long-term changes in the periinfarct region are most likely attributable to severe ischemia during MCA occlusion with early reperfusion preventing irreversible tissue damage.
Theoretically, the decrease in overall excitability in the periinfarct region may be either the result of processes leading to a reduction of viable neurons or to a decreased excitability of individual neurons (or a combination of both). Selective neuronal necrosis is well known to occur in the periinfarct region (Mies et al., 1983; Nedergaard, 1988; Garcia et al., 1996) and may obviously lead to a reduction in the number of neurons. Other processes known to occur in the periinfarct region such as inflammatory reactions (Becker, 1998) and gliosis (Garcia et al., 1997) could potentially affect the excitability of neurons. Finally, selective damage to subcellular neuronal elements such as dendrites (Bolay and Dalkara, 1998) could affect excitability as well. Unfortunately, in the present study we were unable to relate any of these alterations to the severity and the extent of the periinfarct excitability changes because the quality of the histologic sections obtained from the slices after in vitro recording was not sufficient to afford a detailed microscopic analysis of diffuse ischemic changes in the periinfarct region. One unlikely cause of the changes in the periinfarct region is periinfarct depolarizations because they usually affect the entire ipsilateral cortex, whereas the changes reported here were restricted to a rim of about 3 mm when measuring from the lesion border.
The remote effects can be subdivided into changes found ipsi- and contralaterally. Ipsilaterally, the remote effects were restricted to positions close to the midline, i.e., presumably (sensori-)motor cortex, and were only significant in one subgroup (1-hour MCA occlusion, 28-day survival). Possibly, this remote effect is secondary to the degeneration of corticostriatal connections or to pyramidal tract damage. The contralateral remote effects, on the other hand, were already found at 7 days, and they were clearly dependent on the size of the neocortical infarcts, with large infarcts resulting in more prominent alterations compared to smaller infarcts. Here degenerating transcallosal corticocortical connections are likely to be of greater importance than connections between cortex and subcortical structures; the extensive inter-hemispheric connections known to exist in rat cortex are easily compatible with this assumption. Additionally, other mechanisms (unrelated to connectivity) such as cerebral edema, free radical formation, or a general stress response may contribute to the remote excitability changes observed contralaterally.
Several previous studies investigated long-term excitability changes after other types of cortical injury including permanent MCA occlusion in mice (Mittmann et al., 1998), transient global ischemia (Urban et al., 1989; Luhmann et al., 1995) and small photothrombotic cortical infarcts in rats (Domann et al., 1993; Neumann-Haefelin et al., 1995; Buchkremer-Ratzmann et al., 1996), as well as cortical lesion models (Mittmann et al., 1994; Eysel, 1997). All studies reported an increase in excitability in the perilesional region. This contrasts with our findings of a reduced excitability in the periinfarct region. There are two likely explanations for this apparent difference: (1) Excitability was frequently assessed at the single neuron level whereas we investigated overall excitability by measuring field potentials; it is not unlikely that individual neurons would have shown signs of hyperexcitability (despite the reduction of overall excitability) if single-cell measurements had been performed. An overall reduction of excitablity despite hyperexcitability at the single neuron level would be expected when the reduction of neurons (e.g., attributable to selective neuronal cell death) outweighs the increase in excitability of single neurons. (2) The ischemia model used here is in many ways different from typical lesion models where there is no classical penumbra; when using the intraluminal thread model of transient MCA occlusion, however, there is typically a large penumbra which may be salvaged by reperfusion. Reperfusion was initiated relatively early in most animals investigated here; thus, a substantial proportion of the (at least macroscopically) intact periinfarct tissue, which showed the excitability changes, was probably severely ischemic at the time of MCA occlusion. Assuming that ischemia is (directly or indirectly) the cause of the changes in the periinfarct region, we would not expect to find a similar decrease of overall excitability in the vicinity of cortical lesions of other etiology.
