
Abstract
It was evaluated whether postischemic neurodegeneration is apoptosis and occurs with alterations in phosphoinositide-linked metabotropic glutamate receptors (mGluRs) and their associated signaling pathways. A dog model of transient global incomplete cerebral ischemia was used. The CA1 pyramidal cells and cerebellar Purkinje cells underwent progressive delayed degeneration. By in situ end-labeling of DNA, death of CA1 and Purkinje cells was greater at 7 days than 1 day after ischemia, whereas death of granule neurons in dentate gyrus and cerebellar cortex was greater at 1 than at 7 days. Ultrastructurally, degenerating CA1 pyramidal neurons and cerebellar Purkinje cells were necrotic; in contrast, degenerating granule neurons were apoptotic. In agarose gels of regional DNA extracts, random DNA fragmentation coexisted with internucleosomal fragmentation. By immunoblotting of regional homogenates, mGluR1α, mGluR5, phospholipase Cβ (PLCβ), and Gαq/11 protein levels in hippocampus at 1 and 7 days after ischemia were similar to control levels, but in cerebellar cortex, mGluR1α and mGluR5 were decreased but PLCβ was increased. By immunocytochemistry, mGluR and PLCβ immunoreactivity dissipated in CA1 and cerebellar Purkinje cell/ molecular layers, whereas immunoreactivities for these proteins were enhanced in granule neurons. It was concluded that neuronal death after global ischemia exists as two distinct, temporally overlapping forms in hippocampus and cerebellum: necrosis of pyramidal neurons and Purkinje cells and apoptosis of granule neurons. Neuronal necrosis is associated with a loss of phosphoinositide-linked mGluR transduction proteins, whereas neuronal apoptosis occurs with increased mGluR signaling.
The mechanisms for delayed neuronal death (DND) after global cerebral ischemia in the adult CNS are not understood. Structurally and mechanistically, cellular degeneration resulting from hypoxia—ischemia has been thought to be a form of necrosis (Laiho and Trump, 1975; Wyllie et al., 1980; Kerr and Harmon, 1991; Deshpande et al., 1992); but recently postischemic DND has been considered to be a form of apoptosis mediated by programmed cell death (PCD) mechanisms (Héron et al., 1993; MacManus et al., 1995; Nitatori et al., 1995; Krajewski et al., 1995). However, the idea that selectively vulnerable neurons undergo apoptosis after ischemia is very controversial (Deshpande et al., 1992; Martin et al., 1998; Ishimaru et al., 1999), although ischemic DND might be more easily classified according to the concept of the apoptosis—necrosis continuum (MacManus et al., 1995; Portera-Cailliau et al., 1997a,b). The accurate identification of the contributions of apoptosis and necrosis to neuronal death in the adult central nervous system after ischemia has very important therapeutic relevance.
Neuronal cell death after ischemia involves perturbations in intracellular Ca2+ homeostasis and impairments in protein synthesis, possibly resulting from excitotoxic activation of neuronal glutamate receptors (GluRs) (Diemer et al., 1993). The GluRs that modulate intracellular Ca2+ levels within neurons are the ion channel receptors [N-methyl-
Some forms of PCD are Ca2+ dependent (Sen, 1992), and activation of mGluRs and heterotrimeric GTP-binding proteins (G proteins) can regulate neuronal apoptosis in vitro (Copani et al., 1995; Yan et al., 1995). Therefore, we evaluated whether ischemic DND is apoptosis structurally and whether DND is possibly associated mechanistically with abnormalities in phosphoinositide-linked mGluRs and their downstream intracellular signal transduction proteins, including the functionally coupled G proteins and phospholipase C (PLC). Thus, the goal of this study was to provide insight into the possible contribution of apoptosis to neurodegeneration after ischemia and the possible molecular mechanisms for this DND.
METHODS
Model of ischemia
A model of temporary global incomplete cerebral ischemia in dogs was used (Sieber et al., 1995). All animal procedures were conducted in accordance with the NIH Guide for the Care and Use of Laboratory Animals, and protocols were approved by the institutional animal care and use committee. Under sterile conditions, anesthetized (1 to 2% inspired halothane), mechanically ventilated, normothermic (epidural temperature at 37 to 38°C), adult beagles (n = 20) were subjected to transient incomplete global cerebral ischemia by intraventricular infusion of prewarmed sterile artificial cerebrospinal fluid into the lateral ventricle, producing an intracranial pressure of 10 mm Hg below mean arterial blood pressure (Sieber et al., 1995). The intracranial pressure was maintained, keeping cerebral perfusion pressure constant at 10 mm Hg for 20 minutes. To start reperfusion, the cerebrospinal fluid pressure reservoir was disconnected, and intracranial pressure was allowed to normalize. Six dogs were used as nonischemic sham controls.
Neuropathology, TUNEL, and electron microscopy
To evaluate neuropathology, sham control dogs (n = 3) and ischemic dogs surviving for 1 day (n = 5) or 7 days (n = 9) were anesthetized with pentobarbital, vasodilated with sodium nitrite, anticoagulated with heparin, and perfused intraaortically with phosphate-buffered saline followed by a mixture of 4% paraformaldehyde and 0.5% glutaraldehyde. The brains were divided midsagittally, and each hemisphere was cut into 1-cm-thick coronal slabs. In this model of ischemia, neuronal damage is similar bilaterally (Sieber et al., 1995). From the left hemisphere, samples of midhippocampus (containing the septal and temporal hippocampus) and cerebellum (at the level of the vermis) were used for paraffin histology. For quantification of postischemic neurodegeneration in hippocampus and cerebellar cortex (Fig. 1), sections (10 μm thick) were stained with hematoxylin and eosin or the terminal transferase-mediated biotin-dUTP nick end-labeling (TUNEL) method for in situ detection of nuclear DNA fragmentation as described (Portera-Cailliau et al., 1997a,b; Martin et al., 1997a; Al-Abdulla et al., 1998). The TUNEL method was used to identify dying cells after ischemia, although this technique is not specific for apoptosis (Martin et al., 1998).
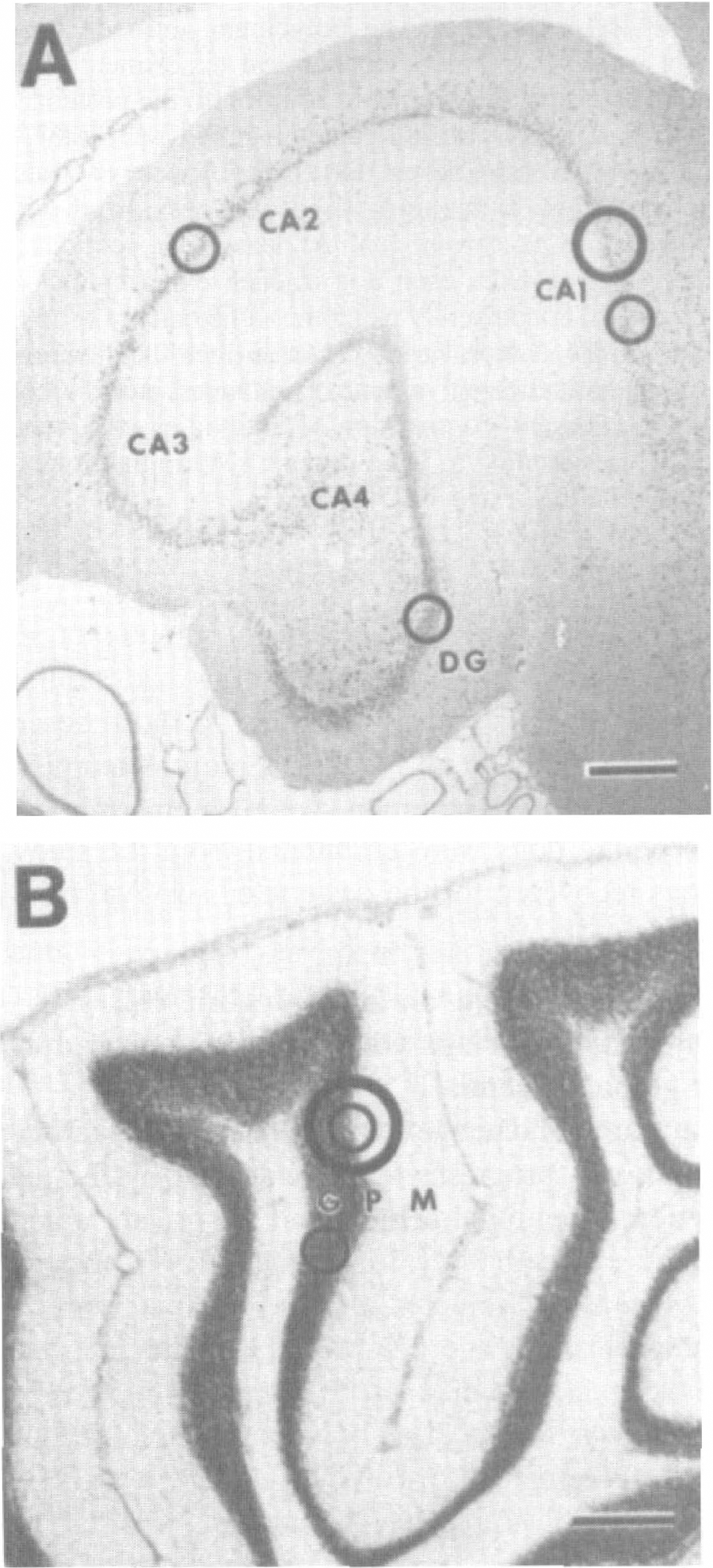
Locations in hippocampus
Profile counting was used to estimate ischemic neuronal damage in hematoxylin and eosin-stained sections in the stratum pyramidale of CA1 (in six microscopic fields at 600x; see Fig. 1A) and in the Purkinje cell layer of the anterior vermis of cerebellar cortex (in four microscopic fields at 400x; see Fig. 1B). Because Purkinje cells are arranged in only a single layer (Fig. 1B), the cerebellar sections were counted at a lower magnification so that a greater number of Purkinje cell could be analyzed per microscopic field. The percentage of neurons with ischemic cytopathology was determined using as criteria microvacuolar change within the soma, peripheralization of Nissl substance, nuclear pyknosis, perinuclear eosinophilia, or peri-karyal shrinkage with cytoplasmic eosinophilia (Sieber et al., 1995; Martin et al., 1997a,b, 1998). The percentage neuronal damage was estimated by identifying the fraction of neurons with ischemic cytopathology relative to the total number of neurons in microscopic fields of the CA1 and cerebellar cortex. The percentage neuronal damage in each region was averaged for each animal, and a group mean was calculated. The number of neurons per square millimeter was calculated for each region in control and ischemic dogs, and the percentage of remaining neurons following ischemia was calculated. Neuropathology scores between groups and regions at each time point were analyzed using the Mann-Whitney test.
