
Abstract
Although profound hypothermia has been used for decades to protect the human brain from hypoxic or ischemic insults, little is known about the underlying mechanism. We therefore report the first characterization of the effects of moderate (30°C) and profound hypothermia (12° to 20°C) on excitotoxicity in cultured cortical neurons exposed to excitatory amino acids (EAA; glutamate, N-methyl-D-aspartate [NMDA], AMPA, or kainate) at different temperatures (12° to 37°C). Cooling neurons to 30°C and 20°C was neuroprotective, but cooling to 12°C was toxic. The extent of protection depended on the temperature, the EAA receptor agonist employed, and the duration of the EAA challenge. Neurons challenged briefly (5 minutes) with all EAA were protected, as were neurons challenged for 60 minutes with NMDA, AMPA, or kainate. The protective effects of hypothermia (20° and 30°C) persisted after rewarming to 37°C, but rewarming from 12°C was deleterious. Surprisingly, however, prolonged (60 minutes) exposures to glutamate unmasked a temperature-insensitive component of glutamate neurotoxicity that was not seen with the other, synthetic EAA; this component was still mediated via NMDA receptors, not by ionotropic or metabotropic non-NMDA receptors. The temperature-insensitivity of glutamate toxicity was not explained by effects of hypothermia on EAA-evoked [Ca2+]i increases measured using high- and low-affinity Ca2+ indicators, nor by effects on mitochondrial production of reactive oxygen species. This first characterization of excitotoxicity at profoundly hypothermic temperatures reveals a previously unnoticed feature of glutamate neurotoxicity unseen with the other EAA, and also suggests that hypothermia protects the brain at the level of neurons by blocking, rather than slowing, excitotoxicity.
Keywords
Induction of hypothermia has long been viewed as a strategy to protect the nervous system from hypoxic, ischemic, traumatic, and inflammatory injuries. Profound degrees of hypothermia have been induced in human subjects for several decades for this purpose, beginning with early experiences with patients suffering from head trauma or central nervous system malignancies (Fay 1940, 1945, 1959). Pioneering work on profound whole-body cooling to protect the brain during cardiovascular and neurosurgical procedures (Bigelow et al., 1950; Lougheed et al., 1955; Rosomoff, 1957; Drake et al., 1964) led to the introduction of deep hypothermia (cooling to 10° to 15°C) into modern clinical neurosurgical practice (Spetzler et al., 1988; Solomon et al., 1991; Solomon, 1994). In recent years, it has become widely appreciated that milder hypothermia (brain cooling of a few degrees Celsius) also confers dramatic neuroprotection in a number of experimental models of brain injury, particularly in animal models of focal and global cerebral ischemia (Busto et al., 1987; Dietrich, 1992; Baker et al., 1992; Ginsberg et al., 1993). However, possibly owing to long-standing clinical experience, profound hypothermia has remained the preferred modality for protecting the human brain in some specialized aspects of cardiovascular and neurosurgical practice.
Despite the repeated therapeutic use of profound hypothermia on human subjects in past decades, the mechanisms by which brain cooling prevents hypoxic or ischemic damage remain unknown. From the onset, reports have confirmed that hypothermia reduces cerebral oxygen metabolism in animals and in humans (Bigelow et al., 1950; Lougheed and Kahn, 1955; Ausman et al., 1993), a feature thought to impart the brain with increased resilience to ischemic insults. To date, many reports have convincingly validated neuroprotection by cooling tissues to 10° to 20°C (Lougheed and Kahn, 1955; Rosomoff, 1957; Michenfelder and Milde, 1992; Gillinov et al., 1993; Shaver et al., 1995; Rokkas et al., 1995), but no cellular studies of deep hypothermia exist to describe the underlying protective mechanisms.
We therefore studied hypothermic protection in cultured cortical neurons. In this system, neurons are separated from the influences of cerebral blood flow and systemic metabolism, which are also temperature sensitive, and are amenable to precise studies of cell viability and physiology under conditions mimicking hypoxia or ischemia and excitotoxicity (Olney, 1978; Choi et al., 1987; Rothman and Olney, 1987; Goldberg et al., 1987, 1989; Goldberg and Choi, 1990). Given the recent finding that cooling of only a few degrees Celsius can protect the brain from ischemia, a number of investigations of mild to moderate hypothermia have been published using similar in vitro systems (Arai et al., 1993; Shuaib et al., 1993; Bruno et al., 1994). However, no reports have examined the effects of profound hypothermia on excitotoxicity, despite the fact that the latter is the modality most commonly applied in clinical settings.
Given the paucity of published data, we sought to examine the behavior of cultured cortical neurons under a range of hypothermic conditions and to study the possible mechanisms by which hypothermia protects against excitotoxicity. A lower limit of 12°C was chosen to reflect the temperatures used in past animal studies and in current clinical practice in human subjects (Spetzler et al., 1988; Solomon et al., 1991; Solomon, 1994). Here we show that moderate (30°C) and profound (20°C) hypothermia blocks, rather than delays, excitotoxicity at the level of individual neurons, though the extent of protection depends on the temperature, the excitatory amino acid (EAA) used, and the EAA challenge duration. Also, we show that prolonged exposures to L-glutamate unmask a temperature-insensitive component of glutamate neurotoxicity unseen with other EAA, and characterize several features of this phenomenon.
METHODS
Tissue culture
Mixed cortical cell cultures containing both neurons and glia were prepared from embryonic Swiss mice at 15 days of gestation as previously described (Hertz et al., 1989; Sattler et al., 1997). Briefly, cerebral cortices taken from 10 to 12 embryos were incubated for 10 to 12 minutes in 0.05% trypsin in EDTA, dissociated by trituration, and plated on poly-L-ornithine-coated 24-well plates (Corning) at a density of 0.43 × 106 cells/well. Only 20 of the 24 wells were plated with cells, leaving 4 wells (one column) available for use as controls, particularly for the purposes of background subtraction of fluorescence data (see below). Cultures used in fluorescence imaging experiments were plated on no. 1 glass coverslips (Faust TMBH, Cologne, Germany) and grown in 35-mm culture dishes. Plating medium consisted of Eagle's minimum essential media (MEM, Earle's salt) supplemented with 10% heat-inactivated horse serum, and 2 mmol/L glutamine, 25 mmol/L glucose, and 26 mmol/L bicarbonate. The cultures were maintained at 37°C in a humidified 5% CO2 atmosphere. After 3 to 5 days in vitro, growth of non-neuronal cells was halted by a 24- to 48-hour exposure to 10 μmol/L FDU-solution (5 μmol/L uridine, 5 μmol/L (+)-5-fluor-2′-deoxyuridine). The cultures were used for experiments after 12 to 16 days in vitro.
Drugs and solutions
The control solution contained (in mmol/L): 121 NaCl, 5 KCl, 20 D-glucose, 10 HEPES acid, 7 HEPES-sodium salt, 3 NaHCO3, 1 sodium pyruvate, 1.8 CaCl2, and 0.01 glycine, adjusted to pH 7.4 with NaOH. Stock solutions of nimodipine (Miles Pharmaceuticals Inc.), 6-cyano-7-nitroquinoxaline (CNQX; Research Biochemicals Inc.), dihydrorhodamine 123 (10 mmol/L; Molecular Probes Inc., Cat# D-632, Lot: 4531-7), fura-2/AM, and calcium green-5N/AM (1 mmol/L; Molecular Probes Inc.) were prepared in dimethyl sulfoxide and kept at −20°C until used. DL-2-Amino-5-phosphonovaleric acid (APV; Sigma Chemical Co., St. Louis, MO, U.S.A.), MK-801 (Research Biochemicals Inc.), and trans-1-aminocyclopentane-1,3-dicarboxylic acid (t-ACPD; Tocris Neuramin) stocks were prepared in distilled water and also stored at −20°C until used. Because dihydrorhodamine (DHR) can undergo photo-oxidation, stocks of this dye were used within 24 hours of preparation. Stock solutions of glutamate, N-methyl-D-aspartate (NMDA), AMPA, and kainate were prepared before each use in control solution. Propidium iodide (PI; 1 mg/mL stock; Molecular Probes Inc.) was prepared in distilled water and dissolved to a final concentration of 50 μg/mL. This concentration of PI produced no observable effects on cell morphology or survival, as demonstrated by the low cell mortality in all control groups. The drugs were always diluted to their final concentrations in control solution. All solutions were sterile-filtered before use. Unless otherwise noted above, all chemicals were obtained from Sigma.
Cell survival assay
Quantitative measurements of cell death were performed in cultures undergoing experimental manipulations as described by Sattler et al. (1997). Briefly, a Cytofluor II fluorescence multiwell plate scanner (PerSpective Biosystems, Framingham, MA, U.S.A.) was modified to allow us to accurately set and maintain the temperature in the scanner's interior to within 0.2°C. Modifications included the addition of insulating material and of thermoelectric heater and cooler assemblies controlled by a precision bipolar thermoelectric controller (Power Puncher BP, Alpha-Omega Instrument Corp. RI, U.S.A.; see Sattler et al., 1997). The fluorescence scanner uses a quartz halogen lamp (spectral range 320 to 700 nm) and appropriate optical filters (530 ± 25 nm excitation, 620 ± 40 nm emission) to excite PI and record its fluorescence emissions.
Outside the multiwell plate scanner, cultures were maintained for the duration of the experiment at 12°, 20°, 30°, or 37°C by placing them in an insulated, humidified chamber (Igloo Cool-Mate™, model 4502, Houston, TX, U.S.A.), in which temperature was also controlled by a thermoelectric heater and cooler powered by a bipolar thermoelectric controller as above.
To measure cell death, the culture medium in each culture well was replaced with control solution containing 50 μg/mL PI and a baseline fluorescence reading was taken in the multiwell scanner. After this, sequential fluorescence readings were taken at appropriate intervals during and for 24 hours after the experimental manipulations. The following equation was then used to calculate the fraction of dead cells in each culture, at any given time:
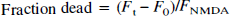
where Ft = PI fluorescence at time t, F0 = initial PI fluorescence at time zero, and FNMDA = background-subtracted PI fluorescence of identical cultures from the same dissection and plating, 24 hours after a 60-minute exposure to 1 mmol/L NMDA. From manual observations at the time of validation of this technique (Sattler et al., 1997), this NMDA exposure routinely produced nearly complete neuronal death in each culture, but had no effect on surrounding glia (also see Bruno et al., 1994; David et al., 1996).
Loading of neurons with indicator dyes
For imaging the free cytoplasmic Ca2+ concentrations ([Ca2+])i, neurons were loaded with Ca2+ indicators at 37°C. The cultures were incubated for 60 minutes in control solution containing either 1 μmol/L fura-2/AM or 5 μmol/L calcium green-5N/AM in a final concentration of 0.2% dimethyl sulfoxide. The cultures were then washed in control solution for a further 20 to 30 minutes to attenuate any background fluorescence from residual extracellular indicator.
For measuring or imaging mitochondrial production of reactive oxygen species (ROS), cultures were loaded at 37°C with 5 μmol/L dihydrorhodamine in control solution for 30 minutes and then used for experiments. This cell-permeant compound is oxidized by ROS into its charged fluorescent derivative rhodamine 123, which localizes preferentially in mitochondria, thereby indicating mitochondrial ROS production (Dugan et al., 1995).
Quantitative assessment of dihydrorhodamine oxidation
Oxidation of DHR in the cultures, induced by EAA at different temperatures, was gauged by measuring the changes in DHR fluorescence with time in the multiwell plate scanner. Baseline DHR fluorescence readings were taken, after which the cultures were exposed to one of five concentrations of an EAA agonist with appropriate antagonists, and fluorescence readings were taken from each well every 5 minutes for 60 minutes. Optical filters for DHR were 485 ± 20 nm excitation, 530 ± 30 emission. The relative degree of DHR oxidation evoked at any time by a given concentration of an EAA acid was expressed as the functional increase in DHR fluorescence: (Ft − F0) / F0, where Ft is the DHR fluorescence recorded at time t and F0 is the baseline fluorescence at time = 0.
Calcium measurements
Cultures loaded with Ca2+ indicator were mounted in a temperature-controlled microscope-stage incubator (Medical Systems Corp., model TC-202) and viewed with an inverted microscope (Nikon Diaphot, equipped with xenon epifluorescence optics) through a fluorite oil-immersion lens (Nikon CF UV-F ×40, NA = 1.3) in contact with the coverslip bottom. A second-generation microchannel-plate intensified CCD-array camera (Quantex Corp., model QX-100) was used to record the fluorescence emissions of the Ca2+ indicator (>510 nm for fura-2, 530 ± 15 nm for calcium green-5N) evoked by excitation through narrow band-pass filters (fura-2: 340 ± 5 nm and 380 ± 6.5 nm; calcium green-5N: 485 ± 11 nm) housed in a computer-controlled filter wheel. Fluorescence images were gathered on an 80486-based personal computer. The system allowed for a maximal time resolution of 2 seconds between successive [Ca2+]i measurements. Four to eight images were averaged at each excitation wavelength. Each average was corrected for background fluorescence and camera dark current by subtracting a frame taken at the beginning of each experiment at each excitation wavelength from an area of the coverslip devoid of cells. Changes in [Ca2+]i in individual neurons were determined by averaging the fluorescence values for all pixels within a boundary traced around the perimeter of the cell soma before starting the experiment. Results were expressed either as the 340/380 nm fura-2 fluorescence ratio, or as the fractional change in calcium green-5N fluorescence: (Ft − F0) / F0 where Ft is the fluorescence at time t, and F0 is the baseline fluorescence. Pilot experiments using zero Ca2+ and high (1 mmol/L) Ca2+ solutions revealed that the fluorescence of fura-2 excited by 340 and 380 nm was unaffected by temperature changes from 20°C to 37°C.
