
Abstract
The mechanisms of ischemic cell damage are still not fully understood. It has been shown that alpha-amino-3-hydroxy-5-methyl-isoxazole-4-propionate (AMPA)/kainate receptor antagonists, such as 6-nitro-7-sulphamoyl-benzo-(f)-quinoxaline-2,3-dione (NBQX), are neuroprotective in models of transient forebrain ischemia, even when applied during recovery, indicating that nonNMDA receptors may play a pivotal role in ischemic cell damage. In the present series of experiments, we studied whether transient cerebral ischemia causes changes in the extent of mRNA editing of AMPA/kainate receptor subunits, a reaction critical for the control of calcium flux through nonNMDA receptor ion channels. Transient cerebral ischemia was produced in rats using the four-vessel occlusion (4-VO) model. After 30 min of ischemia, brains were recirculated for 4, 8, or 24 h. Total RNA was extracted from the cortex, striatum, and hippocampus in order to analyze the extent of mRNA editing of the glutamate receptor subunits GluR2, GluR5, and GluR6. RNA was converted by reverse transcription into cDNA, which was used as a template for subunit-specific polymerase chain reaction (PCR) to amplify a product across the edited base A (A edited to I in the second transmembrane-spanning regions of GluR2, GluR5, and GluR6). PCR products were analyzed with the restriction enzyme Bbv 1, which recognizes the cDNA sequence GC AGC originating from unedited but not that originating from edited GluR2, GluR5, or GluR6 mRNA (GCGGC, the base I is read as G). Restriction digests were electrophoresed, and the bands visualized with ethidium bromide and then photographed. The extent of mRNA editing of the different subunits was quantified using image analysis and appropriate standards. In all control brains studied, GluR2 mRNA was completely edited and remained so after reversible cerebral ischemia. The extent of GluR5 mRNA editing was significantly upregulated in the striatum (from 39 ± 6% in controls to 57 ± 9 and 56 ± 7 after 4 and 8 h of recovery, respectively, p < 0.05 versus control) but not in the cortex and hippocampus. The extent of GluR6 mRNA editing was significantly reduced after 24 h of recovery: in the cortex, from 92 ± 1 to 78 ± 6% (p < 0.01); in the striatum, from 91 ± 2 to 79 ± 1% (p < 0.001); and in the hippocampus, from 90 ± 3 to 80 ± 2% (p < 0.05). A significant reduction was already apparent in the striatum after 4 h of recovery (p < 0.05). Results indicate that mRNA editing is regulated differently in each of the glutamate receptor subunits GluR2, GluR5, and GluR6 after transient cerebral ischemia. The ischemia-induced upregulation of GluR5 mRNA editing observed in the striatum may be indicative of a higher sensitivity to transient ischemia of neurons that exhibit a large fraction of unedited GluR5 mRNA. This assumption is corroborated by the observation (Mackler and Eberwine, 1993) that GluR5 mRNA is completely unedited in neurons of the hippocampal CA1-subfield, a region most vulnerable to transient cerebral ischemia. Whether the decrease in GluR6 mRNA editing observed in all brain structures after ischemia results from a disturbance of the editing reaction or from glial proliferation will have to be established in further experiments. Studying ischemia-induced changes in mRNA editing of glutamate receptor subunits GluR5 and GluR6 may help to elucidate the molecular mechanisms of ischemic cell damage.
The molecular mechanisms of cell damage induced by transient cerebral ischemia are still not fully understood (for reviews, see Kogure et al., 1993). It has been suggested that ischemic cell damage is an excitotoxic process triggered by an overflow of calcium ions into cells; this overflow is induced by glutamate, released during ischemia, which activates calcium fluxes through glutamate receptor ion channels of the N-methyl-D-aspartate (NMDA) type (Benveniste et al., 1988; Choi, 1988). However, NMDA antagonists have failed to be neuroprotective in models of transient forebrain ischemia (Buchan, 1992; for a recent review, see Hossmann, 1994). Recently, it has been shown that antagonists of the nonNMDA glutamate receptors, e.g., 6-nitro-7-sulphamoyl-benzo-(f)quinoxaline-2,3-dione (NBQX), are neuroprotective in different models of transient forebrain ischemia (Sheardown et al., 1990; Buchan et al., 1991; Diemer et al., 1992; Nellgard and Wieloch, 1992). It is noteworthy that protection could be achieved even when these antagonists were applied up to 24 h after ischemia (Sheardown et al., 1993), indicating that pathological processes important for the manifestation of ischemic cell damage take place after ischemia, and that nonNMDA glutamate receptors may play a key role in these processes.