In TUNEL preparations counterstained with cresyl violet, the densities of cells showing DNA fragmentation were determined by counting TUNEL-positive nuclei in six microscopic fields at 1,000× in the stratum pyramidale of CA1 and CA2-superior CA3, the granule cell layer of the dentate gyrus, and the Purkinje cell and granule cell layers of cerebellar cortex (Fig. 1). The TUNEL sections were counted at 1,000× so that TUNEL-positive nuclei could be easily identified and the type of cell could be recognized. The number of TUNEL-positive cells per square millimeter in each region was averaged for each animal, and a group mean was calculated. The TUNEL scores between groups and regions at each time point were analyzed using the Mann-Whitney test.
To determine whether DND has the structure of apoptosis, necrosis, or a hybrid form of degeneration, samples (3 mm3) of hippocampus and cerebellar cortex from the left hemisphere were osmicated immediately, embedded in plastic, and evaluated by electron microscopy. Ischemic DND was evaluated using ultrastructural criteria described previously in models of unequivocal neuronal apoptosis (Portera-Cailliau et al., 1997a,b; Al-Abdulla et al., 1998; Martin et al., 1998, 1999).
DNA isolation and agarose gel electrophoresis
Because the TUNEL method identifies DNA fragmentation independent of mechanisms (Portera-Cailliau et al., 1997b; Martin et al., 1998), we evaluated regional neuropathology based on DNA fragmentation patterns in agarose gels. Genomic DNA was isolated from samples of hippocampus (septal and temporal) and cerebellar cortex from control dogs (n = 2) and dogs at 1 day (n = 2) and 7 days (n = 2) after ischemia. Two additional animals were killed at 6 hours after ischemia. Tissue samples were homogenized in DNA extraction buffer containing 10 mmol/L Tris (pH 7.4), 10 mmol/L NaCl, 25 mmol/L ethylenediaminetetraacetate, 1% sodium dodecyl sulfate, and 1 mg/ml proteinase K and incubated in the same buffer overnight at 37°C. DNA was extracted with an equal volume of salt-saturated phenol/chloroform/isoamyl alcohol (10:10:1), and the recovered aqueous phase was extracted with diethyl ether. The DNA was precipitated with ethanol (2.5 volumes). The DNA pellet was dissolved in 0.1× saline-sodium citrate and incubated (37°C) with DNase-free RNase A (0.1 mg/mL) for 1 hour and then overnight (37°C) with 0.1 mg/mL proteinase K. DNA was reextracted, precipitated, and dissolved in Tris/ ethylenediaminetetraacetate buffer. The DNA samples (~1.0 μg) were 3‘ end-labeled with digoxigenin-11-dideoxy-uridine-5‘-triphosphate using terminal transferase (Boehringer-Mannheim, Indianapolis, IN, U.S.A.), precipitated, resuspended in Tris/ethylenediaminetetraacetate buffer, fractionated by agarose gel (1.2%) electrophoresis, and transferred to nylon membrane followed by ultraviolet cross-linking. Membranes were incubated in 2% nucleic acid blocking reagent (Boehringer-Mannheim) and then in blocking reagent containing 75 mU/mL antidigoxigenin Fab fragments conjugated to alkaline phosphatase (Boehringer-Mannheim). After washing, membranes were reacted with chemiluminescent substrate for alkaline phosphatase detection reagent (Boehringer-Mannheim) and exposed to Kodak X-OMAT film to visualize DNA.
Immunoblotting
To evaluate possible mechanisms for DND, we studied by immunoblotting the expression of the proapoptotic protein Bax as well as mGluRs and their related intracellular signaling proteins. We focused on mGluR1 and mGluR5 because these receptors are G protein-coupled receptors that operate through heterotrimeric GTP-binding proteins and PLC activation, which regulates intracellular Ca2+ by phosphoinositide hydrolysis (Tanabe et al., 1992; Pin and Duvoisin, 1995). Activation of phosphoinositide-linked mGluRs initiates a cascade of events including the activation of PLCβ by Gq proteins, generation of inositol triphosphate and diacylglycerol, and subsequent mobilization of Ca2+ from nonmitochondrial intracellular stores (Pin and Duvoisin, 1995). Control dogs (n = 3) and ischemic dogs at 1 day (n = 2) and 7 days (n = 2) of recovery were anesthetized with pentobarbital, ventilated, and exsanguinated by intraaortic perfusion of ice-cold phosphate-buffered saline. The brains were quickly removed from the skull and placed on ice, and the hippocampus (septal) and anterior lobule of the cerebellum were isolated and frozen in isopentane. The samples were homogenized and subjected to differential centrifugation (Portera-Cailliau et al., 1996), and subcellular fractions were assayed for protein concentration using a Bio-Rad (Hercules, CA, U.S.A.) detection method. For immunoblotting, mitochondria-enriched membrane fractions or synaptic plasma membrane fractions (10, 12, or 15 μg of protein) were subjected to sodium dodecyl sulfate polyacrylamide gel electrophoresis (8, 10, or 15% gels) and transferred to nitrocellulose membrane by electroblotting (Martin et al., 1992, 1997a). Samples of control and ischemic dogs were always run in the same gel. To ensure that equivalent amounts of protein were loaded in each lane and that transfer was comparable, membranes were stained with Ponceau S before immunoblotting. Blots were then blocked with 2.5% nonfat dry milk with 0.1% Tween 20 in 50 mmol/L Tris-buffered saline (pH 7.4) and incubated overnight at 4°C with antibodies to mGluR1α, mGluR5, PLCβ1, or Gαq/11. Affinity-purified rabbit polyclonal antibodies to the C-terminal domains of mGluR1α and mGluR5 were used at a concentration of 0.5 μg IgG/mL (Martin et al., 1992; Blue et al., 1997). Affinity-purified antipeptide rabbit polyclonal antibodies to PLCβ1 and Gαq/11 were obtained from commercial sources (Santa Cruz Biotechnology, Santa Cruz, CA, U.S.A.) and were used at concentrations of 33 or 20 ng IgG/mL, respectively. In the same samples, the levels of proapoptotic protein Bax were measured, using commercial antibodies (Santa Cruz Biotechnology) at 150 ng IgG/mL, to evaluate whether changes in mGluR signaling proteins are associated with alterations in cell death protein levels. After the primary antibody incubation, membranes were washed and incubated with peroxidase-conjugated secondary goat anti-rabbit IgG (0.2 μg/mL) for 1 hour and developed with enhanced chemiluminescence (Amersham, Piscataway, NJ, U.S.A.). All blots were then reprobed with synaptophysin antibody (Boehringer-Mannheim) for a quantitative loading control.
Western blots were quantified densitometrically to evaluate brain regional changes in the levels of these proteins in postischemic animals relative to controls. Films were scanned using a Macintosh Adobe Photoshop program and an Agfa Arcus Plus scanner (Ridgefield Park, NJ, U.S.A.). Densitometric analysis was performed using Signal Analytics IP Lab Gel software (Scanalytics, Fairfax, VA, U.S.A.). Protein levels were expressed as relative optical density measurements, determined by comparing the density and area of the immunoreactive band in each lane scanned with control lanes in the same blot. The relative values for each animal were replicated in at least four immunoblotting experiments. Bax protein levels and signal transduction protein levels were expressed as percentages of control values. Mean percentages were compared among the recovery groups by a Wilcoxon signed rank test with the significance level set at P < 0.05.
Immunocytochemistry
From perfusion-fixed dogs (same as those used for hematoxylin and eosin staining and TUNEL), the right hemisphere was cut into 1-cm-thick coronal slabs, cryoprotected in 20% phosphate-buffered glycerol, and frozen in isopentane. Forebrain and cerebellar samples were cut at 40 μm on a sliding microtome, and sections were placed in antifreeze buffer for storage (−20°C) until immunocytochemical processing. A peroxidase antiperoxidase method (Martin et al., 1992; Fotuhi et al., 1993) was used to detect mGluR1α, mGluR5, and PLCβ1 immunoreactivities in hippocampus and cerebellar cortex of sham and ischemic dogs. The polyclonal antibodies to Bax were not used for immunocytochemical experiments because they recognize several proteins in addition to the proteins at 21 and 23 kDa. Affinity-purified goat anti-rabbit IgG F(ab)2 fragments (Cappel, West Chester, PA, U.S.A.) were used as secondary antibodies. Immunoreactivity was visualized with diaminobenzidene as chromogen. At least four sections from each brain region from each dog in each experimental group were processed concurrently using the same batches of reagents to obviate tissue section variability in antigen localization. Immunocytochemical negative controls included omission of primary antibodies and competition of antibodies with synthetic peptides corresponding to the antigenic C-terminal domains of mGluR1α, mGluR5, and PLCβ1.
RESULTS
Twenty dogs were subjected to 20 minutes of global incomplete cerebral ischemia. All dogs had similar severities of ischemia and were similar to those described previously (Sieber et al., 1995). Epidural temperature during ischemia was maintained at normothermic values. No ischemic dogs were eliminated from the study, and all dogs recovered for the designated survival time after ischemia.