The cultures were perfused with control solution at 1.2 mL/minute. In each experiment, changes in [Ca2+]i evoked by brief (20 to 30 seconds) applications of glutamate and NMDA (30 μmol/L) were imaged in the same neurons at 20°C and at 37°C.
Confocal imaging of dihydrorhodamine fluorescence
Cultures, grown on glass coverslips and loaded with DHR as above, were placed in the microscope-stage incubator at 20°C or at 37°C and were mounted on a Nikon Diaphot 300 microscope. They were viewed through a ×40 oil-immersion lens (Nikon CF UV-F ×40, NA = 1.3) using an inverted laser-scanning confocal microscope equipped with an argon/krypton laser (MRC 1000, Bio-Rad, Hertfordshire, England). The following confocal parameters were used: 488 nm excitation, ⩾515 nm emission wavelengths, pin-hole size set at 2.8 mm, laser intensity attenuated to 1%, gain at 1320, black level at zero. After acquiring a baseline image, NMDA or L-glutamate (300 μmol/L each) was applied to the cultures and images were acquired every 5 minutes for up to 60 minutes.
Experimental procedures
All experimental manipulations for cell survival experiments took place under sterile conditions in a culture hood. The temperature of all solutions was adjusted before use in the temperature-controlled chamber housing the cultures throughout the experiment. Typically, after taking a baseline PI fluorescence reading, the cultures in a given multiwell plate were exposed to one of five concentrations of an EAA agonist with or without antagonists for either 5 or 60 minutes (Table 1). After this, they were returned to control solution, also maintained at the given experimental temperature. Additional PI fluorescence measurements were then taken in the multiwell plate scanner at appropriate intervals. Experiments carried for 24 hours were performed simultaneously in up to 6 multiwell plates, each of which was maintained in the temperature-controlled chamber except when measuring PI fluorescence in the scanner. Thus, several experimental variations (e.g., different EAA) could be performed in many cultures under identical conditions. All experiments were replicated in at least 2 to 4 cultures. In some experiments, 50-μL aliquots of solution were removed from the culture wells at different times, stored at −80°C, and transferred within 24 hours to a commercial laboratory (HSC Biotechnology Service Center, Toronto, Ont., Canada) for high-pressure liquid chromatography determinations of the quantity of glutamate in the medium.
Summary of experiments performed for the present report
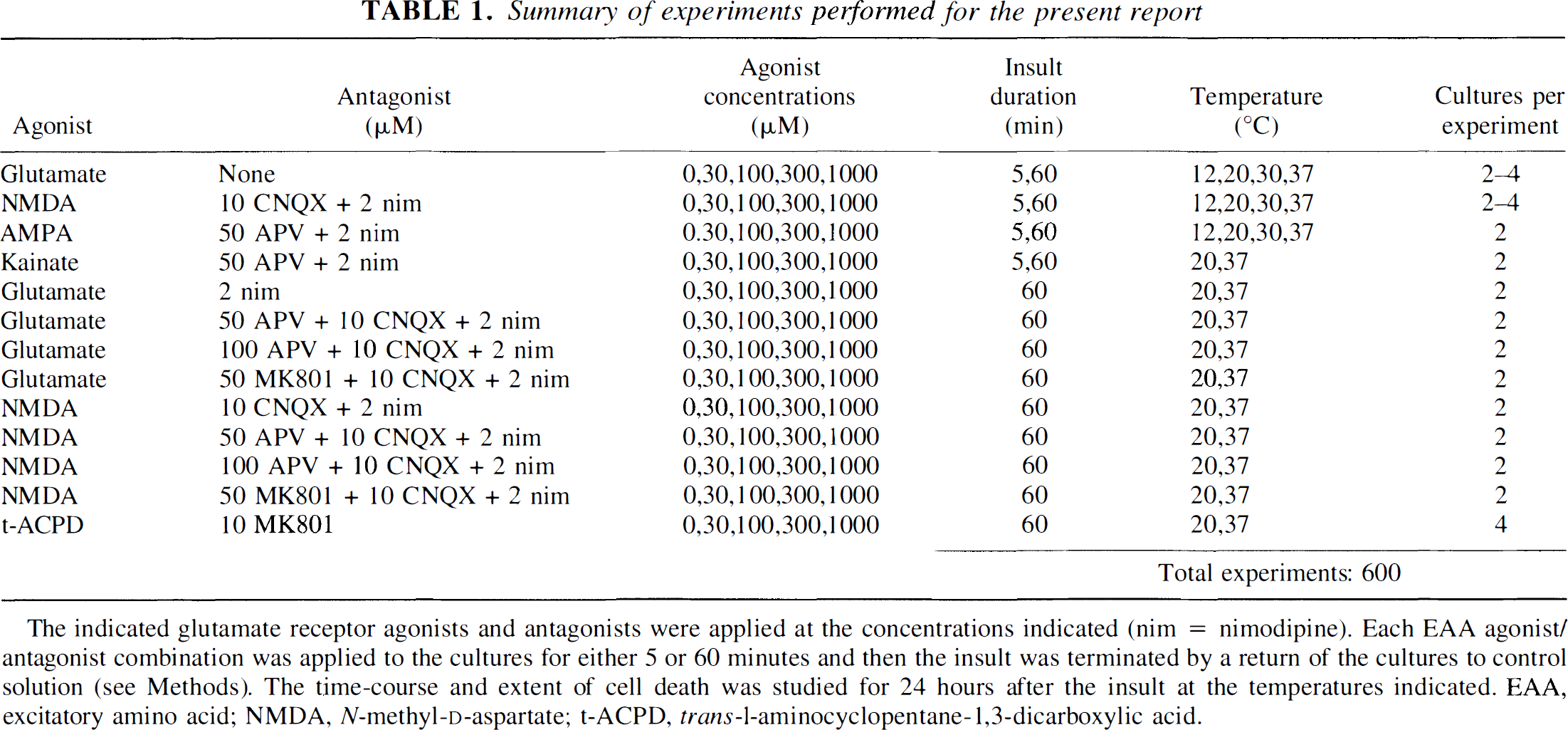
The indicated glutamate receptor agonists and antagonists were applied at the concentrations indicated (nim = nimodipine). Each EAA agonist/antagonist combination was applied to the cultures for either 5 or 60 minutes and then the insult was terminated by a return of the cultures to control solution (see Methods). The time-course and extent of cell death was studied for 24 hours after the insult at the temperatures indicated. EAA, excitatory amino acid; NMDA, N-methyl-D-aspartate; t-ACPD, trans-1-aminocyclopentane-1,3-dicarboxylic acid.
Data analysis
Owing to the large number of inter-related experiments in this report, all data were initially evaluated by multiple linear regression analysis (Armitage and Berry, 1987) to determine the significance of each major experimental parameter (agonist type, agonist concentration, insult duration, and temperature). Subsequent analyses were by Student's t test using the Bonferroni method (Altman, 1991) to evaluate differences between individual group means. Details related to the analysis of a specific data set are included in the appropriate figure legends.
RESULTS
We examined the modulatory effects of a range of temperatures on the time course and extent of excitotoxic injury in cultured cortical neurons (Table 1). Temperatures from 12° to 37°C were studied to encompass both “moderate” (30°C) and “profound” (12° to 20°C) hypothermia, respectively (Busto et al., 1987, 1989; Ginsberg et al., 1993; Bruno et al., 1994). Excitotoxicity was produced using a range of concentrations of NMDA and non-NMDA receptor agonists applied for different durations (5 or 60 minutes). In most experiments (Table 1), appropriate antagonists of glutamate receptors or calcium channels were added to ensure that toxicity was produced specifically by the selected agonist rather than by the secondary activation of glutamate receptors or calcium channels through endogenous EAA release or cell membrane depolarization.
Hypothermia attenuates excitotoxicity triggered by a brief (5 minutes) EAA challenge
Excitotoxicity has been well characterized in past studies at 37°C. Typically, brief (5 to 15 minutes) applications of NMDA or glutamate trigger significant toxicity in cortical neurons during 24 hours (Choi, 1987; Choi et al., 1987; Bruno et al., 1994), whereas longer insults (45 to 60 minutes) accelerate the neurodegeneration seen (Tymianski et al., 1993a, 1993b). Non-NMDA receptor agonists are also toxic, but trigger neurodegeneration more slowly both after brief (Tymianski et al., 1993b) and prolonged (Rothman et al., 1987; Koh et al., 1990) insults. Because little is known about the effects of excitotoxic insults on neuronal survival at lower temperatures, we first examined in detail the effects of brief (5 minutes), and more prolonged (60 minutes) EAA exposures on neuronal survival at temperatures ranging from 12° to 37°C.