A number of recent reports from our laboratory focused on the extent of remote excitability changes after relatively small photothrombotic infarcts (2-mm lesion diameter) affecting the medial somatosensory cortex of the rat (Buchkremer-Ratzmann et al., 1996; Witte and Stoll, 1997; Witte et al., 1997). Paired-pulse inhibition as well as other parameters were found to be altered in widespread regions ipsi- and contralateral to the photothrombotic infarct. When compared to the results presented here the extent of the changes affecting relatively widespread regions found with the photothrombotic model is surprising (particularly the extent of the contralateral changes); one possible explanation may be that the photothrombotic lesions were (by chance) located in strategically important regions connecting to multiple other cortical regions. An alternative explanation would be that the white matter tracts underneath the photothrombotic cortical infarcts had been damaged resulting in more extensive transcallosal deafferentation than explained by the cortical lesion alone. In a separate study we investigated the remote (i.e., contralateral) excitability changes after permanent MCA occlusion (Reinecke et al., 1998); all investigated animals had large infarcts and extensive contralateral impairment of paired-pulse inhibition, indicating that the excitability changes were not just a peculiarity of the photothrombotic infarction model.
At the present time the functional significance of both the decrease of overall excitability in the periinfarct region as well as the remote effects remains speculative. Recently, an in vivo study using recording techniques for motor- and somatosensory evoked potentials reported a persistent transmission failure of cortical synapses in the sensorimotor forelimb area of the rat, i.e., in the penumbra region, after transient MCA occlusion (Bolay and Dalkara, 1998). In combination with our results this is strong evidence for electrical dysfunction affecting the periinfarct region in models with (early) reperfusion both in vivo and in vitro. This dysfunction is likely to have a considerable influence on perilesional processes including perilesional plasticity typically found in classical lesion paradigms (Nudo et al., 1996; Witte, 1998). It is quite conceivable that processes such as long-term potentiation (Hagemann et al., 1998), synaptogenesis, and neurite growth (Stroemer et al., 1995), which are known to occur in the vicinity of cortical infarcts after permanent MCA occlusion and photothrombotic infarction, are impaired in the periinfarct region after transient focal ischemia. In support of this hypothesis is that long-term potentiation has been reported to be impaired after global ischemia (Miyazaki et al., 1993) and after cortical trauma (Reeves et al., 1995).
The remote effects, on the other hand, may be viewed as one (electrophysiologic) form of diaschisis. The term diaschisis was initially coined by von Monakow (1914) to describe transient remote effects after central nervous system injury attributable to the deafferentation of regions connected to the damaged area (for reviews see Feeney and Baron, 1986; Andrews, 1991). Other forms of diaschisis include depressions of glucose metabolism (Dietrich et al., 1994), blood flow, and protein synthesis all of which have been found in animal studies and some in humans. Drugs enhancing intracortical levels of central norepinephrine combined with physical therapy have been shown to promote recovery during the subacute/ chronic stage after cerebral insults and are believed to (at least partially) act by counteracting dysfunction attributable to diaschisis effects (Feeney et al., 1982; Hurwitz et al., 1991). In addition to being the site of negative diaschisis effects, the contralateral hemisphere may be of importance in the recovery process by taking over functions previously occupied by the lesioned hemisphere as suggested by positron emission tomography studies (Chollet and Weiller, 1994). Whether the electrophysiologic remote effects observed in the present study play a role in these functionally important processes is presently unclear.
In summary, we have delineated the extent of the excitability changes both in the periinfarct and in remote brain regions after transient MCA occlusion. The marked reduction of overall excitability in the periinfarct region indicates that there may be relatively widespread and possibly persisting functional disturbances in this region. This type of excitability change is likely to be specific to or at least particularly prominent in models of transient arterial occlusion. The remote excitability changes, on the other hand, appear to be secondary to transhemispheric and corticostriatal deafferentation and are encountered in models of both transient and permanent arterial occlusions as well as in typical cortical lesion models. Both the periinfarct and the remote excitability changes may turn out to be valuable indicators of functional disturbances which could become targets of therapeutic interventions in the subacute to chronic stage after stroke.
Footnotes
Acknowledgments
The authors would like to express their gratitude to C. Bruehl and M. Lutzenburg for helpful discussions and to D. Steinhoff for technical assistance.