CA1 pyramidal neuron and Purkinje cell degeneration overlaps with granule neuron death after global ischemia
The neuronal damage after transient global incomplete ischemia was progressive, as evidenced by the delayed reduction in neuronal density and the greater amount of ischemic cytopathology in neurons at 7 than at 1 day after ischemia (Table 1). At 1 day after ischemia, CA1 pyramidal neuron and cerebellar Purkinje cell densities were not significantly different from control values (Table 1). At 7 days after ischemia, 43% of CA1 pyramidal cells remained, while Purkinje cell densities were 78% of control (Table 1). The percentage of CA1 pyramidal neurons with ischemic cytopathology increased from 13% at 1 day of recovery to 52% at 7 days of recovery (Table 1). In cerebellum, the percentage of Purkinje cells with cytopathology was 24% at 1 day and 69% at 7 days after ischemia (Table 1). In hematoxylin and eosin sections of control animals, background neuronal injury (possibly due to the anesthesia, perfusion—fixation, brain compression during removal from the skull, or histological processing) was ≤3% in CA1 and cerebellum (Table 1).
Neuropathology in dog hippocampus and cerebellum after global incomplete ischemia
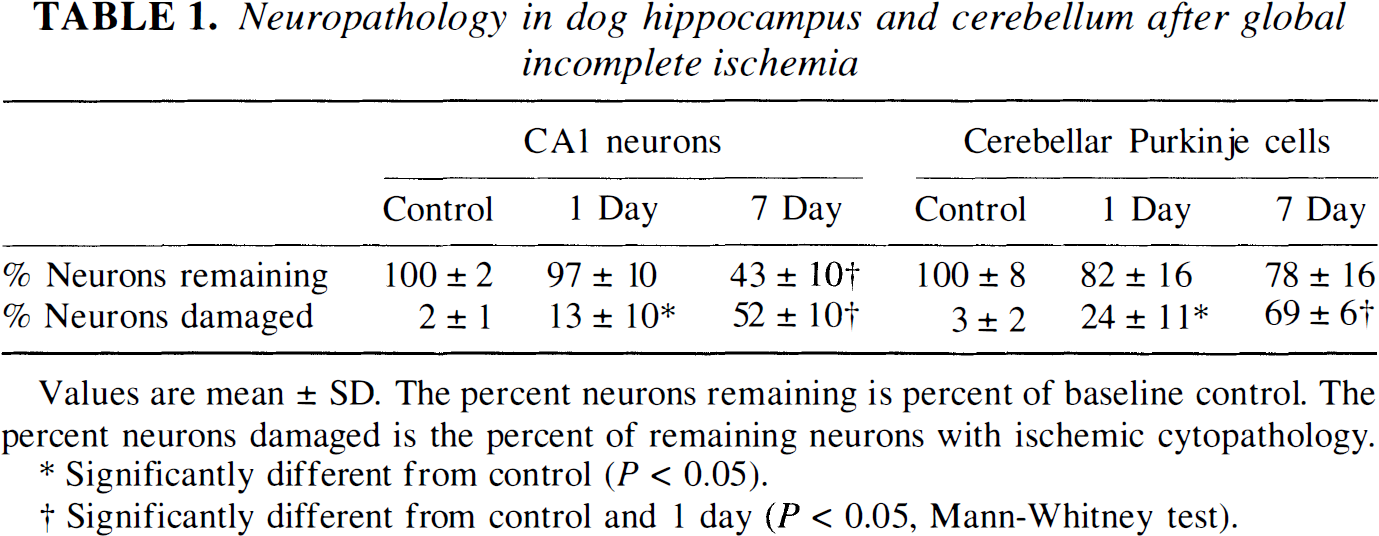
Values are mean ± SD. The percent neurons remaining is percent of baseline control. The percent neurons damaged is the percent of remaining neurons with ischemic cytopathology.
Significantly different from control (P < 0.05).
Significantly different from control and 1 day (P < 0.05, Mann-Whitney test).
Neurons in hippocampus and cerebellar cortex were undergoing nuclear DNA fragmentation at 1 and 7 days after ischemia, as determined by TUNEL (Figs. 2 and 3). In hippocampus, subsets of neurons in the pyramidal cell layer of CA1 and CA2–superior CA3 (CA3a) were TUNEL-positive as well as subsets of granule neurons in the hippocampal dentate gyrus. In CA1, the number of TUNEL-positive cells/mm2 was significantly greater (P < 0.05) at 7 than at 1 day, whereas more dentate gyrus granule neurons were TUNEL-positive at 1 than at 7 days (Fig. 3). In cerebellar cortex, the number of TUNEL-positive Purkinje cells at 7 days after ischemia was approximately twofold higher than at 1 day, whereas TUNEL-positive cells in the granule cell layer were nearly twofold higher at 1 than at 7 days (Fig. 3).
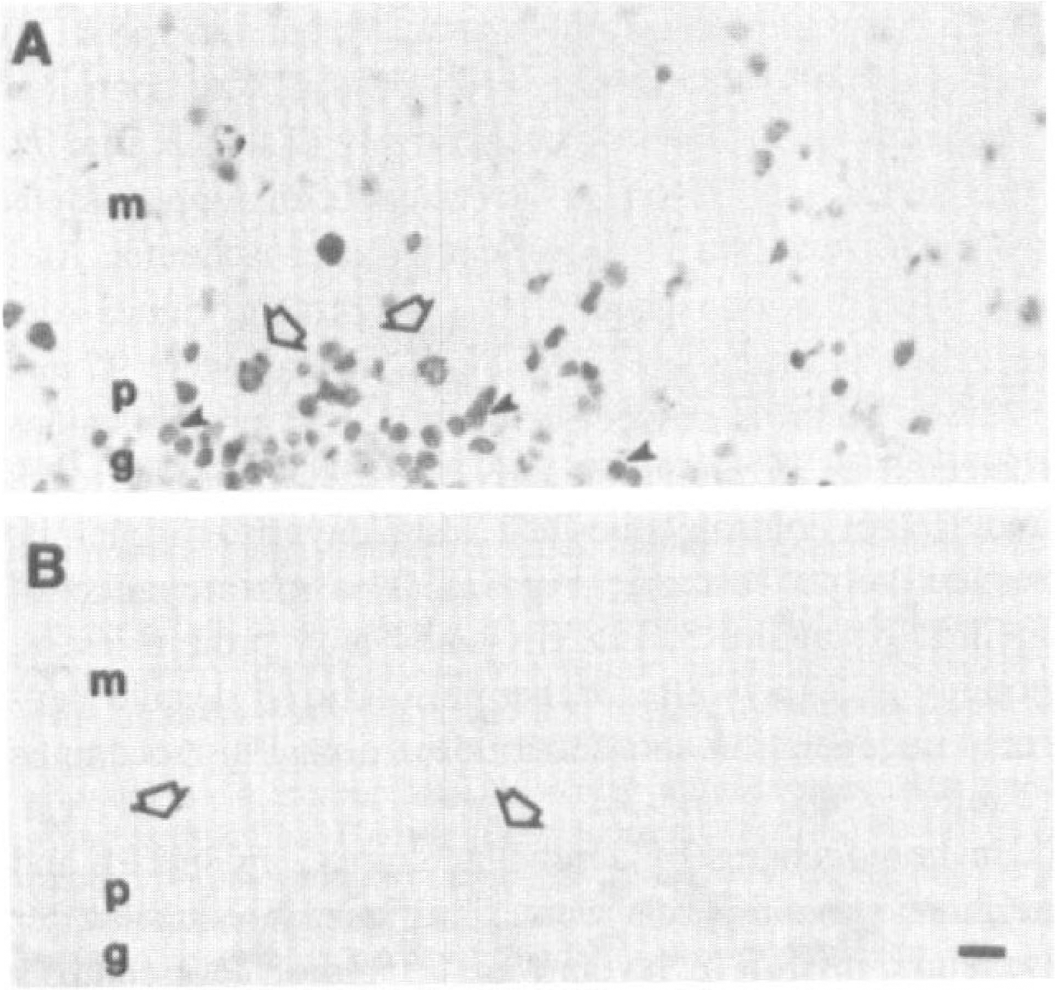
Nuclear DNA fragmentation in dog cerebellar cortex after global incomplete ischemia, m, molecular layer; p, Purkinje cell layer; g, granule cell layer.