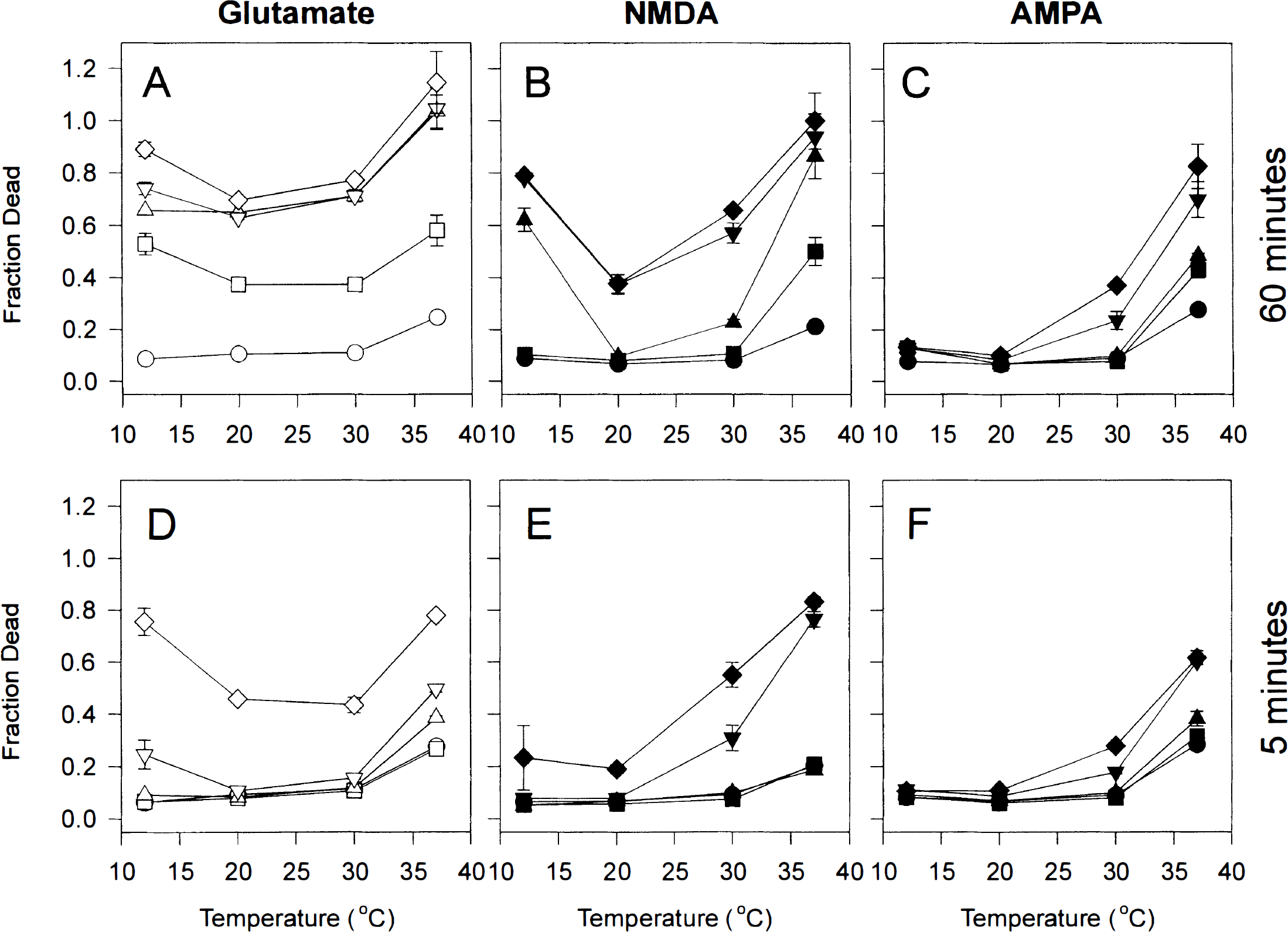
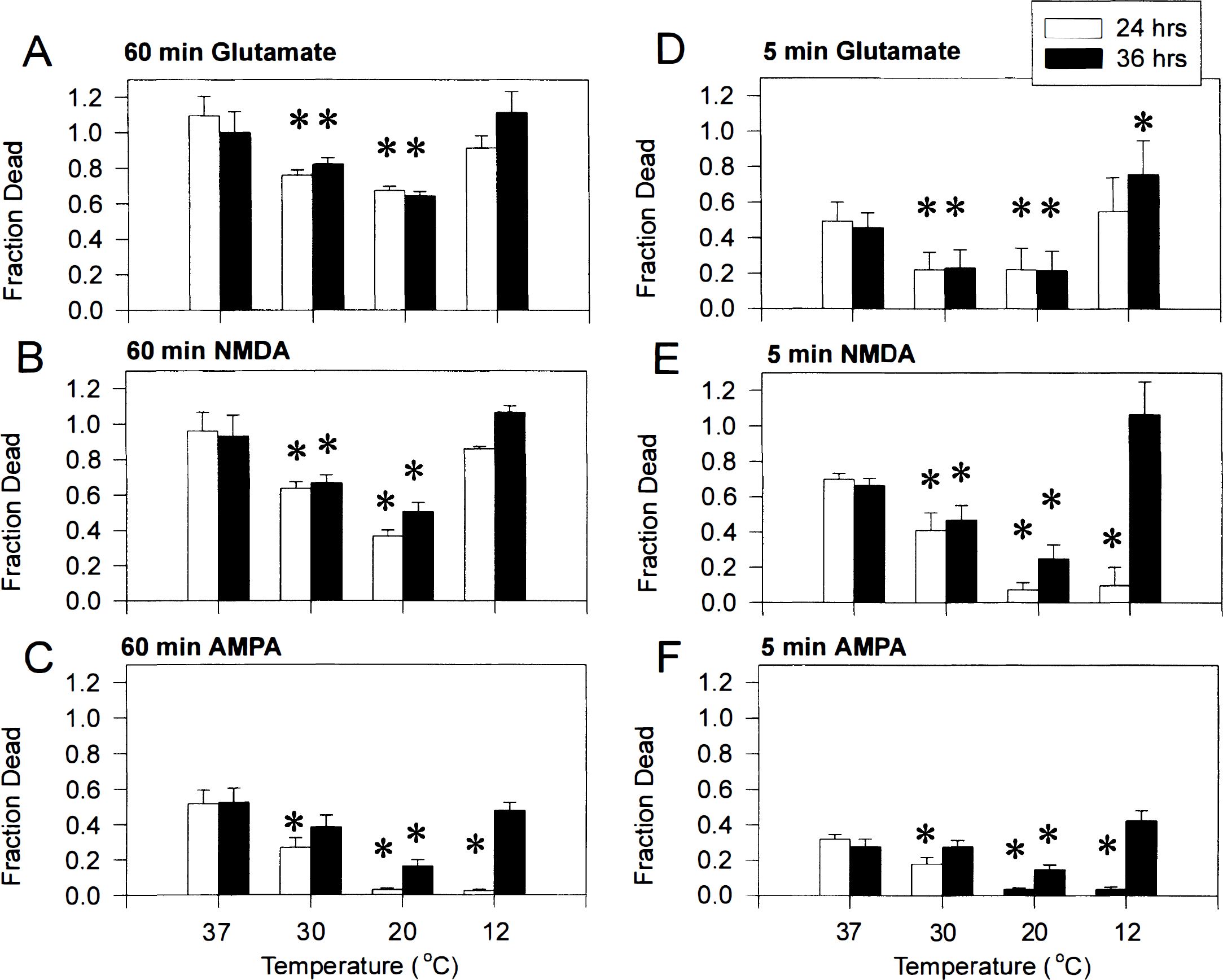
Representative effects of 5-minute EAA applications at 37° and 20°C are shown in Fig. 1A. At 37°C, cultures exposed to a range of glutamate concentrations (0 to 1,000 μmol/L) exhibited a time- and concentration-dependent increase in cell death that was highest with 1,000 μmol/L glutamate (diamonds in Fig. 1A, panel a; almost 80% cell death by 24 hours). Exposure to NMDA (Fig. 1A, panel b) and AMPA (Fig. 1A, panel c) also triggered neurodegeneration in a time- and concentration-dependent manner. Lowering the temperature to 20°C conferred dramatic protection against all three EAA across the entire concentration-response range (Fig. 1A, panels d-f). Protection was complete in AMPA-challenged cultures, in which no toxicity was evident even at the highest agonist concentration used (1,000 μmol/L; Fig. 1A, panel f). Cell death was also markedly attenuated in cultures challenged with glutamate and NMDA, in which toxicity was evident only with the highest agonist concentrations (Fig. 1A, panels d and e).
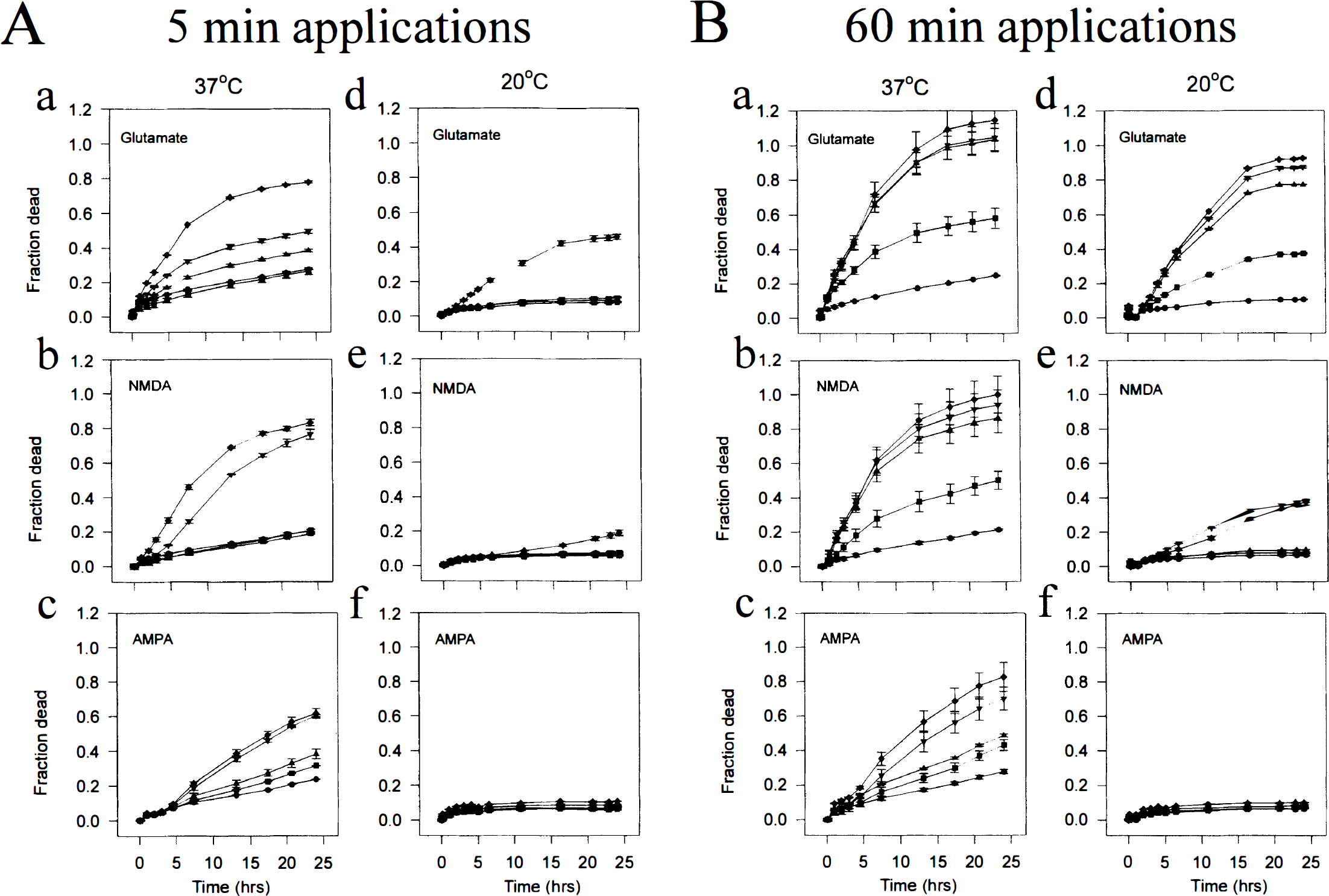
Representative experiments showing the effects of 5-minute
Hypothermia also protects against prolonged excitatory amino acid insults (60 minutes), although the extent of protection varies with the excitatory amino acid agonist
Representative effects of a longer (60 minutes) application of glutamate, NMDA, or AMPA on the time course and extent of neurotoxicity are shown in Fig. 1B. These longer EAA exposures were more neurotoxic than the 5-minute insults for each EAA, each concentration, and each temperature (e.g., compare Fig. 1A, panels a-c and 1B, panels a-c). As in the brief EAA exposure experiments, lowering the temperature to 20°C conferred dramatic protection against the toxicity of NMDA and of AMPA (Fig. 1B, panels e and f). However, unlike NMDA and AMPA toxicity, the toxicity produced by a 60-minute glutamate insult was relatively resistant to the effects of hypothermia (Fig. 1B, panel d).
The impact of hypothermia on excitatory amino acid toxicity is U shaped
The effects of the agonist type (glutamate, NMDA, or AMPA), concentration, and temperature were evaluated as shown in Fig. 1 using a total of 360 cultures. Multiple regression analysis revealed each of these variables to be a significant independent determinant of cell death (multiple linear regression, n = 360, F = 89.8, P < 0.00001 for each variable). The data were then further subdivided into groups according to the EAA exposure duration, the agonist used, and agonist concentration (Fig. 2A–F). Multiple pairwise comparisons were used to determine significant differences in cell survival at different temperatures (Table 2).
Effects of temperature on 24-hour neuronal survival after an excitatory amino acid challenge
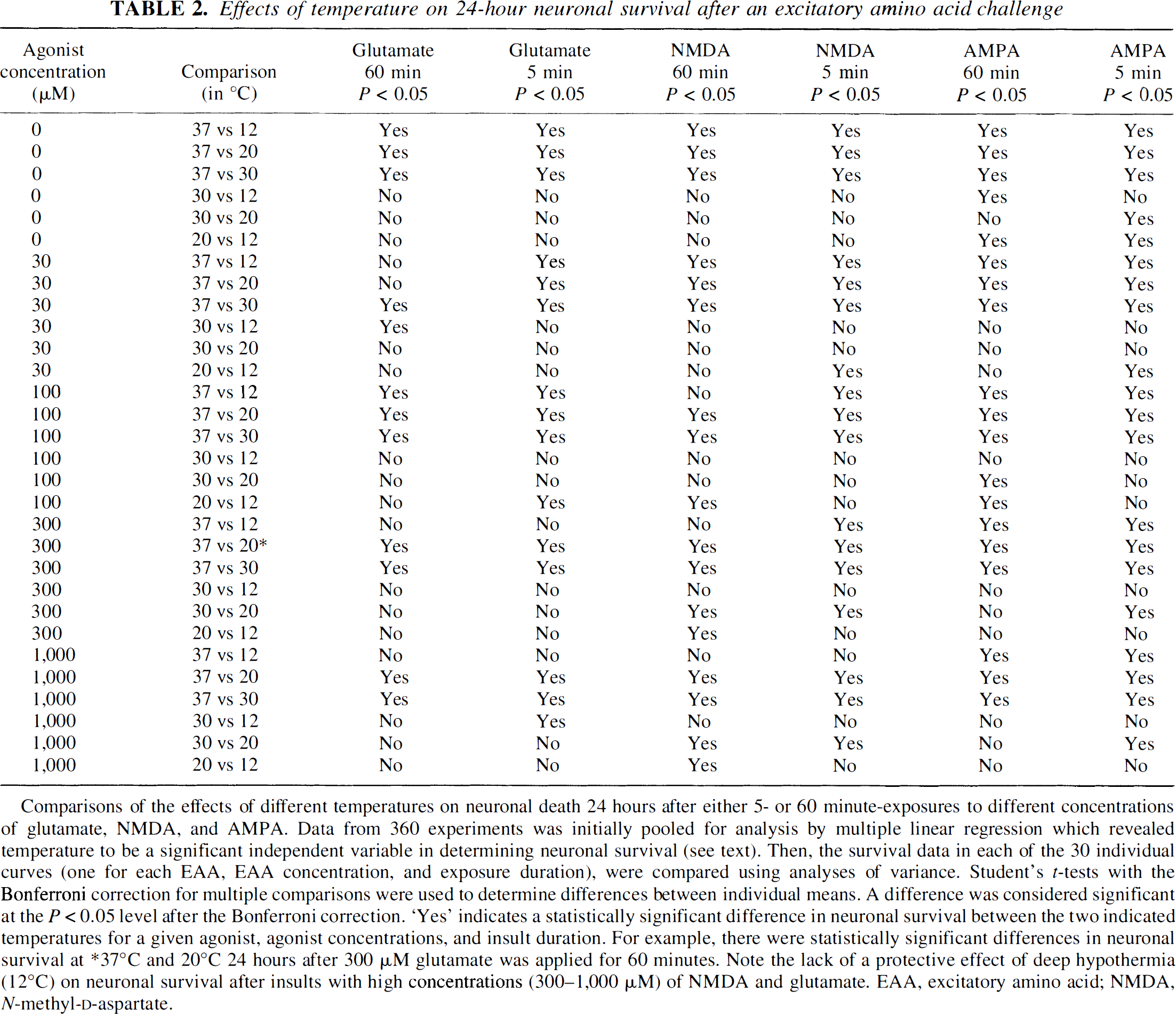
Comparisons of the effects of different temperatures on neuronal death 24 hours after either 5- or 60 minute-exposures to different concentrations of glutamate, NMDA, and AMPA. Data from 360 experiments was initially pooled for analysis by multiple linear regression which revealed temperature to be a significant independent variable in determining neuronal survival (see text). Then, the survival data in each of the 30 individual curves (one for each EAA, EAA concentration, and exposure duration), were compared using analyses of variance. Student's t-tests with the Bonferroni correction for multiple comparisons were used to determine differences between individual means. A difference was considered significant at the P < 0.05 level after the Bonferroni correction. “Yes” indicates a statistically significant difference in neuronal survival between the two indicated temperatures for a given agonist, agonist concentrations, and insult duration. For example, there were statistically significant differences in neuronal survival at ∗37°C and 20°C 24 hours after 300 μM glutamate was applied for 60 minutes. Note the lack of a protective effect of deep hypothermia (12°C) on neuronal survival after insults with high concentrations (300-1,000 μM) of NMDA and glutamate. EAA, excitatory amino acid; NMDA, N-methyl-
The protective effects of hypothermia are U shaped. Cultures were challenged for either 60 minutes
For any given glutamate concentration (30 to 1,000 μmol/L), cell death at 24 hours was a U-shaped function of temperature (Fig. 2A and D). Thus, lowering the temperature from 37° to 30°C imparted a protective effect, but lowering it further to 20°C did not (Table 2). Furthermore, at 12°C, glutamate neurotoxicity was higher than at either 20° or 30°C, resembling in extent that seen at normothermia. A similar U-shaped response to temperature was observed with cultures challenged with NMDA (Fig. 3B and E), except that lowering the temperature from 30° to 20°C conferred a further degree of protection not seen with glutamate. The increased toxicity of both NMDA and glutamate at 12°C as compared with 20° to 30°C was highly significant (multiple linear regression, n = 120, F = 58.8, P < 0.0001).