It is not known, at the present time, how nonNMDA glutamate receptor ion channels—alpha-amino-3-hydroxy-5-methyl-isoxazole-4-propionate (AMPA) and kainate receptors—might contribute to the development of ischemic cell damage. The known open therapeutic window for antagonists of these glutamate receptor ion channels provides evidence that the pathological process in which AMPA/kainate receptors are involved must operate after ischemia. Key findings were that the extent of calcium ion influx into neurons of the hippocampal CA1 subfield induced by stimulating the perforant path is significantly increased after cerebral ischemia and that these calcium spikes can be blocked almost completely with the AMPA/kainate receptor antagonist, NBQX, and, to a lesser extent, by the NMDA antagonist, Mk-801 (Andinè et al., 1992). These observations may be interpreted as indicating that a calcium conductance through AMPA/kainate receptor ion channels contributes to the development of ischemic cell damage.
Traditionally, it is believed that calcium conductance can be elicited only by activating NMDA receptor ion channels, whereas activation of AMPA/kainate receptors elicits a conductance for monovalent ions. Based on in vitro experiments, it has been established recently that even a calcium conductance can be elicited by activating AMPA/kainate receptors (Hollmann et al., 1991; Sommer et al., 1991; Burnashev et al., 1992; Köhler et al., 1993). The passage of calcium ions through AMPA/kainate receptor ion channels is controlled by a single amino acid in the second transmembrane spanning region of the receptor molecule; when this position is occupied by glutamine (codon CAG), calcium can pass, but calcium conductance is blocked when this position is occupied by arginine (codon CGG). In all AMPA/kainate glutamate receptors, the genetic sequence subjected to editing in the second transmembrane region is CAG, while the mRNA for the sub units GluR2, GluR5, and GluR6 exists in two forms, namely CAG and CGG (Sommer et al., 1991). This change in the base sequence at the mRNA level is termed RNA editing. Recent results suggest that, during the editing reaction, A is deaminated to I (Rueter et al., 1995; Yang et al., 1995), which is then identified as G during protein synthesis or during the analytical process used in the present study (reverse transcription and PCR, see below). Thus, the gating of calcium ions by AMPA/kainate glutamate receptors is controlled by mRNA editing of the respective receptor sub unit mRNA.
A postischemic activation of calcium fluxes through AMPA/kainate receptors (as evidenced from the observation of Andiné et al., 1992, see above) could be elicited by either of two different means: (a) selective decrease in the proportion of edited AMPA/kainate receptor subunits (e.g., GluR2, which is completely edited in the physiological state and normally blocks calcium conductance) or (b) selective increase in the proportion of unedited subunits. In fact, a selective decrease in GluR2 relative to GluR1, GluR3 and GluR4 mRNA levels has been found in the hippocampal CA1 subfield after ischemia (Pellegrini-Giampietro et al., 1992), and it has been shown by Pollard et al. (1993) that the GluR2 flip/flop expression is dramatically reduced in the CA1-subfield within 24–48 h after ischemia. Other workers, however, were unable to confirm a selective decrease of GluR2 mRNA levels in the hippocampal CA1-subfield after ischemia (Diemer et al., 1994). We, therefore, decided to study whether transient forebrain ischemia changes the extent of mRNA editing of the AMPA/kainate receptor subunits GluR2, GluR5, or GluR6. The extent of editing was quantified with the aid of a newly developed analysis system, including restriction analysis of subunit-specific PCR products with the enzyme Bbv 1 (Paschen et al., 1994b).
MATERIALS AND METHODS
Male Wistar rats weighing 230–300 g were used for the present study. Transient forebrain ischemia was produced by using the four-vessel occlusion (4-VO) model described by Pulsinelli and Brierly (1979), modified as described by Schmidt-Kastner et al. (1989). Animals were anesthetized with 1.5% halothane in 70% N2O and 30% O2 (vol/vol). On the first day, both vertebral arteries were coagulated. On the following day, both common carotid arteries were exposed. Electroencephalograms were (EEGs) continuously recorded. After stabilization of physiological parameters, both common carotid arteries were occluded. Only those animals in which EEGs flattened completely upon vascular occlusion were included in the present series of experiments. After 30 min of almost complete ischemia, clips were removed and the common carotid arteries were viewed under the microscope to check that the blood was circulating freely.