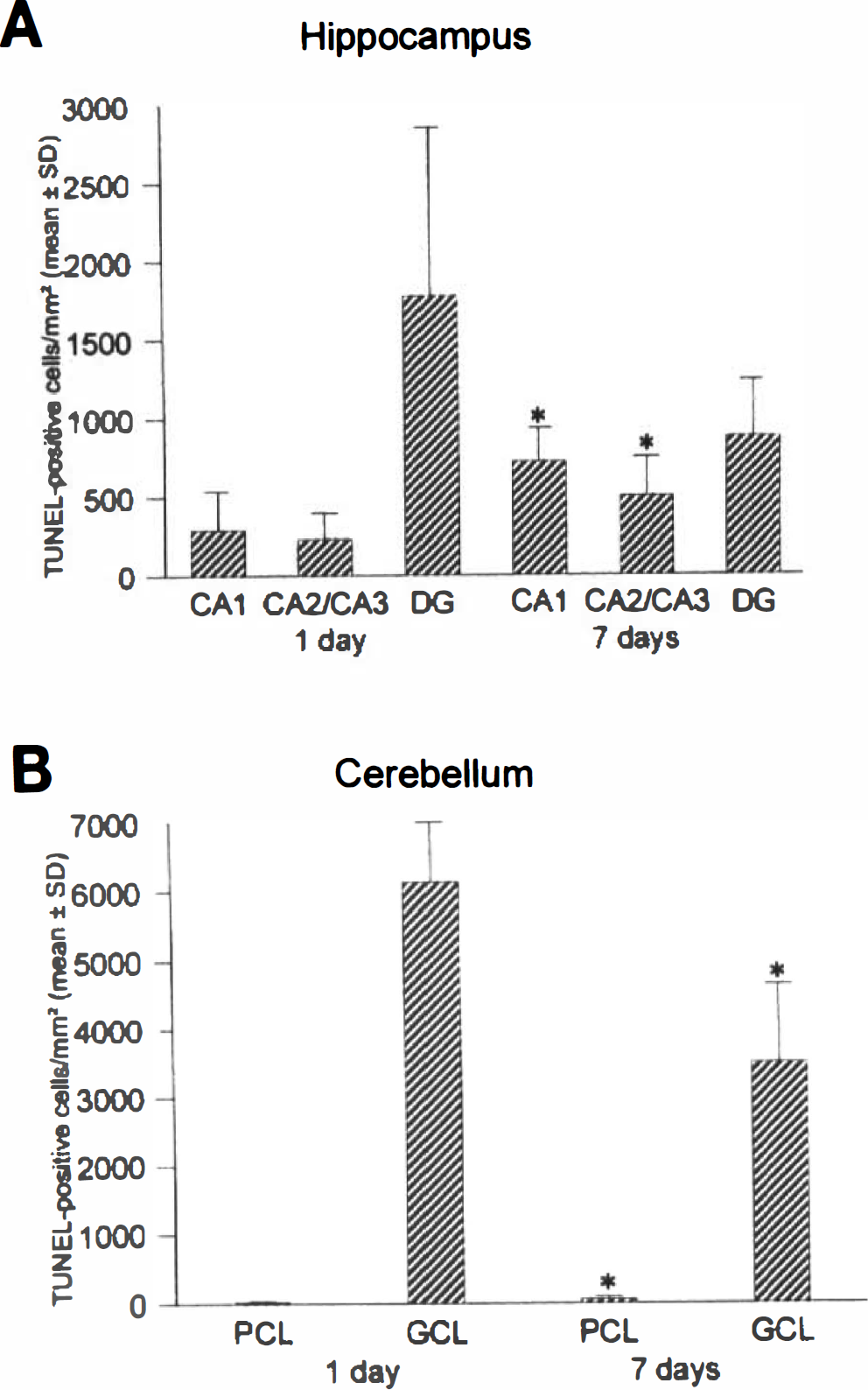
Quantification of terminal transferase-mediated nick end-labeling (TUNEL)-positive cells in hippocampus
In pyramidal neurons of hippocampus and in Purkinje cells, the TUNEL-positive nuclei usually had reaction product that was random in distribution, with irregularly shaped aggregates, or had uniform and nonaggregated labeling, unlike the organized spherical clumping or crescentic capping of chromatin that was found in granule neurons (Figs. 2A and 5F-5H) and is characteristic of neuronal apoptosis (Portera-Cailliau et al., 1997a,b; Al-Abdulla et al., 1998; Martin et al., 1999).
In agarose gels, random (smearing) and internucleosomal fragmentation of DNA was detected in genomic DNA extracts of dog hippocampus and cerebellar cortex after ischemia (Fig. 4, lanes 2 and 4). These different patterns of DNA degradation coexisted, most prominently at 7 days after ischemia. The internucleosomal fragmentation was similar to the pattern detected during developmental PCD of neurons (Fig. 4, lane 1). No DNA fragmentation was detected in control dog hippocampus and cerebellar cortex (Fig. 4, lanes 3 and 5), and no fragmentation of DNA was observed at 6 hours after ischemia (data not shown).
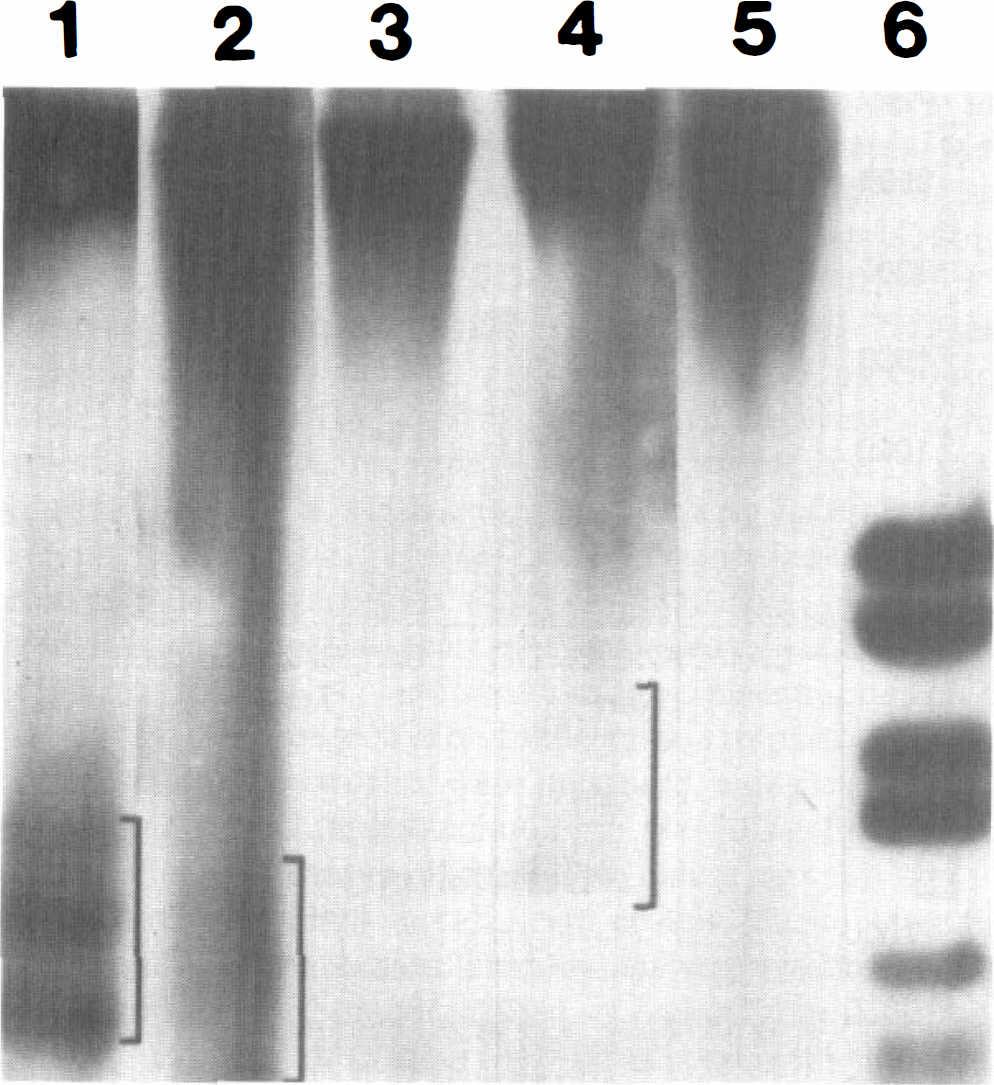
Analysis of DNA fragmentation patterns in dog hippocampus and cerebellar cortex by agarose gel electrophoresis. Genomic DNA was isolated and fractionated from ischemic dog hippocampus (lane 2) and cerebellar cortex (lane 4) at 7 days of recovery and from control dog hippocampus (lane 3) and cerebellar cortex (lane 5). Programmed cell death in developing rat retina at postnatal day 19 served as a positive control for internucleosomal fragmentation of DNA (lane 1). Brackets delineate internucleosomal fragments (lanes 1, 2, and 4). Smearing of DNA is also seen in ischemic dog hippocampus and cerebellar cortex (lanes 2 and 4). Base pair molecular mass markers (lane 6) are (top to bottom): 2,176, 1,766, 1,230, 1,033, 653, 517, and 453.
CA1 pyramidal neuron and Purkinje cell death is necrosis, but granule neuron death is apoptosis after ischemia
With use of criteria for neuronal apoptosis established in previous experiments (Portera-Cailliau et al., 1997a,b; Al-Abdulla et al., 1998; Martin et al., 1998, 1999), the degeneration of CA1 pyramidal neurons and Purkinje cells after ischemia was not apoptosis (Fig. 5). The degeneration of these neurons was consistent with necrosis (Figs. 5A–5C). Whereas nuclear pyknosis and chromatin condensation into irregular clumps were observed in these dying neurons, this pattern was very dissimilar to that found in unequivocal neuronal apoptosis (Portera-Cailliau et al., 1997a,b; Al-Abdulla et al., 1998; Martin et al., 1998, 1999). These ultrastructural observations are consistent with the hematoxylin and eosin staining and TUNEL patterns found in CA1 pyramidal neurons and cerebellar Purkinje cells, indicating a nonapoptotic structure, as shown previously (Martin et al., 1997a, 1998). In contrast, granule neuron degeneration in the dentate gyrus and cerebellar cortex was apoptosis (Fig. 5D—H). For example, early structural changes were chromatin margination and crescentic capping of chromatin at the nuclear envelope and dispersion of many ribosomes within the cytoplasm (Fig. 5F and G). These changes occurred in the presence of maintained mitochondrial morphology (Fig. 5G). Later in the progression of apoptosis, granule neurons underwent condensation of chromatin into large, round clumps and convolution of the nuclear envelope and then budding (Fig. 5H).