Neurotoxicity produced by 60-minute insults with glutamate is relatively temperature insensitive when compared with that of NMDA. Each bar represents the fraction of dead neurons (mean + SD of two to four experiments) 24 hours after a 60-minute insult with a range of glutamate or NMDA concentrations (0 to 1,000 μmol/L). Experiments were performed at 12° to 37°C
Lowering the temperature from 37° to 30°C and then to 20°C also produced significant stepwise reductions in AMPA toxicity, which was abolished at 20°C (Fig. 2C and F). Unlike with NMDA and glutamate, reducing the temperature to 12°C did not enhance AMPA toxicity throughout the 24-hour observation period.
Neurotoxicity from prolonged glutamate insults is relatively temperature insensitive
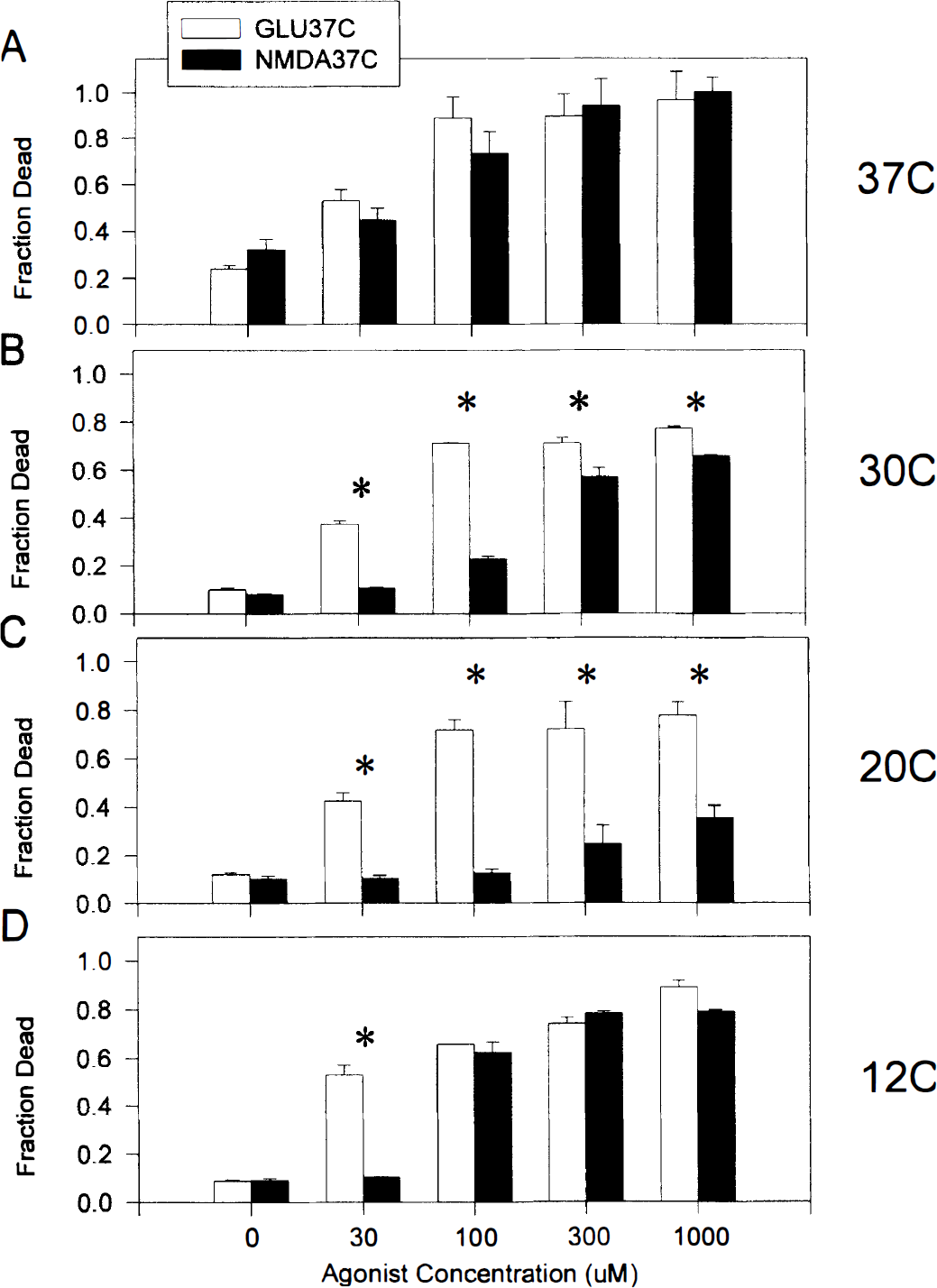
A surprising finding is the relative insensitivity of a 60-minute glutamate insult to the protective effects of hypothermia (compare Fig. 1B, panels a and d). This was investigated further in a total of 180 cultures exposed for 60 minutes to a range of glutamate or NMDA concentrations at 12°, 20°, 30°, or 37°C (Fig. 3). Under normothermic conditions (37°C; Fig. 3A), equimolar concentrations of glutamate and NMDA were equally toxic. However, lowering the temperature to 30°C had a minor impact on the toxicity of glutamate, whereas NMDA toxicity declined substantially, particularly at the lower agonist concentrations (Fig. 3B). The disparity between glutamate and NMDA toxicity was further accentuated at 20°C, such that NMDA toxicity was almost completely abolished whereas glutamate toxicity was unaffected. This phenomenon disappeared at 12°C owing to the enhancement of NMDA toxicity at this temperature (see above).
Sixty-minute challenges with AMPA and kainate, and brief (5 minutes) challenges with glutamate, NMDA, AMPA, and kainate were all less toxic at lower temperatures (Table 3 and Table 4; 60 cultures and 140 cultures, respectively). Unlike NMDA toxicity, AMPA and kainate toxicity was not enhanced at the lowest temperature (12°C). At 37°C, the toxicity of high AMPA (300 to 1,000 μmol/L for 60 minutes) exceeded that of equimolar quantities of kainate (Table 3).
Effects of temperature on neuronal survival after a 60-minute challenge with non-NMDA receptor agonists
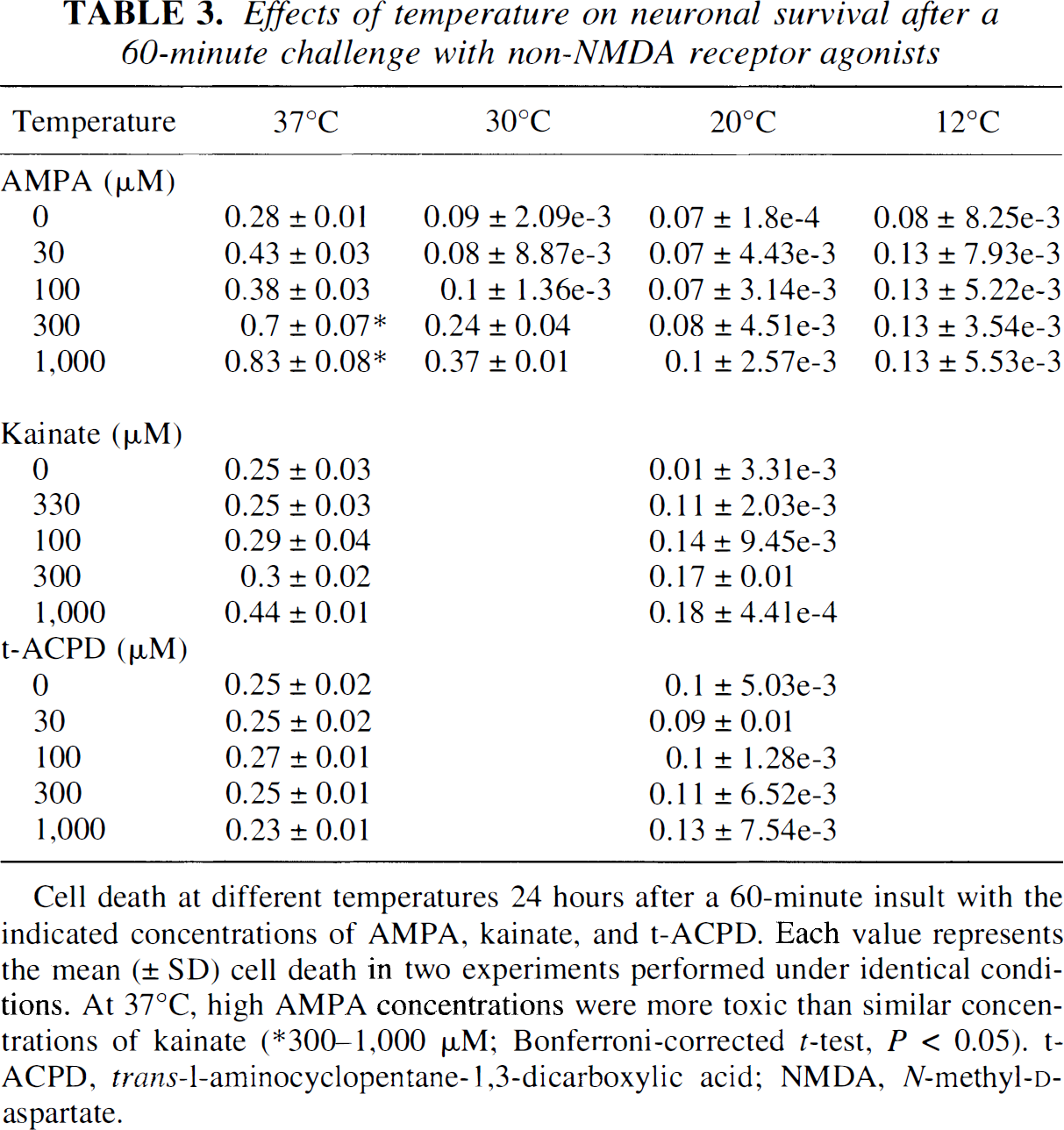
Cell death at different temperatures 24 hours after a 60-minute insult with the indicated concentrations of AMPA, kainate, and t-ACPD. Each value represents the mean (± SD) cell death in two experiments performed under identical conditions. At 37°C, high AMPA concentrations were more toxic than similar concentrations of kainate (∗300–1,000 μM; Bonferroni-corrected t-test, P < 0.05). t-ACPD, trans-1-aminocyclopentane-1,3-dicarboxylic acid; NMDA, N-methyl-
Glutamate toxicity was attenuated by hypothermia in the brief (5 minutes) exposure experiments, and the phenomenon of temperature insensitivity of toxicity seen with the 60-minute glutamate exposures was not observed (Fig. 1A; Table 4). No differences were found between the toxicity of glutamate and NMDA at any of the temperatures after the 5-minute application (multiple linear regression, n = 80, F = 30.0, P = 0.1823).