After postischemic recovery periods of 4, 8, or 24 h, animals were reanesthetized and decapitated. Brains were taken out as quickly as possible and put into a pH 7.4, phosphate-buffered saline solution, prechilled in ice-water. Brains were dissected on a prechilled glassplate, first into the two hemispheres and then into the striatum, hippocampus, and cortex. Immediately after dissection, brain samples were put into prechilled tubes, frozen in liquid nitrogen, weighed, and stored at −80°C until analysis.
Biochemical reagents were obtained from New England Biolabs, Beverly, MA, U.S.A. (Bbv 1), Boehringer Mannheim, Germany (taq polymerase, ladder V), GIBCO (Gaithersburg, MD, U.S.A.) (reverse transcriptase kit), and Sigma (St. Louis, MO, U.S.A.).
Total RNA was extracted from brain samples as described by Chomczynski and Sacchi (1987). In this method, ∼100 mg of tissue was homogenized in 1 ml 4 M guanidinium thiocyanate solution, supplemented with 0.5% sarcosyl and 25 mM 2-mercaptoethanol. Each 500 H.1 sample of homogenate was added to 500 μl phenol, 100 μl chloroform/isoamylalkohol (49:1, vol/vol), and 50 μl 2 M ammonium acetate solution, pH 4.0, and the total RNA was extracted. After repetitive precipitation of RNA with isopropanol and a final wash with 75% ethanol, RNA was solubilized in Tris/EDTA (TE) buffer. The quality and quantity of isolated RNA was assessed by extinction (E) readings. Only those preparations in which the E ratio of 260/280 was >1.8 were used for the present study.
Total RNA was converted by reverse transcription into cDNA (GIBCO BRL). Five μg of total RNA was dissolved in 13 μl water, a mixture of oligo deoxyuridine monophosphate (dT) and random hexamer primers added, and the solution heated to 70°C for 10 min. After quick chilling on ice, cDNA synthesis was performed by adding synthesis buffer (10 mM Tris–HCl, pH 8.4, 25 mM KCl, 1.25 mM MgCl2, and 50 μg/ml bovine serum albumin (BSA), 500 μM deoxynucleosidetriphosphate (dNTP) mix, 5 mM dithiothreitol (DTT), and 10 U reverse transcriptase. After incubation at room temperature for 10 min, the solution was heated to 42°C for 50 min. The reaction was then terminated by heating to 95°C for 5 min. The RNA template was destroyed by incubation with RNase H (2 U per reaction) at 37°C for 20 min.
cDNA solution (1 μl) (equivalent to 250 ng of total RNA) was used for amplifying a product across the edited base located in the second transmembrane domain of each of the three glutamate receptor subunits GluR2, GluR5, and GluR6. The polymerase chain reaction (PCR) was performed in a total of 50 μl of solution containing 250 μM each of the four deoxynucleotides, 1.5 mM MgCl2, 50 mM KCl, 0.01% gelatin in 10 mM Tris–HCl buffer, pH 8.3, and 0.5 U taq polymerase (Boehringer). Reactions were run for 35 cycles, beginning with an initial denaturation step at 94°C for 5 min, annealing at 60°C for 1 min, synthesizing at 72°C for 1 min, denaturing at 94°C for 1 min, and, finally, synthesizing step at 72°C for 10 min. The following primers were used: for amplification of GluR2 genomic DNA, upper strand primer 5′-AGCAGATTTAGCCCCTACGAG-3 lower strand primer 5′-TTGAAGCAGGTGAGTGACCAA-3′, amplification product length was 174 bp; for GluR2 cDNA, upper strand primer 5-AGCAGATTTAGCCCCTACGAG-3′, lower strand primer 5′-TAAGTTAGCCGTGTAG-GAGGA-3′, amplification product length was 231 bp; for GluR5 cDNA, upper strand primer 5′-GTTTGTGATTGCGAGGTTCACA-3′, lower strand primer 5′-CAGGTTGGCCGTGTAGGATGA-3′, amplification product length was 233 bp; and for GluR6 cDNA, upper strand primer 5-GACTCAGACGTGGTGGAAAAC-3′, lower strand primer 5′-CGTCCTCCACTGCTCCATACT-3′, amplification product length was 271 bp. Before analyzing the samples from transient ischemic brains, it was ascertained, by restriction analysis, that the correct products had been amplified (Paschen and Djuricic, 1994, 1995).