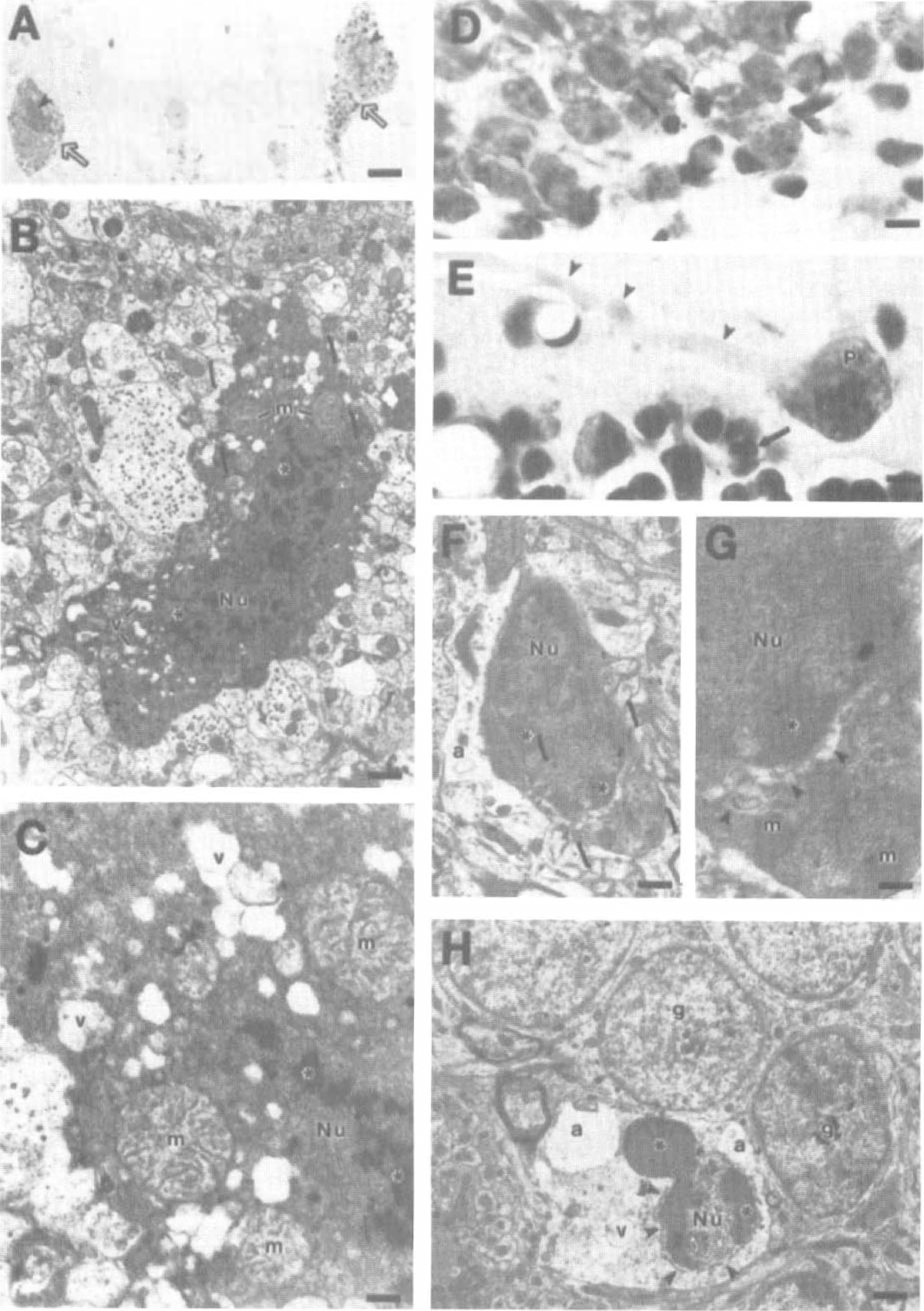
Necrosis and apoptosis occur in distinct populations of neurons after ischemia.
To further evaluate the presence or absence of indicators for neuronal apoptosis after cerebral ischemia, immunoblotting was used to evaluate the levels of Bax protein expression in hippocampus and cerebellum. A representative immunoblot of Bax expression in control and ischemic dogs is shown in Fig. 6. Bax levels at 1 and 7 days after ischemia did not differ significantly from control levels in homogenates of dorsal hippocampus, as quantified by comparing the density and area of the immunoreactive band in each lane of ischemic dog homogenate scanned with values of control dog lanes in the same blot. Bax protein levels (in relative optical density units, means ± SD) in hippocampus were 32.4 ± 6.2 (control), 27.9 ± 8.0 (1 day after ischemia), and 32.1 ± 10.5 (7 days after ischemia). In contrast, Bax protein expression was significantly (P < 0.05) increased to 128 ± 5% of control cerebellum values at 7 days after ischemia but not at 1 day after ischemia.
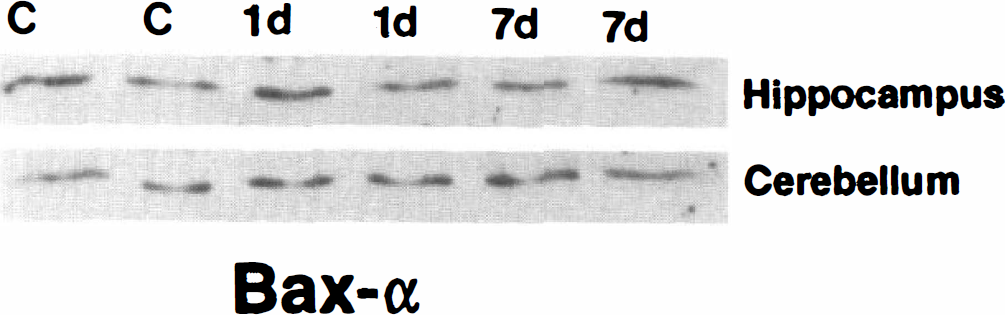
Immunoblot analysis of Bax expression in mitochondria-enriched subcellular fractions of hippocampus (15 μg of protein/ lane) and cerebellar cortex (12 μg of protein/lane) from sham control (c) and ischemic dogs at 1 day (1 d) and 7 days (7 d) of recovery. In hippocampus, Bax levels are unchanged at 1 and 7 days after ischemia. In cerebellar cortex, Bax levels are increased at 7 days afer ischemia.
Alterations in metabotropic glutamate receptor signaling pathways occur in hippocampus and cerebellum after ischemia
In homogenates of whole septal hippocampal formation, mGluR1α protein levels (detected as an immunoreactive band at 142 kDa) were unchanged, with values 108 and 114% of control at 1 and 7 days after ischemia, respectively (Table 2; Fig. 7); similarly, mGluR5 protein levels (detected as an immunoreactive band at 148 kDa) were unchanged, with values 105 and 112% of control at 1 and 7 days of recovery, respectively (Table 2; Fig. 7). The PLCβ1 and Gαq11 protein levels in hippocampus were also not changed significantly after ischemia. At 1 and 7 days after ischemia, PLCβ1 protein values (detected as an immunoreactive 80-kDa band) were 95 and 122% of controls, respectively, and Gαq11 protein values (detected as an immunoreactive 45-kDa band) were 98 and 81% of control values at 1 and 7 days after ischemia, respectively (Table 2; Fig. 7). The maintenance of mGluR1α, mGluR5, PLCβ1, and Gαq11 protein levels, notably at 7 days after ischemia, occurred despite neuronal degeneration and death in the dorsal hippocampus (Table 1; Figs. 2 and 3).
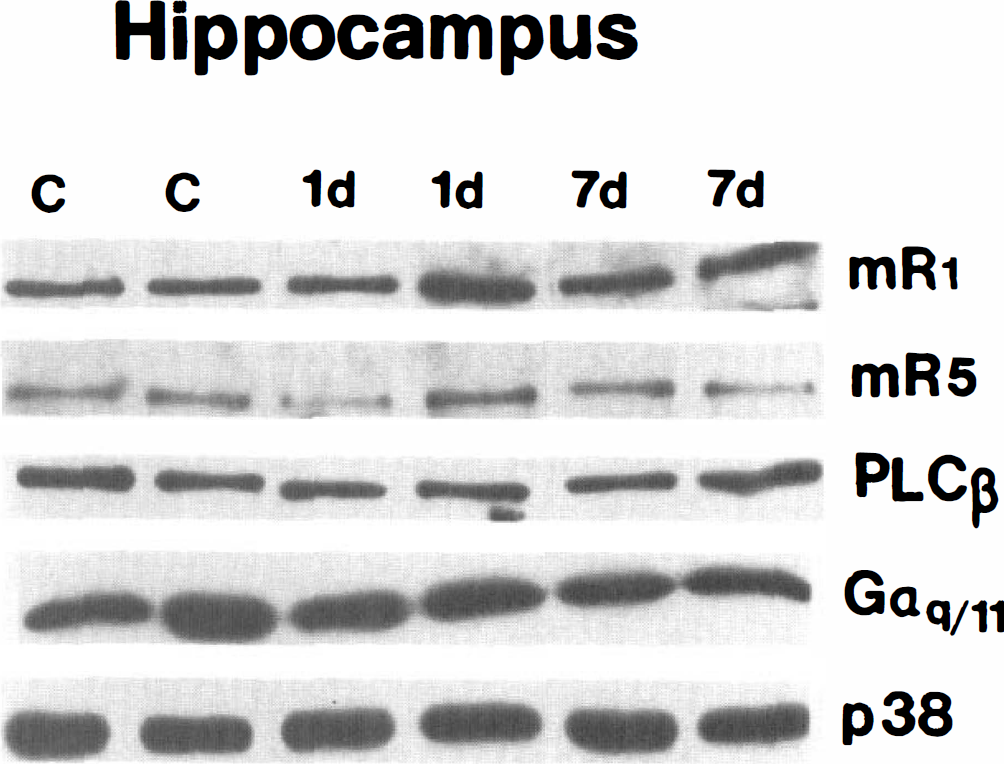
Immunoblot analysis of metabotropic glutamate receptor (mGluR) 1α (mR1), mGluR5 (mR5), phospholipase Cβ (PLCβ), and Gαq/11 proteins in synaptic membrane fractions (10 μg of protein/lane) of hippocampus from sham control (c) and ischemic dogs at 1 day (1 d) and 7 days (7 d) of recovery. Blots were probed for synaptophysin (p38) as a loading control.