Effects of temperature on neuronal survival after a 5-minute EAA challenge
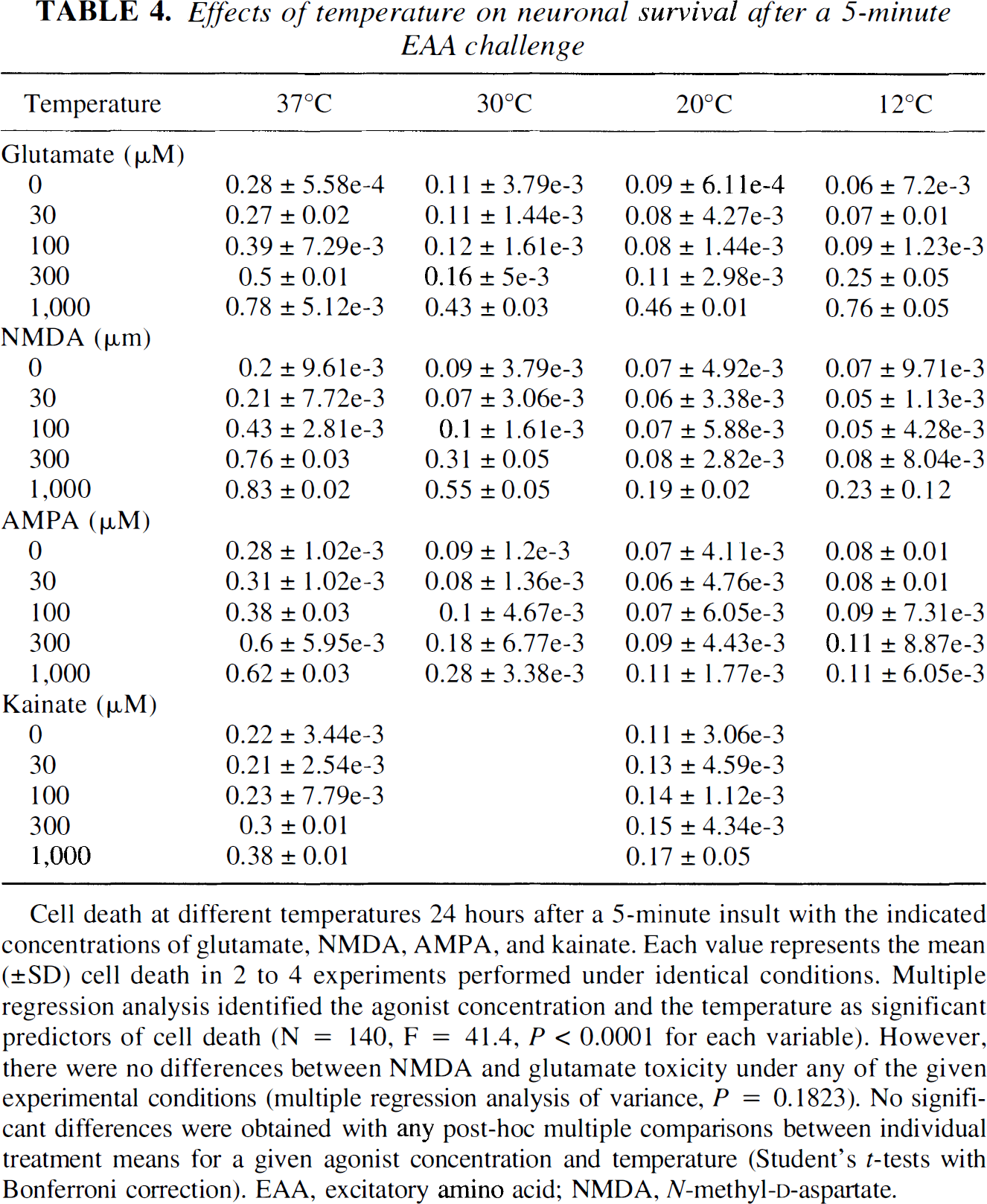
Cell death at different temperatures 24 hours after a 5-minute insult with the indicated concentrations of glutamate, NMDA, AMPA, and kainate. Each value represents the mean (±SD) cell death in 2 to 4 experiments performed under identical conditions. Multiple regression analysis identified the agonist concentration and the temperature as significant predictors of cell death (N = 140, F = 41.4, P < 0.0001 for each variable). However, there were no differences between NMDA and glutamate toxicity under any of the given experimental conditions (multiple regression analysis of variance, P = 0.1823). No significant differences were obtained with any post-hoc multiple comparisons between individual treatment means for a given agonist concentration and temperature (Student's t-tests with Bonferroni correction). EAA, excitatory amino acid; NMDA, N-methyl-D-aspartate.
The protective effects of hypothermia persist after rewarming to 37°C
To determine whether the protective effects of hypothermia persist on rewarming, the cultures from experiments shown in Fig. 4 and Table 3 and Table 4 were returned to the 37°C incubator after the 24-hour observation period at the lower temperatures. After an additional 12 hours, cell death was measured again.
Persistence of neuroprotection by hypothermia after rewarming to 37°C for 12 hours. After the 24-hour time point, all cultures used in the experiments shown in Figs. 1, 2 and 3 and Table 3 and Table 4 were transferred from the experimental temperature (12°, 20°, 30°, or 37°C) to a 37°C incubator for an additional 12 hours. A final measurement of cell death was then made. The panels show results of experiments in which glutamate
At all temperatures greater than 12°C, there were no increases in cell death throughout the 12-hour rewarming period (Fig. 4). At 12°C, cultures challenged with NMDA agonists for 60 minutes were not protected, and thus toxicity remained after rewarming (Fig. 4A and B). Neurons challenged with AMPA for 5 or 60 minutes at 12°C were apparently initially protected. However, cell death returned to control levels on rewarming (Fig. 4C and F). Furthermore, in neurons challenged with glutamate and NMDA for 5 minutes (Fig. 8D), cell death actually exceeded control levels after rewarming from 12°C (Fig. 8D and E). Thus, neuroprotection by hypothermia to 20° to 30°C persisted after rewarming, whereas protection after cooling to 12°C did not. This suggests that hypothermia acts by aborting the initiation or propagation of neurotoxic cascades rather than by simply slowing down the neurodegenerative processes triggered by excitotoxicity. However, excessive cooling may abrogate this process or trigger its own toxic sequelae.
The temperature-insensitive component of glutamate neurotoxicity is mediated via NMDA receptors, not via ionotropic non-NMDA receptors or metabotropic receptors
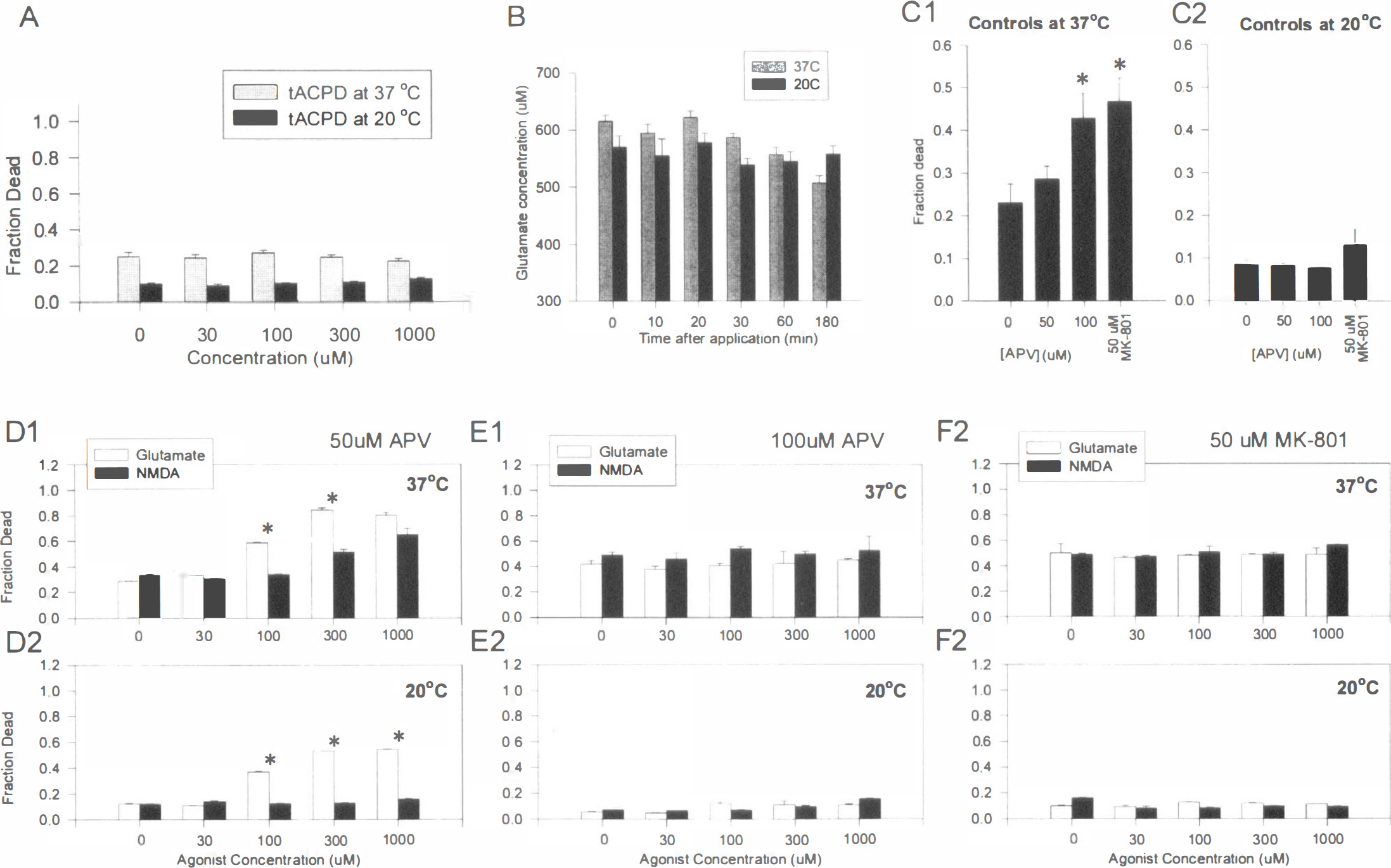
Glutamate, the endogenous excitatory neurotransmitter, activates all ionotropic and metabotropic glutamate receptors whereas NMDA activates only the one receptor subtype. The excess toxicity of glutamate at hypothermic temperatures could not have been caused by upregulating non-NMDA ionotropic receptor toxicity, because the latter was reduced, not increased, by hypothermia (Figs. 1 and 2; Table 3 and Table 4). However, metabotropic glutamate receptors (mGluR) are also activated by L-glutamate and may participate in neurotoxic events by affecting endogenous glutamate release, intracellular Ca2+ mobilization, phospholipase activation, or cAMP formation (Watkins and Collinridge, 1994; Nicoletti et al., 1996). To determine whether mGluR can contribute to the neurodegeneration seen here, we challenged cultures at 37°C and at 20°C with t-ACPD (0 to 1,000 μmol/L), a group I and II mGluR agonist (mGlu1–3 and mGlu5; Watkins and Collinridge, 1994). MK-801 (10 μmol/L), an NMDA receptor antagonist, was added throughout the experiment to prevent any secondary activation of NMDA receptors, which could trigger toxicity through other pathways. However, t-ACPD was nontoxic at either temperature (Fig. 5A), excluding a role for t-ACPD-sensitive mGluR in glutamate toxicity in this culture model.
The temperature-insensitive component of glutamate toxicity is mediated by NMDA receptors.
Another way to collectively examine the role of mGluR in glutamate toxicity is to activate them with glutamate in the presence of ionotropic glutamate receptor antagonists. To this end, the cultures were challenged with a range of glutamate concentrations in the presence of DL-APV or MK-801, competitive and noncompetitive NMDA receptor antagonists, respectively, and CNQX, an antagonist of AMPA and kainate receptors (Table 1; Fig. 5D–F). Nimodipine, a Ca2+ channel blocker, was also included in all solutions to attenuate any secondary contribution of voltage-sensitive Ca2+ channels.
At 37°C, glutamate neurotoxicity in the presence of 50 μmol/L DL-APV was unaffected across a range of glutamate concentrations (Fig. 5, panel D1), whereas NMDA toxicity was significantly attenuated. At 20°C, 50 μmol/L DL-APV completely blocked NMDA toxicity, but had a significantly lesser impact on glutamate toxicity (Fig. 5, panel D2). This persisting disparity between the toxicity of glutamate and NMDA at lower temperatures was finally abolished by increasing the concentration of DL-APV to 100 μmol/L (Fig. 5, panel E1 and E2). The same result was also obtained when 50 μmol/L MK-801 was used (Fig. 5, panels F1 and F2). In other experiments, lesser concentrations of MK-801 (5 to 10 μmol/L) produced similar results (not shown).
Because the temperature-insensitive component of glutamate neurotoxicity could be completely blocked by sufficient concentrations of NMDA receptor antagonists, it is likely that this phenomenon is mediated by NMDA receptor activation, and not via ionotropic non-NMDA receptors or via metabotropic receptors.