PCR products were precipitated, washed with ethanol, and dissolved in 40 μl of a solution containing 50 mM NaCl, 10 mM Tris–HCl, 10 mM MgCl2, and 1 mM DTT, pH 7.9 (later used for incubating PCR products with the restriction enzyme Bbv 1, see below). To semiquantify the content of the PCR product, 5 μl of this solution was run on a 2% agarose gel; semiquantification enabled us to take similar amounts of the PCR products for restriction analysis, gel electrophoresis, and image analysis (see below), irrespective of the total yield of the PCR product.
The extent of RNA editing was analyzed by incubating the PCR products with the restriction enzyme Bbv 1 (New England Biolabs). This enzyme recognizes the sequence GC(A/T)GC and cuts the DNA strand eight bases downstream of the recognition site. GCAGC is the sequence of unedited GluR2, GluR5, and GluR6 mRNA around the edited base A (A edited to I, which is then converted to G during reverse transcription and the PCR). Thus, by incubating the PCR products with Bbv 1, the fraction of the amplification product originating from unedited mRNA is split into two smaller fragments (138 and 93 bp for GluR2, 139 and 94 bp for GluR5, and 190 and 81 bp for GluR6), while the fraction of the PCR product originating from edited mRNA is left intact. For restriction analysis with Bbv 1, reactions were run in 20 μl of reaction buffer (see preceding paragraph). After adding 1.3 U of Bbv 1, incubation was continued for 120 min at 37°C. Thereafter, solutions were heated to 60°C for 10 min and put on ice prior to electrophoresis.
Restriction digests were electrophoresed together with standards (see below) using a 2% agarose gel supplemented with ethidium bromide. After electrophoresis, gels were washed with water to remove excess ethidium bromide and then transilluminated with ultraviolet light. The emitted fluorescence light was recorded photographically using an appropriate barrier filter. Films were developed by standard techniques, negatives scanned with a rotating microdensitometer (Scandig 3, Joyce Loebl, Gateshead, UK), and processed by the computer program NIH-Image of Wayne Rasband (National Institutes of Health, Bethesda, MD, U.S.A.) run on a Macintosh II computer.
The extent of mRNA editing was quantified by creating standard curves (OD ratio versus % editing) with Plasmids carrying cDNA inserts originating from unedited or edited GluR5 or GluR6 mRNA. PCR reactions were run with these plasmids as templates and PCR products from the respective edited and unedited versions were mixed to obtain ratios equivalent to different extent of editing. These mixtures were processed in exactly the same way as were the brain samples (Bbv 1 incubation and image analysis) and used for creating the standard curve. Two standards were run on each gel, and these standards were used to quantify the extent of editing of samples run on the same gel. For quantification, the OD of the fraction of the PCR product not cut by Bbv 1 (originating from edited mRNA) and that of the larger restriction fragment (originating from unedited mRNA) were evaluated, and the logarithm of the ratio of these ODs correlated to the extent of editing.
There were four animals in each group. All values given are means ± SD. Statistical analysis was performed by one-way analysis of variance (ANOVA) followed by Scheffe's F-test.
RESULTS
A standard curve for the quantification of the extent of GluR6 mRNA editing is given in Fig. 1. Fig. 1A shows a gel wherein PCR reactions had been run with plasmids with cDNA inserts originating either from edited or unedited GluR6 mRNA. These PCR products were mixed to obtain ratios equivalent to different extents of editing (95–10% editing), as indicated in the legend to Fig. 1A. The gel was photographed and the negative used to create the standard curve shown in Fig. 1B, as described in the Materials and Methods section. The best relationship between ODs and % editing was obtained when the logarithm of the OD ratio (OD ratio of the band of the uncut PCR product to that of the larger restriction fragment) was correlated to the extent of editing (r = 0.99). Using this approach, the extent of mRNA editing could be assessed reliably, as indicated by the high correlation coefficient (Fig. 1B). For quantification of the extent of editing in brain samples, two standards were run on each gel, equivalent to 80 and 20% editing (for GluR5) or 95 and 80% editing (for GluR6).