Quantitative immunoblot analysis of phosphoinositide-linked mGluR signal transduction proteins after global incomplete ischemia
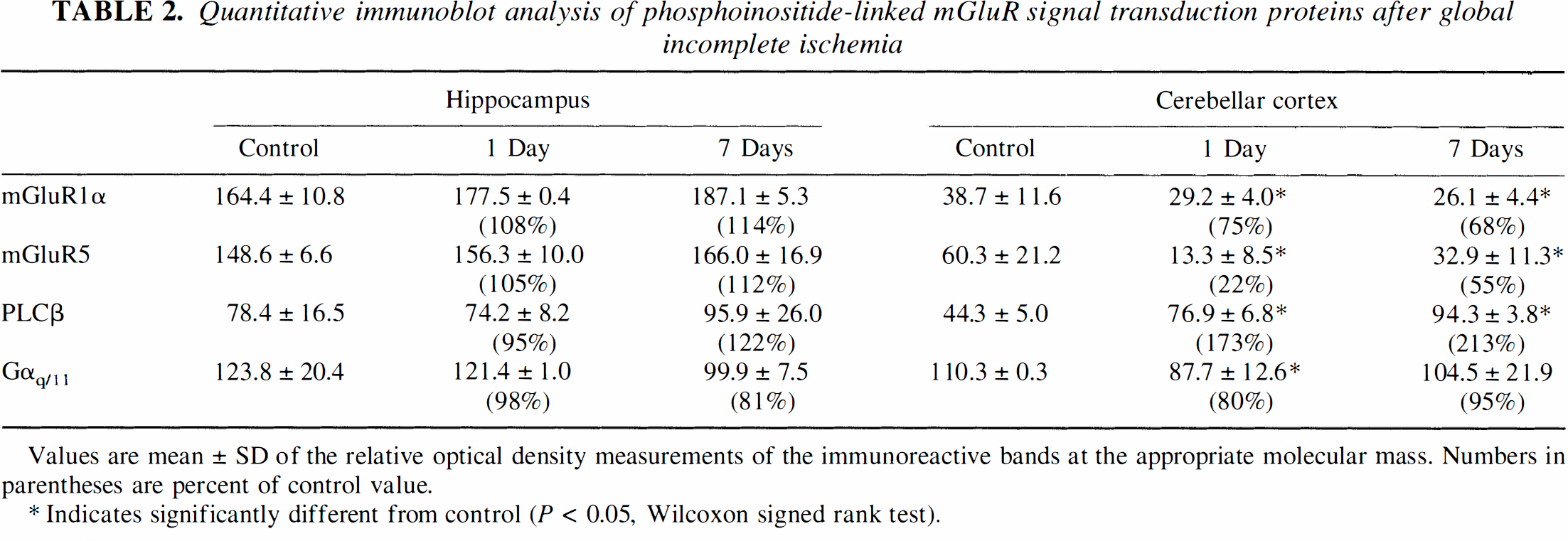
Values are mean ± SD of the relative optical density measurements of the immunoreactive bands at the appropriate molecular mass. Numbers in parentheses are percent of control value.
Indicates significantly different from control (P < 0.05, Wilcoxon signed rank test).
In homogenates of cerebellar cortex, mGluR1α and mGluR5 protein levels were significantly reduced after ischemia. mGluR1α levels were 75 and 68% of controls at 1 and 7 days after ischemia, respectively (Table 2; Fig. 8), whereas mGluR5 was reduced to 22% of control at 1 day and then returned to 55% of control levels by 7 days (Table 2; Fig. 8). In contrast, PLCβ1 levels in cerebellar cortex were increased significantly to 173 and 213% of control at 1 and 7 days after ischemia, respectively, whereas Gαq11 was reduced to 80% of control at 1 day and then returned to control levels by 7 days (Table 2; Fig. 8).
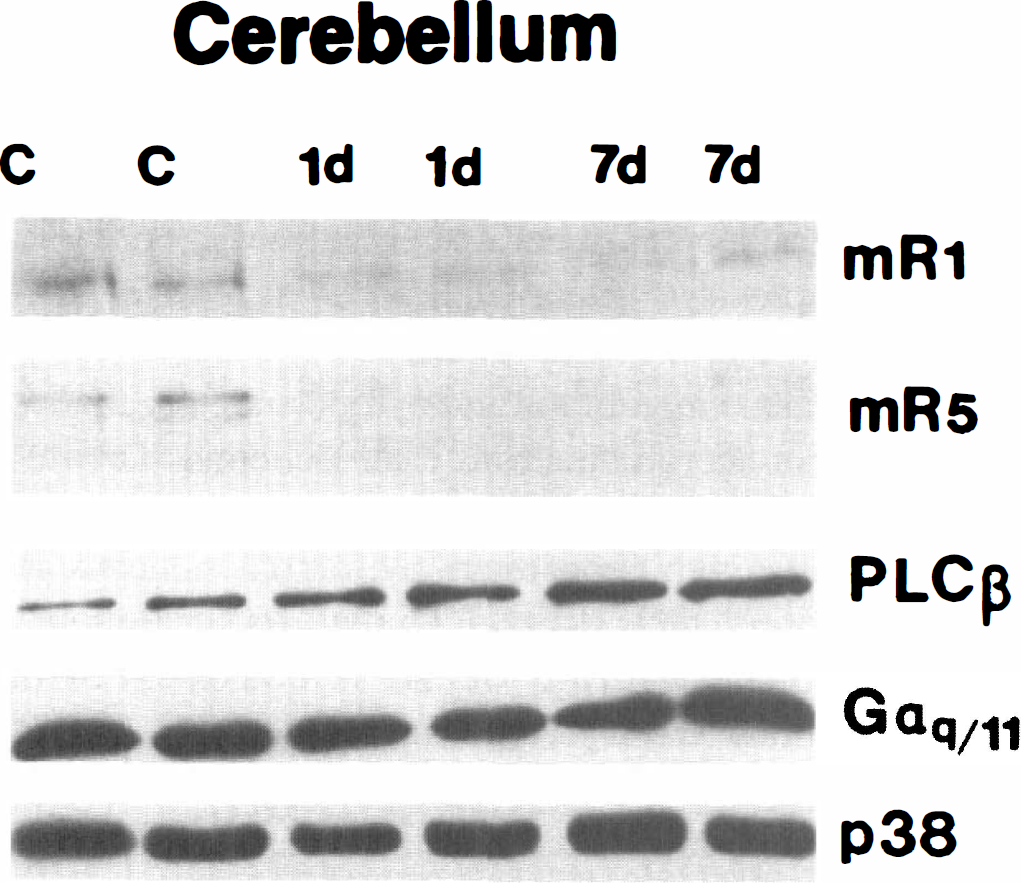
Immunoblot analysis of metabotropic glutamate receptor (mGluR) 1α (mR1), mGluR5 (mR5), phospholipase Cβ (PLCβ), and Gαq/11 proteins in synaptic membrane fractions (10 μg of protein/lane) of cerebellar cortex from sham control (c) and ischemic dogs at 1 day (1 d) and 7 days (7 d) of recovery. Blots were probed for synaptophysin (p38) as a loading control.
The maintenance of some protein levels in regional homogenates, despite neuronal degeneration, suggested the possibility that changes occur in the cellular localization of mGluR1α, mGluR5, and PLCβ1 after ischemia. We used immunocytochemistry to determine the localization of mGluR1α, mGluR5, and PLCβ1 in hippocampus and cerebellum. In control dog hippocampus, mGluR1α is localized primarily within the stratum oriens of CA1 and in the dentate gyrus hilus, with only faint immunolabeling of granule cells (Fig. 9A). This pattern of mGluR1α localization in dog hippocampus is similar to the pattern shown in rat brain (Martin et al., 1992). In ischemic dogs, granule cell immunolabeling is augmented compared with controls (Fig. 9B), whereas the intensity of mGluR1α immunoreactivity in the neuropil of CA1 is diminished. In control dogs, mGluR5 is highly enriched in CA1 pyramidal cell bodies and in the neuropil of stratum oriens and stratum radiatum (corresponding to the dendritic fields of CA1 pyramidal neurons), whereas dentate gyrus granule neurons are less intensely immunoreactive than CA1. After ischemia, mGluR5 immunoreactivity in the CA1 pyramidal cell body and dendritic layers is reduced as compared with controls, but the dentate gyrus molecular layer, subiculum, and parahippocampal gyrus are more intensely immunoreactive compared with control (data not shown). In normal hippocampus, PLCβ1 is highly enriched in cell bodies and proximal dendrites of pyramidal neurons and in the neuropil of CA1 (Fig. 9C) and is localized less intensely in the granule cells of the dentate gyrus (Fig. 9E). After ischemia, PLCβ1 immunoreactivity is lost in the pyramidal cell body and dendritic (radiatum) layers of CA1 (Fig. 9D); in contrast, PLCβ1 immunoreactivity is increased in the granule cell and molecular layers of the dentate gyrus (Fig. 9F) and in other locations corresponding to the distribution of the perforant path (not shown), suggesting an augmented presynaptic localization of PLCβ1 after ischemia.