NMDA receptor antagonists possess an inherent, temperature-dependent toxicity
About 75% to 80% of control cultures maintained in control solution at 37°C survived for 24 hours. This was increased to more than 90% at 20°C (see controls in Fig. 3A and D). Interestingly, at 37°C, the survival of neurons in control (EAA-untreated) cultures was reduced in the presence of DL-APV or MK-801 in a concentration-dependent manner (Fig. 5, panel C1). This NMDA antagonist toxicity was temperature-sensitive, as hypothermia (20°C) markedly attenuated the toxicity of both NMDA receptor antagonists (Fig. 5, panel C2). It is uncertain whether the NMDA antagonist toxicity seen here in vitro corresponds to the toxicity of these same agents reported in rat brains after in vivo administration (Olney et al., 1989; Olney, 1994; Fix et al., 1993, 1995). However, such in vitro toxicity is apparently a property of high concentrations of NMDA antagonists (Freeman and Goldberg, 1994), which were necessary to affect the temperature-insensitive component of glutamate neurotoxicity (Fig. 5D–F).
Impairment of glutamate reuptake by hypothermia does not explain the temperature insensitivity of glutamate neurotoxicity
Synaptic transmission at central mammalian excitatory synapses is largely terminated by a reuptake mechanism, which clears glutamate from the synaptic cleft. Glutamate transport is a sodium- and potassium-coupled process that exists in both neurons and in astroglia (Rothstein et al., 1994). If hypothermia attenuates this process (Williamson and Neale, 1992), then impaired glutamate clearance could potentially contribute to the persistence of glutamate toxicity seen in this study. To explore this directly, we measured the effect of lowering temperature on the clearance of L-glutamate from the culture medium (Fig. 5B). When 1 mmol/L glutamate was added to cultures, the measured glutamate concentration remained unchanged in the experimental solution for up to 3 hours. Cultures maintained at 37°C were no better at clearing this endogenous EAA from the medium than were cultures at 20°C. This is contradictory to thermodynamic arguments, on the basis of which glutamate reuptake should slow down at 20°C. However, in addition to reducing uptake, hypothermia may also reduce the release of endogenous glutamate that, at 37°C, contributes significantly to neurodegeneration (Tymianski and Sattler, unpublished data). This could potentially nullify the effect of hypothermia on glutamate reuptake, and may account for the result obtained (Fig. 5B). Regardless of the interpretation, given that temperature insensitivity of glutamate neurotoxicity was evident when 1 mmol/L glutamate was used (Fig. 4), it is unlikely that impairment of glutamate reuptake by hypothermia alone explains this phenomenon.
Hypothermia attenuates EAA-evoked [Ca2+]i increases
Toxicity to NMDA, but not to glutamate, differed at 37° and 20°C. However, both ligands acted via the NMDA receptor (Fig. 5). One possibility is that hypothermia decreases the affinity of NMDA, but this effect is less marked with glutamate as the ligand. This could be examined using binding assays (Yoneda and Ogita, 1991), but these would miss any effect of temperature on ion channel kinetics that could differ between the two ligands. The alternative is to measure the end result associated with neurotoxicity, which is Ca2+ loading. Under this hypothesis, both NMDA and glutamate should cause similar Ca2+ loading at 37°C, but NMDA-induced Ca2+ loading should decrease preferentially at the lower temperatures. To examine this, we measured changes in [Ca2+]i evoked by two EAA at 37° and 20°C in the same cells.
Initial measurements were performed using fura-2, a ratiometric Ca2+ indicator that would obviate any potential confounding effects of temperature on dye loading and retention, cell geometry, and light path. The results were adjusted for the effects of temperature on the affinity of fura-2 for Ca2+ (see Fig. 6 legend). Twenty-second applications of 30 μmol/L glutamate or NMDA were used because these concentrations produced non-saturating [Ca2+]i responses that did not decay after several applications to the same cells. Also, at 37°C, these concentrations produced the same degree of toxicity in the current preparation (see Fig. 3). The [Ca2+]i measurements were performed in the same neurons at both 20° and 37°C.
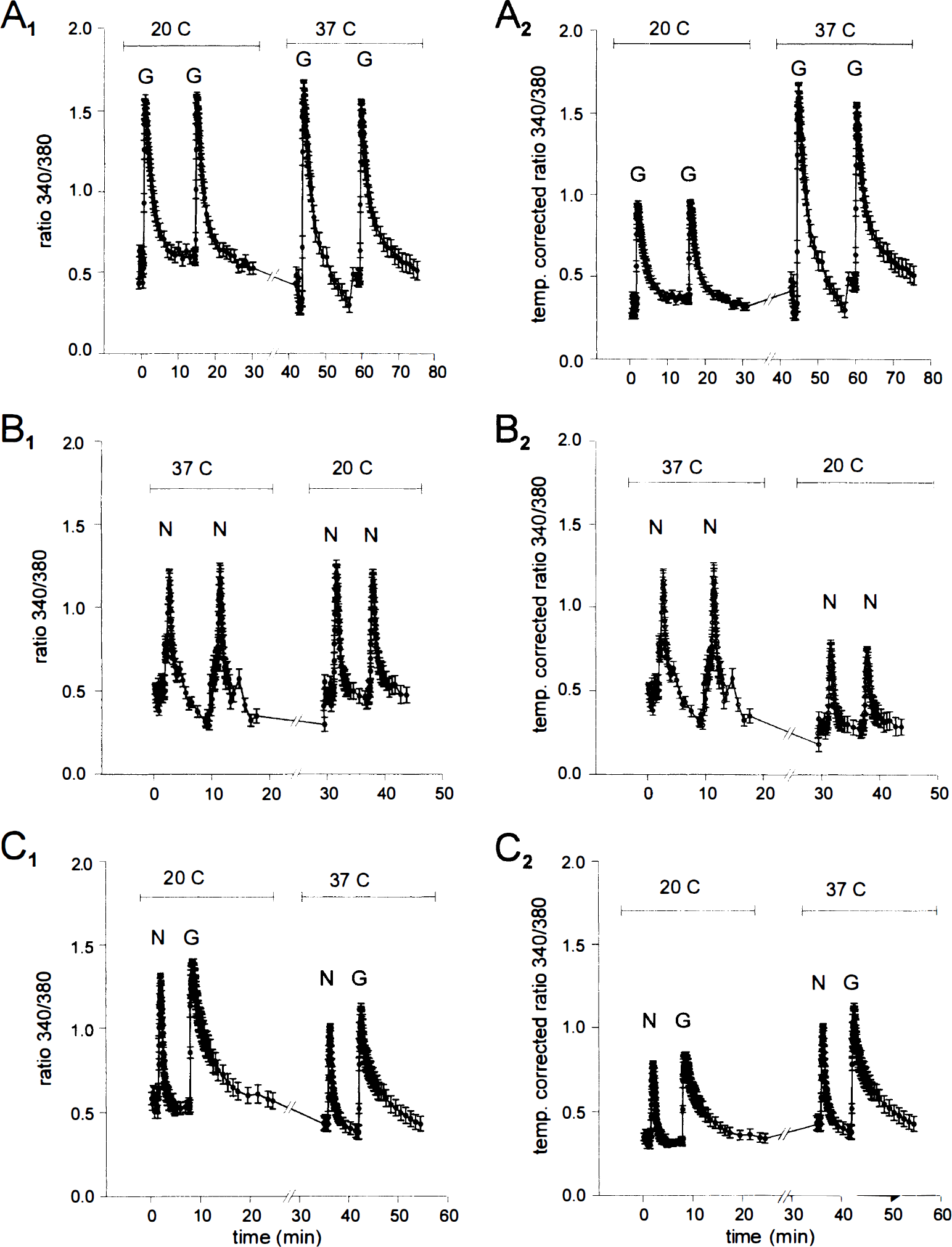
[Ca2+]i imaging with fura-2 suggests that hypothermia equally attenuates glutamate- (G; 30 μmol/L) and NMDA- (N; 30 μmol/L) evoked [Ca2+]i increases. Both EAA were applied for 20 seconds. [Ca2+]i transients were evoked at 20° and 37°C in the same neurons in each experiment. Changes in [Ca2+]i are presented as 340/380 nm fura-2 fluorescence ratios (panels
Glutamate applications produced transient, fully reversible [Ca2+]i increases at both temperatures (Fig. 6, panel A1) that, when corrected for the effects of temperature on fura-2, were larger at 37° than at 20°C (Fig. 6, panel A2). A similar effect was seen with NMDA applications (Fig. 6B). However, no preferential attenuation of NMDA-evoked [Ca2+]i elevations was detected at the lower temperature (Fig. 6C). Thus, hypothermia apparently attenuates [Ca2+]i increases evoked by both EAA. However, measurements with fura-2 failed to reveal any differences between the effects of NMDA and of L-glutamate.
A low-affinity Ca2+ indicator reveals a disparity between glutamate and NMDA-evoked [Ca2+]i increases
Measurements of [Ca2+]i with fura-2 may underestimate actual stimulus-evoked changes in [Ca2+]i. For example, measurements of depolarization-evoked [Ca2+]i transients in cardiac myocytes revealed that the peak [Ca2+]i transient amplitude measured with furaptra (magfura, a low-affinity Ca2+ indicator with an affinity for Mg2+ ions) was four- to sixfold greater than that measured with fura-2 (Berlin and Konishi, 1993). Similar discrepancies in values reported by low- and high-affinity Ca2+ indicators have been described in cerebellar granule cell presynaptic terminals (Regehr and Atluri, 1995). Such discrepancies may arise from Ca2+ buffering by fura-2, which prevents [Ca2+]i from rising to high levels, or from saturation of the indicator dye. Both artifacts could be minimized by using a low-affinity Ca2+ indicator. Therefore, we used calcium green-5N, an indicator having a 10-fold or more lower Ca2+ affinity than fura-2 (Kd for Ca2+ about 3.3 μmol/L), to measure whether differences between glutamate- and NMDA-evoked neuronal [Ca2+]i increases exist in our preparation at 37° and 20°C. Calcium green-5N is a nonratiometric dye, and the effects of temperature on its Ca2+ affinity are less well characterized. Nevertheless, it is useful for comparing the effects of the two NMDA receptor agonists on [Ca2+]i relative to each other at each temperature. Brief applications of glutamate and NMDA (30 μmol/L each) were used as before. Figure 7A and B reveals that at both 20° and 37°C glutamate produced considerably larger [Ca2+]i changes than similar applications of NMDA, a finding consistent with binding assays (Yoneda and Ogita, 1991) and electrophysiological studies (Lester and Jahr, 1992) that confirm that NMDA receptors have a greater affinity for L-glutamate than for NMDA. It is likely therefore, that the lower affinity indicator better reflects the ability of the given EAA agonist (glutamate or NMDA) to raise [Ca2+]i than the fura-2 measurements (Fig. 6). However, the ratio of the glutamate- to NMDA-evoked [Ca2+]i transient peaks was similar at 20° and 37°C. Thus, this result also failed to reveal any temperature-dependent differences in the relative abilities of NMDA and glutamate to raise [Ca2+]i.
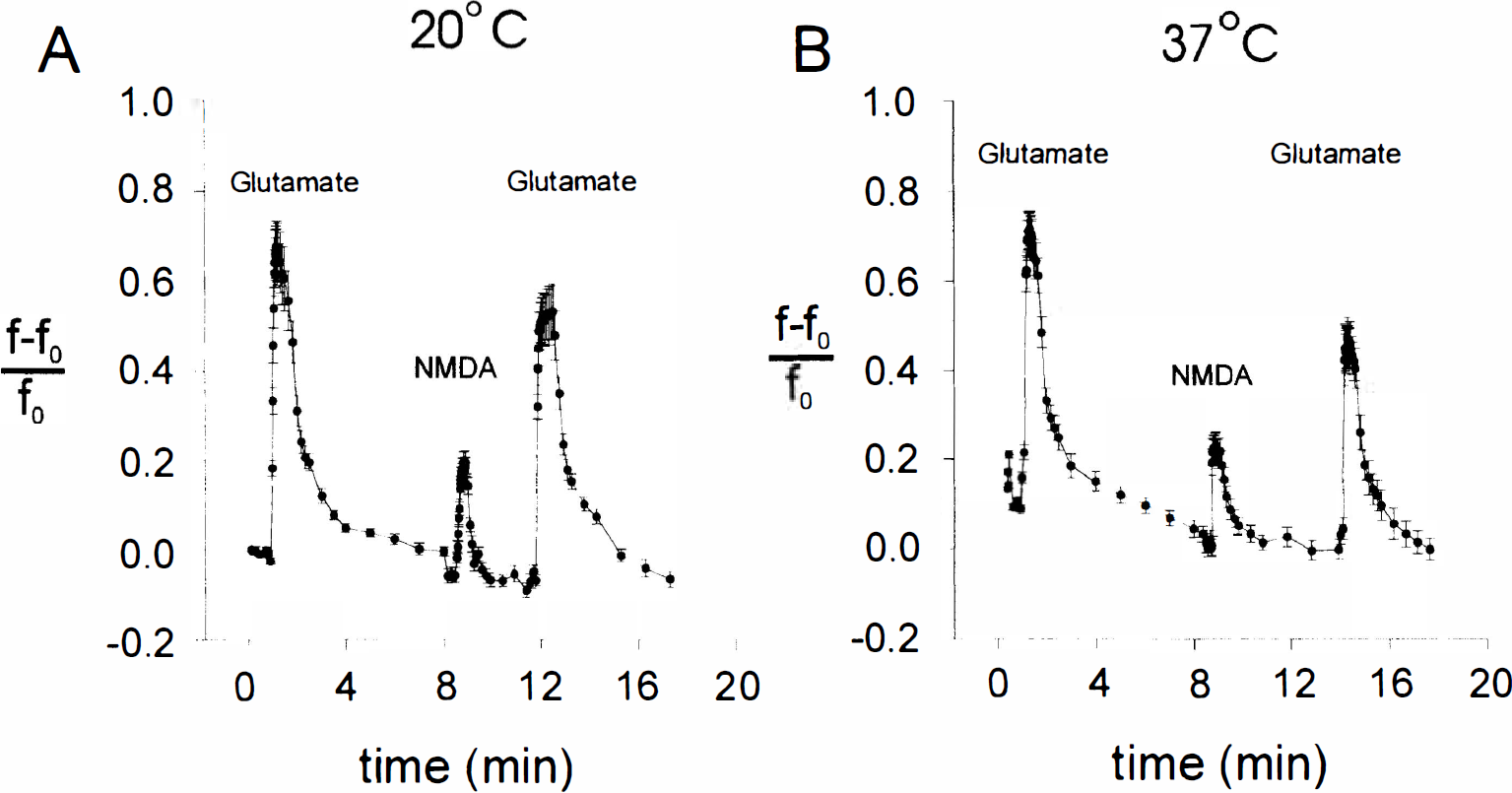
[Ca2+]i measurements with a low-affinity Ca2+ indicator calcium green-5N (Kd ≈ 3.3 μmol/L at 22°C) suggest that glutamate (30 μmol/L) evokes greater [Ca2+]i increases than NMDA (30 μmol/L) at both 20° and 37°C. [Ca2+]i transients were elicited using 20-second exposures to either glutamate or to NMDA. Changes in [Ca2+]i are expressed as the fractional increase in calcium green-5N fluorescence intensity (see Methods). Each tracing represents the mean [Ca2+]i response (± SD) obtained from the number of neurons (n) indicated in each panel legend.
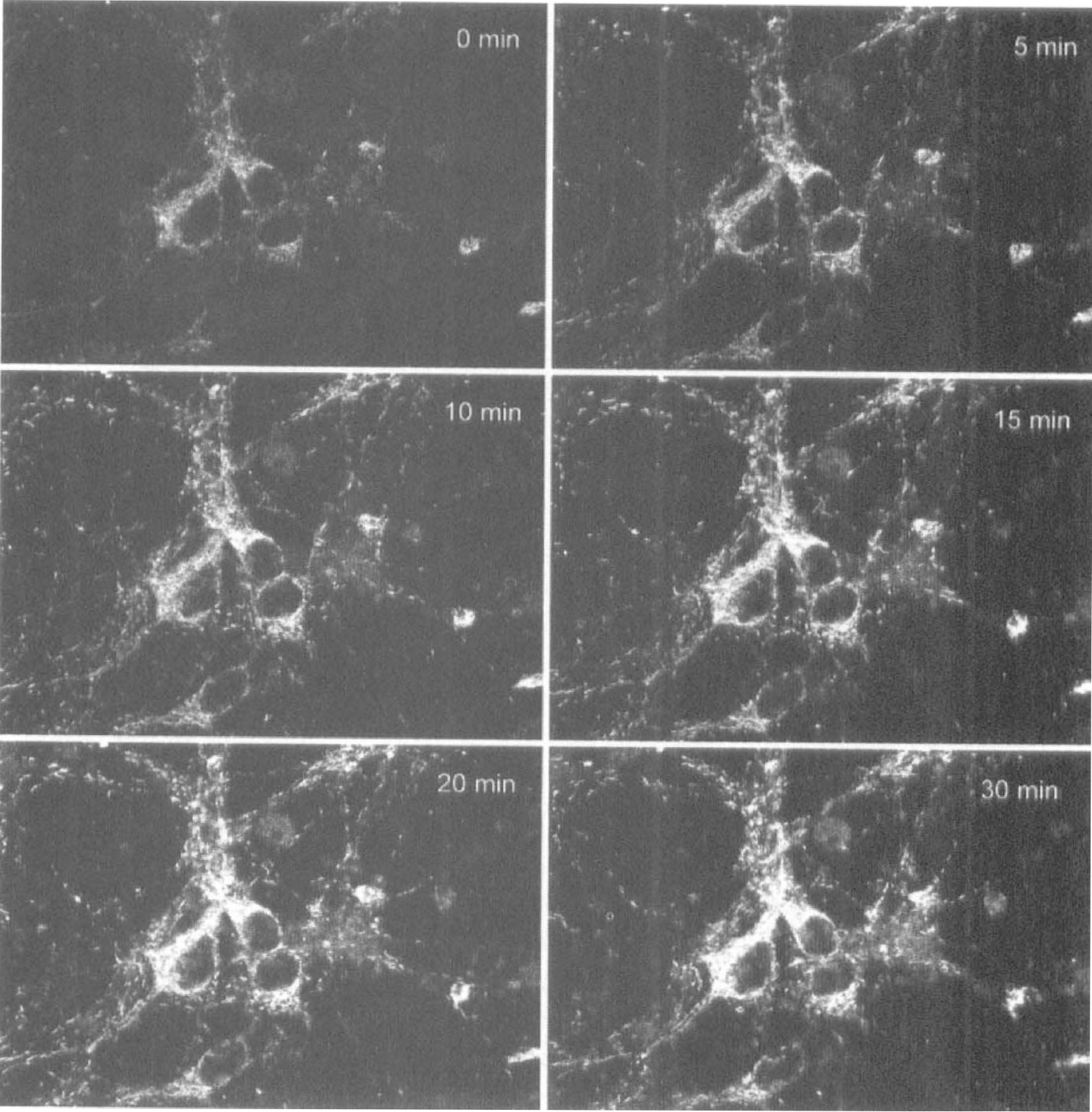
Confocal micrographs of dihydrorhodamine fluorescence in three cultured cortical neurons immediately before (0 minutes) and every 5 minutes for 30 minutes after exposure to 300 μmol/L NMDA. Increases in DHR fluorescence were restricted to punctate structures compatible with mitochondria in neurons and were not observed in the underlying glial monolayer (not shown in this confocal plane). Scale bar is 25 μm. DHR, dihydrorhodamine; NMDA, N-methyl-D-aspartate.
Hypothermia does not affect excitatory amino acid-evoked mitochondrial production of reactive oxygen species
The [Ca2+]i measurements have not provided an explanation for the relative temperature insensitivity of glutamate neurotoxicity. We therefore examined whether the disparity in glutamate and NMDA toxicity at lower temperatures could be explained by an EAA-activated process occurring subsequent to both NMDA receptor activation and Ca2+ influx. We examined the production of ROS, as this phenomenon is known to be triggered by NMDA receptors and has frequently been implicated as a mediator of neurotoxicity (Pellegrini-Giampietro et al., 1990; Lafon-Cazal et al., 1993; Coyle and Puttfarcken, 1993; Reynolds and Hastings, 1995). We studied EAA-evoked mitochondrial ROS production with the oxidation-sensitive indicator dihydrorhodamine 123 (DHR), because this process was reported to be selectively associated with Ca2+ influx through NMDA receptors and could not be triggered by cell membrane depolarization with KCl or by non-NMDA receptor agonists (Dugan et al., 1995). Specifically, we tested the hypothesis that hypothermia would preferentially attenuate NMDA-evoked ROS production more than that evoked by glutamate.
First, we tested whether loading our neurons with DHR produces a mitochondrial localization of the dye and an increase in mitochondrial fluorescence attributable to EAA application. Cultures grown on glass coverslips were loaded with DHR and viewed with a laser-scanning confocal microscope set for high gain and low laser intensity to minimize photo-oxidation of the dye (see Methods). Fig. 8 is representative of four experiments at 20°C in which cultures were exposed to 300 μmol/L NMDA after the acquisition of a baseline image (t = 0 minutes). The punctate distribution of the dye and the absence of fluorescence from nuclear areas indicate that DHR fluorescence likely originated from mitochondria (Dugan et al., 1995). Application of NMDA caused a progressive increase in DHR fluorescence during 30 minutes, which was confined primarily to neurons and their processes. Similar changes were not observed in the absence of NMDA, nor in the underlying glial monolayer, which was visualized in a different confocal plane (not shown). Similar increases of DHR fluorescence could be produced by applying L-glutamate (300 μmol/L; four experiments, not shown). Experiments at 36.5°C produced similar results with both NMDA and L-glutamate (eight experiments, not shown).
Dihydrorhodamine fluorescence can be quantified from the confocal images (Dugan et al., 1995). However, this approach is labor intensive, produces data from only a few neurons at a time, and may involve a subjective component as it requires one to manually define cell boundaries in each fluorescence image. Having shown that the DHR signal was EAA sensitive and originated from mitochondria-like structures (Fig. 8), we chose to use the multiwell plate fluorescence scanner (Sattler et al., 1997) to measure the time course and extent of changes in DHR fluorescence evoked by NMDA, L-glutamate, and kainic acid applied at several concentrations (0 to 1,000 μmol/L). Temperature inside the scanner was maintained at either 37°C (Fig. 9A–C) or 20°C (Fig. 9D–E) throughout.
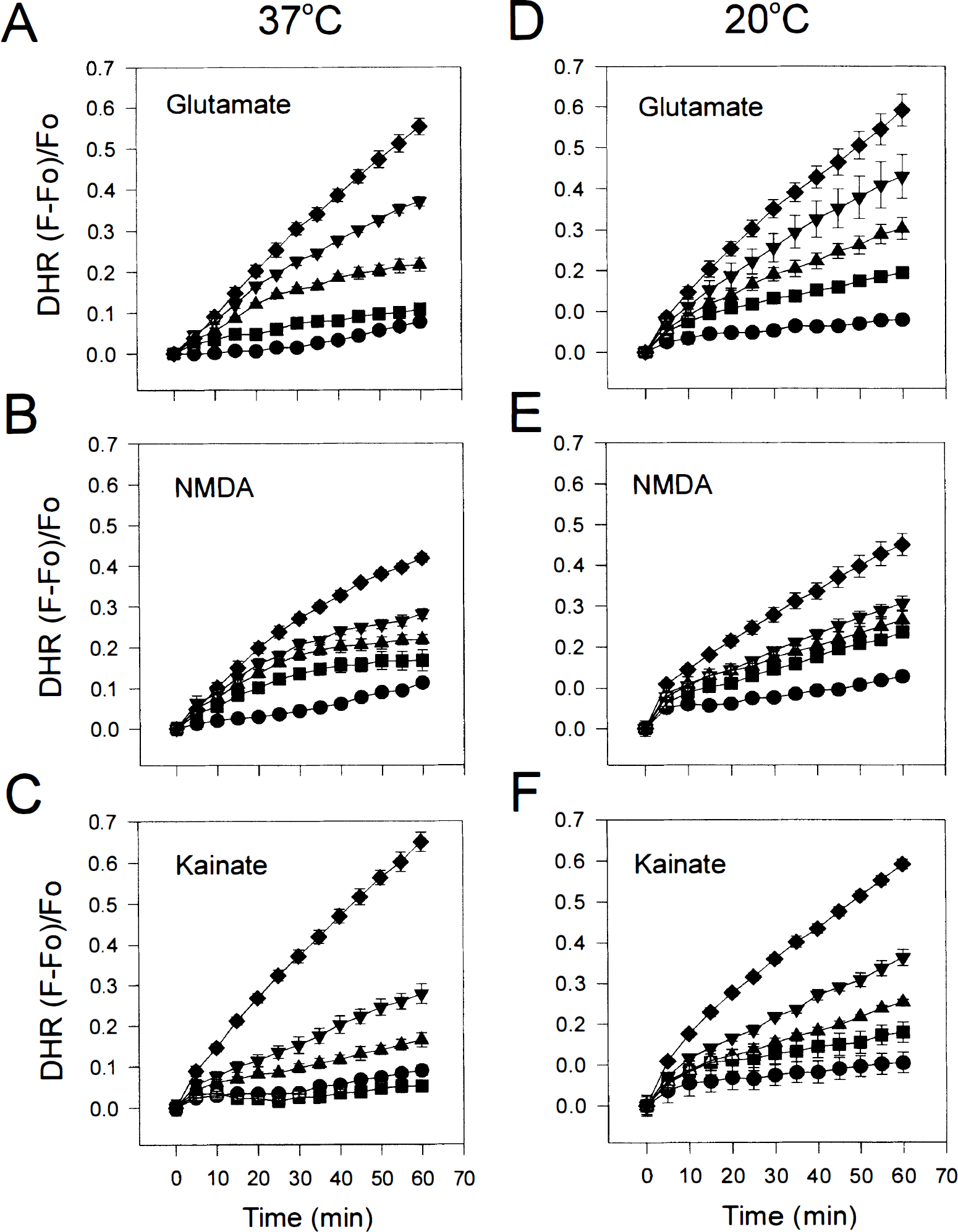
Hypothermia does not attenuate EAA-evoked increases in DHR fluorescence. Applying glutamate
All EAA cause a concentration-dependent increase in DHR fluorescence (Fig. 9). Consistent with the ability of glutamate to evoke greater [Ca2+]i increases in the neurons than NMDA (Fig. 7), glutamate also produced greater increases in DHR fluorescence (compare Fig. 9A and D with Fig. 9B and E). However, lowering the temperature from 37° to 20°C had no impact whatsoever on either the rate or the extent of DHR oxidation produced by any of the EAA tested (compare Fig. 9A–C with Fig. 9D–E). Thus, ROS production as measured by DHR oxidation did not parallel the toxicity of these EAA agonists and did not reflect any temperature sensitivity of one EAA over another. It is conceivable that ROS production from mitochondria provides only a small proportion of the ROS that trigger neurotoxicity or that the role of ROS in neurotoxicity has been overestimated.