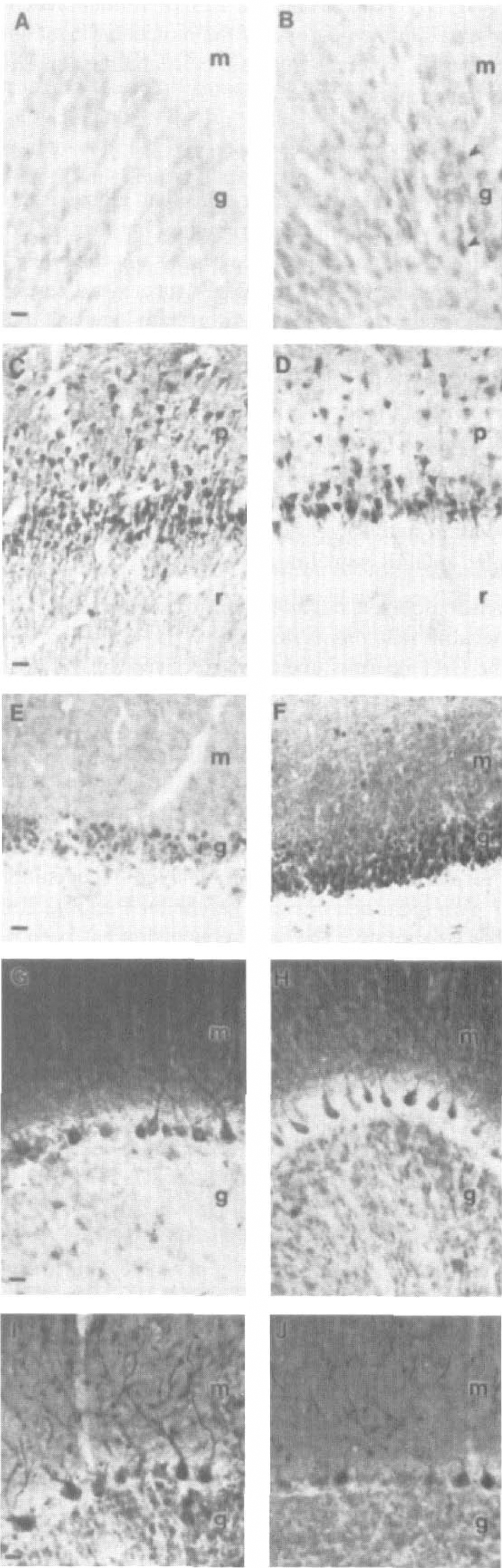
Alterations occur in the regional and cellular localization of metabotropic glutamate receptor (mGluR) 1α and phospholipase Cβ (PLCβ) in hippocampus and cerebellar cortex at 7 days after global ischemia in dog. Photomicrographs on the left (
In control dog cerebellar cortex, mGluR1α is enriched in the neuropil of the molecular layer and in Purkinje cell bodies, but in the granule cell layer, occasional Golgi type II cells are immunopositive and granule cells are only weakly immunoreactive for mGluR1α (Fig. 9G). This pattern of mGluR1α localization in dog cerebellum is similar to the pattern shown in rat brain (Martin et al., 1992). After ischemia, mGluR1α immunoreactivity is reduced in the neuropil of the molecular layer but is increased in the granule cell layer (Fig. 9H). Like mGluR1α, mGluR5 staining in the cerebellum was less intense in the molecular layer after ischemia compared with control (data not shown). The PLCβ1 level in control cerebellum is highly enriched in Purkinje cell bodies and dendrites extending throughout the molecular layer, which also contains diffuse neuropil immunoreactivity, and the granule cell layer is not uniformly immunoreactive (Fig. 9I). After ischemia, PLCβ1 immunoreactivity is increased in the neuropil of the molecular layer and in granule cells, whereas Purkinje cell labeling for PLCβ1 is prominently decreased (Fig. 9J).
DISCUSSION
These experiments demonstrate that global ischemia results in two temporally overlapping, but distinct, forms of neuronal cell degeneration: necrosis of selectively vulnerable neurons (CA1 pyramidal neurons and cerebellar Purkinje cells) and apoptosis of granule neurons in dentate gyrus and cerebellum. Neuronal cell death in these regions is accompanied by differential changes in phosphoinositide-linked mGluR transduction mechanisms. Necrosis of CA1 neurons and Purkinje cells occurs in association with a loss of PLC-coupled mGluR signaling proteins, whereas apoptosis of granule neurons occurs in association with increases in proteins involved in PLC-coupled mGluR signaling.
Ischemic delayed degeneration of selectively vulnerable neurons is necrosis
The CA1 neurons undergo selective DND after ischemia in rodents (Ito et al., 1975; Kirino, 1982) and in dogs (Sato et al., 1990). The DND of cerebellar Purkinje cells is not studied commonly in rodent forebrain models of ischemia, but our study in dogs provided a novel opportunity to compare similarities among neuronal degeneration in hippocampus and cerebellum after ischemia. Our results show that ischemic delayed degeneration of CA1 pyramidal neurons and cerebellar Purkinje cells is similar structurally. The same conclusion has been made using a model of global complete ischemia in cats (Martin et al., 1998). Furthermore, our data demonstrate that the DND of selectively vulnerable neurons following global ischemia is cellular necrosis rather than apoptosis or a hybrid of apoptosis and necrosis. This degeneration of CA1 pyramidal neurons and Purkinje cells after ischemia is very distinct from neuronal apoptosis (Portera-Cailliau et al., 1997a; Al-Abdulla et al., 1998; Martin et al., 1998, 1999); it is also distinct from non-NMDA GluR-mediated excitotoxic neuronal apoptosis (Portera-Cailliau et al., 1997b; Martin et al., 1998), but it is very similar to NMDA receptor-mediated excitotoxic neuronal necrosis (Portera-Cailliau et al., 1997b; Martin et al., 1998; Ginsberg et al., 1999). Both the nucleus and the cytoplasm undergo ultrastructural perturbations consistent with cellular necrosis, with the main features being irregular clumping of chromatin, swelling and degeneration of organelles, extensive cytoplasmic vacuolation, destruction of plasma membrane integrity, and eventual dissolution of the cell. Moreover, dying CA1 neurons do not bud to form discrete, round nuclear fragments, as in apoptosis (cf. Fig. 4C and H) (Wyllie et al., 1980; Kerr and Harmon, 1991; Martin et al., 1998). The nuclear pyknosis with condensation of chromatin into many small, irregularly shaped clumps in ischemic neurons contrasts with the formation of few, uniformly dense, and regularly shaped chromatin aggregates that occurs in neuronal apoptosis (Portera-Cailliau et al., 1997a; Al-Abdulla et al., 1998; Martin et al., 1998, 1999).
The cell plasma membrane and cytoplasmic organelles are damaged in necrotic neurons after ischemia. The immunocytochemically identified loss of mGluR-PLC signaling proteins in hippocampal pyramidal neurons in CA1 and Purkinje cells (as well as the loss of immunolabeling in their dendritic fields) is consistent with plasma membrane damage. The mitochondria swell and the inner mitochondrial membrane undergoes cristaeolysis. These mitochondrial abnormalities have been demonstrated in cellular necrosis (Laiho et al., 1971; Laiho and Trump, 1975) and can be produced when plasma membrane function and ATP synthesis are impaired (Laiho and Trump, 1975). Our observations are also consistent with previous findings showing that organelles that function in protein synthesis and posttranslational modification become structurally abnormal early in the course of ischemic neurodegeneration and are persistently abnormal during the process of neurodegeneration (Kirino et al., 1984; Martin et al., 1998). These structural changes are consistent with the finding that total protein synthesis is severely reduced by 6 hours after transient global forebrain ischemia and is reduced persistently in CA1 neurons, with the vast majority of pyramidal neurons never regaining their normal biosynthetic activity (Thilmann et al., 1986; Araki et al., 1990; Johansen and Diemer, 1990; Furuta et al., 1993). Cytoskeletal disintegration also occurs early after ischemia, particularly in dendrites, before the degeneration of neuronal cell bodies (Kitagawa et al., 1989; Yamamoto et al., 1990). This rapid disassembly and proteolysis of the cytoskeleton after ischemia (Kitagawa et al., 1989) contrast with the organized structure of the cytoskeleton and the cytoskeletal accumulation in neurons undergoing apoptosis (see Fig. 4G) (Al-Abdulla et al., 1998; Martin et al., 1998, 1999).
Nevertheless, previous studies have suggested that DND in hippocampus is apoptosis. The first observation suggesting this possibility was the finding that systemic treatment with protein synthesis inhibitors protected against CA1 neuron loss after global ischemia (Goto et al., 1990; Shigeno et al., 1990), although the hypothermic effects of protein synthesis inhibitors were not considered. However, other experiments have shown that protein and RNA synthesis inhibitors do not ameliorate postischemic DND in CA1 and that this neurodegeneration is not PCD (Deshpande et al., 1992). More recent studies have focused on DNA fragmentation. By in situ DNA end-labeling methods, many studies have shown that selectively vulnerable populations of neurons undergo nuclear DNA fragmentation after global cerebral ischemia in the adult brain (Héron et al., 1993; MacManus et al., 1995; Nitatori et al., 1995). However, in situ end-labeling methods for DNA fail to discriminate among apoptotic and necrotic cell deaths (Grasl-Kraupp et al., 1995; Portera-Cailliau et al., 1997b; Martin et al., 1998) and can also detect DNA fragments during DNA synthesis (Lockshin and Zakeri, 1994); thus, these results per se cannot be interpreted as solely apoptosis. DNA integrity after global ischemia in adult brain has also been studied by gel electrophoresis. In DNA extracts of adult rat or gerbil brain, internucleosomal fragmentation has been found after transient global forebrain ischemia and has been interpreted as apoptosis occurring by PCD mechanisms (Okamoto et al., 1993; Héron et al., 1993; MacManus et al., 1995; Nitatori et al., 1995; Bhat et al., 1996). However, it is also uncertain whether internucleosomal DNA fragmentation is specific for apoptosis, because it occurs in ischemic liver necrosis (Fukuda et al., 1993), in NMDA receptor-mediated excitotoxic neuronal necrosis in adult brain (Portera-Cailliau et al., 1997b) and in culture (Gwag et al., 1997; Sohn et al., 1998), and in cells undergoing necrosis induced by calcium ionophores and heat shock (Collins et al., 1992). The analysis of DNA fragmentation patterns (and protein levels) in brain extracts is further confounded by tissue homogenization of heterogeneous cell systems, which precludes the evaluation of cell death on a cell-by-cell basis. Nonneuronal cells (e.g., astrocytes, oligodendrocytes, inflammatory cells, and vascular cells) also die following central nervous system injury, including ischemia-reperfusion and axotomy-target deprivation, and some of these nonneuronal cells die apoptotically (Martin et al., 1997a, 1998; Al-Abdulla et al., 1998). The interpretation of DNA fragmentation data is limited by the specificity and the caveats of the assay system, and, as concluded in previous studies (Kerr and Harmon, 1991; Collins et al., 1992; Portera-Cailliau et al., 1997b; Martin et al., 1998), cell structure is still the best indicator for classifying cell death in vivo. In our experiments, we found coexisting random and internucleosomal DNA fragmentation, and we attribute these different patterns to necrosis of pyramidal neurons and Purkinje cells and apoptosis of granule neurons within these regions. Our electron microscopic data support this conclusion.