DISCUSSION
Moderate (30°C) and profound (20°C) hypothermia reduced excitotoxicity at the level of individual neurons in a manner dependent on the temperature, the EAA, and the EAA challenge duration. Hypothermia reduced excitotoxicity in cultures subjected to both brief (5 minutes) and prolonged (60 minutes) insults with glutamate, NMDA, AMPA, and kainate (Figs. 1, 2 and 3; Table 2, Table 3 and Table 4. However, excessive cooling (to 12°C) was toxic (Figs. 2 and 3). The protective effects of hypothermia at 20° to 30°C persisted after rewarming to 37°C, but rewarming from 12°C was deleterious (Fig. 4). Neurons briefly challenged with all EAA responded similarly to hypothermia. However, hypothermia had surprisingly little effect on the toxicity of 60-minute glutamate applications, whereas the toxicity of prolonged insults with the other EAA, such as NMDA, was dramatically reduced. The temperature-insensitive component of glutamate neurotoxicity was still mediated by NMDA receptors (Fig. 5D–F), not by the activation of ionotropic or metabotropic non-NMDA receptors (Fig. 5A) nor, in any simple manner, by inhibition of glutamate reuptake by hypothermia (Fig. 5B). The disparity in the toxicity of NMDA and glutamate at lower temperatures was not explained by effects of hypothermia on EAA-evoked [Ca2+]i increases (Figs. 6 and 7), nor by effects on mitochondrial ROS production (Figs. 8 and 9). However, using a low-affinity Ca2+ indicator, it was evident that equimolar quantities of NMDA and glutamate produce markedly different changes in [Ca2+]i not detectable by indicators having a lower Ca2+ affinity.
Presynaptic or postsynaptic mechanisms?
Bruno et al. (1994) studied the effects of mild to moderate hypothermia on cortical neurons exposed to oxygen-glucose deprivation or EAA. They showed that cooling to 30°C virtually abolished anoxia-induced glutamate release into the extracellular medium. This, and similar findings in vivo (Mitani and Kataoka, 1991; Ginsberg et al., 1993; Rokkas et al., 1995; Baker et al., 1995; Globus et al., 1995), suggests that a major mechanism by which mild to moderate hypothermia protects neurons against anoxia or ischemia is by attenuating endogenous glutamate release and subsequent excitotoxicity. Cooling to 30°C protected neurons against brief (10 minutes) challenges with EAA only if hypothermia was maintained beyond the initial insult, because much of the damage produced was subsequent to the release of endogenous glutamate (Bruno et al., 1994).
Given that mild to moderate hypothermia already abolishes endogenous EAA release, lowering the temperature further should not confer additional neuroprotection by this mechanism. However, in the case of both NMDA and AMPA toxicity in this report, cooling from 30° to 20°C conferred additional protection across a range of EAA concentrations and exposure durations (Figs. 3 and 4; Table 3 and Table 4). Thus, postsynaptic mechanisms must also play a role in the protection of neurons by profound hypothermia.
Potential postsynaptic mechanisms of neuroprotection by hypothermia
Many cellular processes are attenuated by cooling. Some are maximally attenuated at temperatures lower than those that are conventionally considered as mild to moderate hypothermia (30° to 33°C).
Transmembrane ionic fluxes and ion homeostasis Ionic channels are temperature sensitive. For example, single channel recordings of NMDA receptor activity show that single NMDA channel conductance increases steeply at temperatures in excess of 20°C (Feldmeyer and Cull-Candy, 1993; Chung and Kuyucak, 1995). Ionic currents are decreased and the mean channel open times increased at lower temperatures, with Q10 values in excess of 3 for either parameter between 14° and 24°C. Interestingly, these Q10 values may vary with the specific NMDA agonist used (McLarnon and Curry, 1990), possibly accounting for the observed discrepancy between the neurotoxic effects of NMDA and glutamate in the present study. Thus, temperature reductions of several degrees Celsius, as seen with profound hypothermia (e.g., from 37° to 20°C) will have a significantly greater impact on ionic fluxes through glutamate receptor channels than would the lesser temperature decrements of mild hypothermia.
Hypothermia also slows ionic pumps and exchangers. Thus, cooling may slow down the reverse operation of the Na/Ca exchanger during anoxia and excitotoxicity (Mattson et al., 1989; Stys et al., 1991a, 1991b; Kiedrowski et al., 1994; White and Reynolds, 1995), which, in turn, would reduce cellular Ca2+ loading and resultant injury (Knerr and Lieberman, 1993). The Ca-ATPase, another important mechanism for cellular Ca2+ extrusion and sequestration, is also temperature sensitive: higher temperatures lead to a more fluid cell membrane, which allows faster rotational motion of the ATPase protein in the lipid membrane, which correlates with greater Ca-ATPase activity (Bigelow et al., 1986). There appears to be a temperature threshold, around 20°C, for the increased Ca-ATPase activity: greater than 20°C, the activation energies for Ca-ATPase activity, lipid fluidity, and protein rotational motion are essentially equal, whereas these are decreased at lower temperatures (Squier et al., 1988). Also, less than 20°C Ca-ATPase protein aggregation is increased, making rotational motion of the protein units cumbersome. ATPase protein aggregation is minimized at higher temperatures (20° to 40°C), such that a larger population of smaller protein units (presumably capable of faster rotation and more efficient activity) exists.
The suggestion that the temperature sensitivity of glutamate receptor channels and ionic pumps is maximal around 20°C implies that profound hypothermia may have significantly greater impact on cellular ionic fluxes than mild hypothermia. This may explain why previous measurements of EAA-evoked [Ca2+]i increases using fluorescent calcium indicators at 30° to 33°C have not revealed significant differences from measurements at 37°C (Arai et al., 1993; Bickler et al., 1994; Bruno et al., 1994). In further support of the possibility that the efficiency of [Ca2+]i modulatory mechanisms may differ at temperatures less than 30°C is the finding by Konig et al. (1994), who reported that AMPA-evoked [Ca2+]i elevations in cultured rat brain stem cells returned to baseline values at 24°C, but evoked complex secondary calcium increases when the temperature was raised to 27°C.
Cellular energy production Early theories of the mechanisms of protection by hypothermia focused on reductions in cerebral metabolic rate (Bigelow et al., 1950; Lougheed and Kahn, 1955; Michenfelder and Milde, 1992; Ausman et al., 1993). However, in vivo studies of brain ischemia at mild to moderate hypothermia do not consistently report a significant salvage from ATP depletion (Ginsberg et al., 1993; Verhaegen et al., 1995). In vitro studies also report variable effects of mild to moderate hypothermia on cellular ATP: in the studies of Bruno and colleagues (1994), moderate hypothermia (30°C) attenuated slightly cellular ATP loss observed during 60 minutes of oxygen-glucose deprivation. However, it had no effect on ATP loss measured 1 hour after a 10-minute NMDA insult or a 1-hour AMPA or kainate insult. Additional in vitro investigations suggest that ATP depletion is slowed by mild to moderate hypothermia only in the first few minutes of the insult. For example, in rat and gerbil brain slices, ATP depletion rates after anoxic insults at 34° or 31°C were reduced in the first 2 to 3 minutes, but not after 5 to 15 minutes of anoxia (Paschen and Djuricic, 1995). Similarly, after excitotoxic injury to isolated embryonic retinas, the rate of ATP loss was slowed by cooling from 37° to 30°C during the first several minutes, but by 10 minutes ATP levels were comparable at the two temperatures (Zeevalk and Nicklas, 1993).
Presently, little is known about the effects of deeper levels of hypothermia on high-energy metabolite production during anoxia, ischemia, or excitotoxicity. In one study, mitochondrial respiration and ATP synthesis were preserved in rat brain, liver, and kidney rendered ischemic at 4°C (Baumann et al., 1989). However, whether deep hypothermia prevents mitochondrial dysfunction or ATP loss more than mild to moderate hypothermia is unknown.
The production of toxic reaction products It has been suggested that the attenuation of production of free radical species after neuronal injury is a potential protective mechanism of hypothermia. Experiments in vivo have shown that moderate hypothermia (30°C) is sufficient to attenuate hydroxyl radical production after brain ischemia and trauma (Globus et al., 1995; Kil et al., 1996). Mild hypothermia (33°C) may also attenuate postischemic production of nitric oxide (Kader et al., 1994). It is unknown whether such processes are further enhanced at even lower temperatures.
Experiments in cultured cerebellar granule cells have examined whether cooling attenuates increases in oxidative metabolism triggered by applying a Ca2+ ionophore. The oxidation of intracellular dichlorofluorescein, a fluorescent marker of cytoplasmic ROS production (Reynolds and Hastings, 1995), was increased after the application of ionomycin at 36°C. This increase was not affected by cooling neurons to 26°C, but was markedly reduced at 16°C and virtually abolished at 4°C (Oyama et al., 1994). The authors attributed this effect to a reduction in oxidative metabolism, as ionomycin-induced increases in the fluorescence of fluo-3, a Ca2+ indicator, were reportedly unaffected by the temperature reduction.
The mechanism by which a reduction in ROS might occur is uncertain. No evidence exists to suggest that the mitochondrial electron transport chain, a source of free radical species, is selectively targeted by hypothermia (Baumann et al., 1989). However, mild temperature reductions (to 30° to 35°C) have been shown to attenuate astroglial release of arachidonic acid metabolites after hypoxia or hypoglycemia (Haun et al., 1993) and the production of some prostaglandins after challenges with calcium ionophores and ischemia in skeletal muscle and rat brains, respectively (Patel et al., 1994; Majumdar et al., 1995). Whether such mechanisms are also operative at the level of individual neurons of the CNS remains to be elucidated.
Other mechanisms Other potential targets for the protective effects of hypothermia exist. Among these are cellular protein synthesis (Widmann et al., 1993; Bergstedt et al., 1993; Paschen and Djuricic, 1995), the activity of innumerable cellular enzymes such as Ca2+/calmodulin-dependent protein kinase II (Hu et al., 1995) and protein kinase C (Busto et al., 1994), cell membrane fluidity (Squier et al., 1988), action potential propagation (Kiyosue et al., 1993), and the ischemic induction of heat-shock proteins (Shaver et al., 1995). Further studies are needed to address the potential impact of moderate and deep hypothermia on these processes.
Potential mechanisms of toxicity of hypothermia
Mild to moderate hypothermia appears to be well tolerated by the brain in vivo, and by individual neurons and glia in vitro, as there have been no reports of toxicity attributable to mild temperature reductions. However, as demonstrated in this study, more profound hypothermia (12°C) has the potential to damage neurons (Figs. 2, 3 and 4). In studies of traumatically injured spinal neurons, Lucas and colleagues showed that cooling to 17°C for 2 hours and rewarming at 37°C for 22 hours was well tolerated by control (uninjured) neurons and protected injured neurons. However, neurons cooled below 17°C experienced swelling, and many died on rewarming (Lucas et al., 1990). Hypothermic swelling and death could be reduced by pretreatment with NMDA antagonists (Lucas et al., 1990, 1994). Ultrastructural studies of the damage induced in spinal neurons cooled to 10°C revealed cytoplasmic vacuolation, mitochondrial abnormalities, and cytoskeletal disruption. Many neurons exhibited beading and vacuolation of dendrites and fragmentation of microtubules (Emery and Lucas, 1995). These dendritic changes could be mimicked by the application of NMDA and prevented with NMDA antagonists (see also Stewart et al., 1991; Bindonkas and Miller, 1995).
These observations may be explained by the temperature sensitivity of certain cytoskeletal elements in mammalian tissues. For example, a proportion of microtubules, whose size and shape depends on an equilibrium between assembly and disassembly, depolymerize on cooling to the temperatures indicated above. Assembly and disassembly rates of microtubules are modulated not only by temperature, but also by ionic conditions, such that Ca2+ or Mg2+ excess considerably accelerates microtubule disassembly (Barton et al., 1987; Gal et al., 1988). Thus, the combination of Ca2+ influx with hypothermia further favors an equilibrium shift toward microtubule disassembly. These conditions were met in the current report through the application of EAA to the cultures at low temperatures. Also consistent with this was the finding that control neurons, which were spared EAA-evoked Ca2+ increases, were not damaged by lowering the temperature to 12°C (Fig. 3). Under this hypothesis, hypothermia in the absence of a calcium load was insufficient to trigger cytoskeletal damage.
CONCLUSION
This wide survey of the effects of moderate and profound cooling of neurons on excitotoxicity reveals that hypothermia, within a certain range (down to 20°C), imparts a resilience to EAA that persists on rewarming. Also, it unmasks a temperature-insensitive component of glutamate neurotoxicity that is unseen with the other EAA. This feature, unique to glutamate, could potentially confound many studies of excitotoxic phenomena and should therefore be a subject of further study.
Footnotes
Acknowledgements
M.T. is a clinician-scientist of the Medical Research Council of Canada. R.S. is a student of the National Centers of Excellence of Canada.