This and previous ultrastructural studies illustrate that ischemic neurodegeneration in selectively vulnerable regions is not apoptosis (Kirino, 1982; Kirino and Sano, 1984; Kirino et al., 1984; Deshpande et al., 1992; Martin et al., 1998). These studies demonstrate that ischemic neurodegeneration has the typical features of cellular necrosis, consistent with the acute parenchymal cell death found in other organs after ischemia (Wyllie et al., 1980). Yet, other studies with electron microscopic data have asserted that DND of CA1 pyramidal neurons after ischemia is apoptosis (Nitatori et al., 1995), although unambiguous ultrastructural evidence of apoptosis in these neurons in the adult brain has not been demonstrated. This discrepancy regarding whether DND fits the pattern of apoptosis is likely to be due to differences in criteria for identifying neuronal apoptosis and to the detail of the electron microscopic analysis. Alternatively, DND after ischemia may fall along a structural and mechanistic apoptosis—necrosis continuum (MacManus et al., 1995; Portera-Cailliau et al., 1997a,b; Martin et al., 1998). For example, GluR-mediated excitotoxic death of neurons occurs along an apoptosis—necrosis continuum, and the structure of excitotoxic neuronal degeneration is influenced by the subtype of GluR that is activated (Portera-Cailliau et al., 1997a,b). In the adult rat brain, the degeneration of neurons caused by NMDA receptor activation is structurally necrotic; however, the neuronal death produced by non-NMDA GluR activation is distinct from that caused by NMDA receptor stimulation. This non-NMDA receptor-mediated neuronal death in adult brain has some cytoplasmic and nuclear features reminiscent of neuronal apoptosis (Portera-Cailliau et al., 1997b). Thus, like non-NMDA GluR-mediated excitotoxicity, DND after ischemia could be a hybrid of apoptosis and necrosis, with the death of these neurons not strictly apoptosis or necrosis, according to a traditional binary classification of cell death, but occurring as intermediate or hybrid forms with coexisting characteristics that lie along a structural continuum with apoptosis and necrosis at the extremes. Because this continuum is influenced by the subtype of glutamate receptor that is activated, DND after ischemia may not be identical in every neuron, possibly because of the high diversity in the expression, localization, and function of GluR subtypes or the diversity in second messenger systems in the central nervous system. Surprisingly, however, we did not find ultrastructural evidence that ischemic DND resembles non-NMDA GluR-mediated excitotoxic neuronal death, but rather it is structurally very similar to NMDA receptor-mediated excitotoxic necrosis (Portera-Cailliau et al., 1997a,b; Martin et al., 1998).
Granule neurons undergo apoptosis after ischemia
The degeneration of granule neurons in dentate gyrus and cerebellum after global ischemia sharply contrasts with the degeneration of CA1 neurons and Purkinje cells. Granule neuron death after ischemia closely resembles apoptosis. Thus, granule neurons provide an internal standard for classic apoptosis with which CA1 pyramidal neuron and cerebellar Purkinje cell degeneration after ischemia can be compared to demonstrate that the death of these latter neurons is not apoptosis. As with apoptosis in nonneuronal tissues (Kerr and Harmon, 1991), in developing brain during naturally occurring PCD of neurons (Portera-Cailliau et al., 1997a), and in some neuronal groups after axotomy-target deprivation (Al-Abdulla et al., 1998; Martin et al., 1999), apoptosis of granule neurons was characterized morphologically by nuclear and cytoplasmic condensation. The most prominent alterations were condensation of chromatin into few, large, round clumps or crescentic caps, aggregation and lamination of the cytoskeleton, and cellular shrinkage. Membranous organelles, including mitochondria, remained intact until the late stages of apoptosis. Neurodegeneration after global ischemia involves entire neural systems, and neuronal connectivity as well as metabolic factors possibly dictate the pattern of selective vulnerability (Martin et al., 1997b). Granule neuron death may be a direct consequence of the ischemia, or it may be secondary to necrotic degeneration of hippocampal pyramidal neurons and cerebellar Purkinje cells and thus is a form of target deprivation-induced apoptosis (Martin et al., 1998, 1999; Al-Abdulla et al., 1998), because the targets of dentate granule neurons include hippocampal pyramidal neurons and the targets of cerebellar granule neurons are Purkinje cells. This possibility is supported by in vitro studies showing that, in response to serum deprivation, cerebellar granule neurons undergo transcription-dependent apoptosis (D'Mello et al., 1993; Watson et al., 1998) by activation of a pathway involving c-Jun phosphorylation (Watson et al., 1998). Similar mechanisms seem to be operative in a related in vitro paradigm (i.e., low potassium) of cerebellar granule neuron apoptosis (D'Mello et al., 1993; Watson et al., 1998), and, interestingly, activation. of phosphoinositide-linked mGluRs blocks this neuronal apoptosis (Copani et al., 1995). Thus, augmented mGluR expression in cerebellar and hippocampal granule neurons after cerebral ischemia may signify a physiological attempt at neuroprotection rather than an event mediating the apoptosis.
Contribution of programmed cell death mechanisms in ischemic neurodegeneration
Gene products that regulate PCD of mammalian cells have been studied after ischemia. Bax mRNA is increased in both vulnerable and less vulnerable regions after ischemia (Chen et al., 1996). Changes in mRNA after cerebral ischemia are difficult to interpret in light of damage to organelles that function in protein synthesis and posttranslational processing, thereby potentially rendering the translation and formation of mature products inefficient. By immunoblotting, Bax protein in hippocampus is increased transiently at 6 hours after ischemia but then returns to control levels (Krajewski et al., 1995). We found no changes in Bax protein levels in hippocampus at 1 and 7 days after ischemia, but we observed an increase in Bax in cerebellum at 7 days after ischemia. A possible explanation for a lack of detection of changes in Bax protein levels in hippocampus by immunoblotting is that apoptotic death of dentate gyrus granule neurons is less frequent than granule neuron death in cerebellum (see Fig. 3). Also, granule neurons compose a greater population of cells in cerebellum than in hippocampus (West et al., 1991; Korbo et al., 1993). However, a sustained postischemic increase in Bax protein levels has been shown in hippocampus but not cerebellum (Chen et al., 1996), although the contributions of different cells to this observation have not been identified. Important in this regard is the finding that mRNA and protein levels for the proapoptotic cysteine protease ICE interleukin-1 converting enzyme (caspase-1) are increased in hippocampus after global ischemia in gerbils, but this change is associated with inflammatory cells rather than selectively vulnerable CA1 pyramidal neurons (Bhat et al., 1996). In addition, after global ischemia in rat, caspase-3 mRNA levels are progressively elevated in CA1 at 24 to 72 hours, although most of the neurons are already lost by 72 hours, whereas caspase-3 mRNA is transiently elevated in dentate gyrus granule cells at 8 hours after ischemia (Chen et al., 1998). This latter pattern would be more consistent with a role for caspase-3 in the granule neuron apoptosis that we have identified. On the other hand, Bcl-2 overexpression in transgenic mice reduces hippocampal pyramidal neuron degeneration after global ischemia (Kitagawa et al., 1998), although this effect of Bcl-2 cannot yet be specifically ascribed to antiapoptotic activity, because this protein has additional functions in injured neurons (Chen et al., 1997). It is still uncertain, however, whether the absence of a classic apoptotic structure in selectively vulnerable neurons after ischemia is sufficient evidence to exclude the possibility that PCD may be operative, because all forms of PCD may not occur via apoptosis (Schwartz et al., 1993).
Metabotropic glutamate receptor signaling pathways after ischemia
The distinct forms of degeneration in different populations of hippocampal and cerebellar neurons after ischemia were accompanied by differential changes in proteins involved in phosphoinositide-coupled mGluR signal transduction. These changes were more easily identified by immunocytochemistry rather than by immunoblotting (except for PLCβ in cerebellum), perhaps because of overlapping and differential contributions of distinct populations of neurons that are not divisible in gel analyses of regional homogenates. Necrosis of CA1 neurons and Purkinje cell is associated with a loss of mGluRs and PLCβ. In contrast, apoptosis in granule cell populations in hippocampus and cerebellum is paralleled by increased expression of these proteins. Our results are consistent with the reported loss of mGluR1 and mGluR5 mRNA in CA1 after global ischemia (Iversen et al., 1994). This observation is not surprising when considering the postsynaptic, somatodendritic localization of these proteins (Martin et al., 1992; Blue et al., 1997) and the rapid damage to dendritic membranes and the cytoskeletal proteolysis that occur in these neurons after ischemia (Kitagawa et al., 1989; Yamamoto et al., 1990). These abnormalities further support our conclusion that hippocampal pyramidal neurons and Purkinje cells undergo necrosis after ischemia. In contrast, in granule neurons of dentate gyrus and cerebellum, immunoreactivity for mGluRs and PLCβ is enhanced. This finding is consistent with the concept that apoptosis results from the de novo expression or activation of a PCD program (Sen, 1992), but it is not clear whether changes in PLC-coupled mGluRs participate directly in the mechanisms for apoptosis in these neurons or whether these changes reflect the activation of compensatory survival signaling pathways (Copani et al., 1995).
The possible roles for PLC-coupled mGluRs in the mechanisms for necrotic degeneration of selectively vulnerable neurons after ischemia are uncertain. Activation of mGluRs is necessary for Purkinje cell survival in vitro (Mount et al., 1993) and protects neurons from oxidative stress (Sagara and Schubert, 1998). Thus, a loss of somatodendritic phosphoinositide-linked mGluRs in Purkinje cells and CA1 neurons may have a role in the necrosis of these neurons after ischemia. Alternatively, this loss is possibly a consequence of necrotic degeneration of these neurons. It is possible that augmented activity of PLC-coupled mGluR signal transduction proteins does not participate in the primary mechanisms for DND of CA1 pyramidal neurons and cerebellar Purkinje cells after ischemia. This conclusion is supported by the finding that neither pharmacological blockade of mGluRs nor ablation of the mGluR1 gene reduces ischemic and excitotoxic brain injury in vivo (Ferraguti et al., 1997). However, other observations suggest that endogenous activation of mGluR1 contributes to ischemic and excitotoxic neuronal degeneration (Bruno et al., 1999). Additional work is necessary to precisely identify the role of mGluR signaling in neurodegeneration after ischemia.
Footnotes
Acknowledgments
The authors thank Ann Price and Dawn Spicer as well as the technical staff of the Department of Pathology electron microscope core facility (Marilyn Miller, Barbara Plantholt, and Gerald Horne) for their assistance.