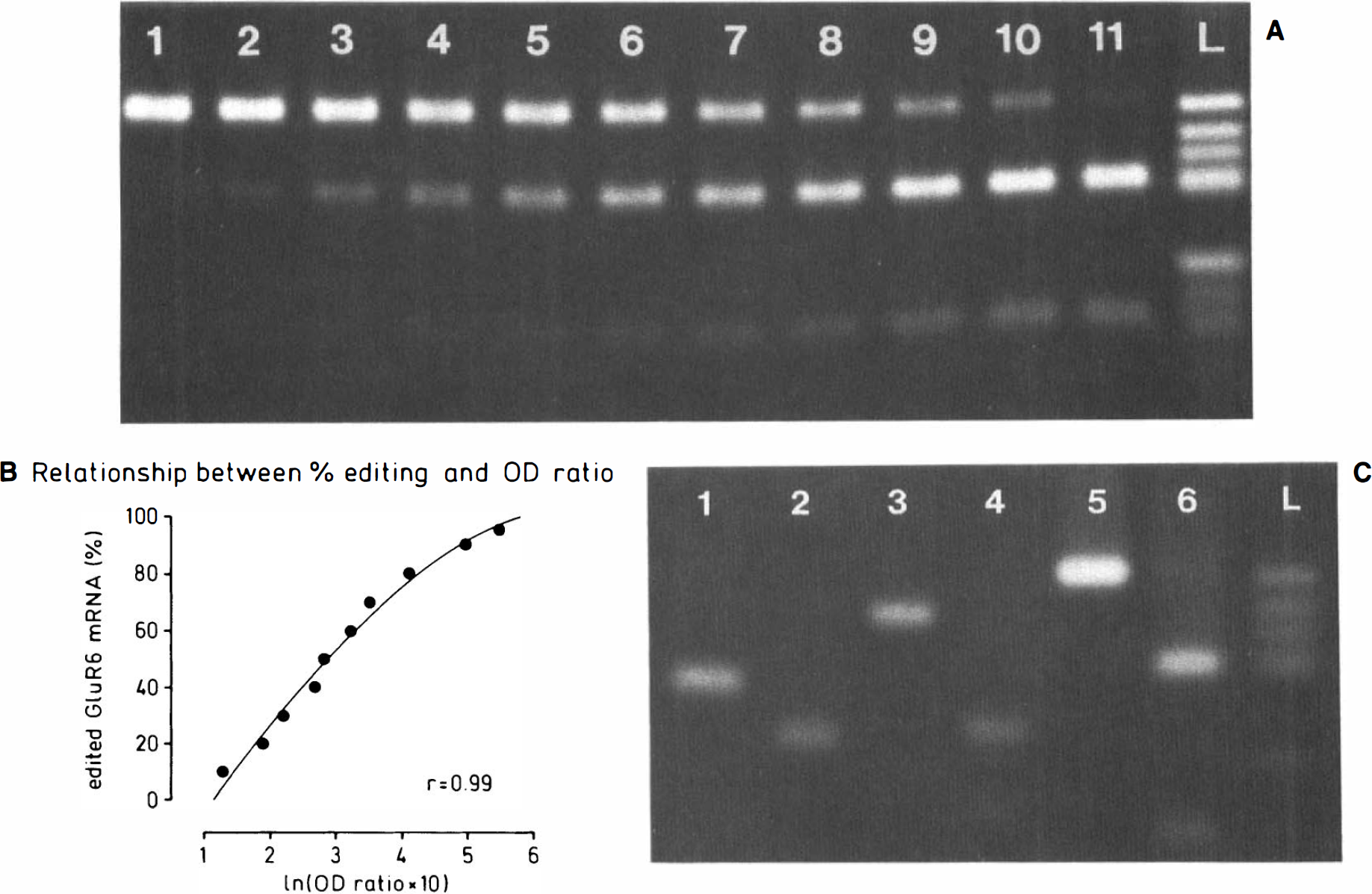
For the quantification of GluR6 mRNA editing
Fig. 1C illustrates how the restriction enzyme Bbv 1 cuts PCR products originating from unedited RNA. PCR was run with genomic DNA (with GluR2-specific primers) or with plasmids carrying GluR5 or GluR6 cDNA as inserts originating from unedited GluR5/GluR6 mRNA. PCR products amplified from all three glutamate receptor subunits were clearly cut into smaller fragments upon incubation with Bbv 1. This indicates that our system of analysis, which is based on the fact that PCR products from unedited mRNAs are cut into two smaller fragments while those from edited mRNAs are not, works reliably for all three subunits.
GluR2 mRNA was completely edited in all three of the brain structures studied. We found no decrease in the extent of GluR2 editing after ischemia. In Fig. 2A, the position at which the larger restriction fragment would have appeared had unedited GluR2 mRNA been present is highlighted by an arrow. To illustrate the lower detection limit for unedited GluR2, gels were overloaded with PCR products (prepared from RNA isolated from the cortex, striatum, and hippocampus of animals subjected to 30 min forebrain ischemia followed by 24 h of recovery) so that 1% of the PCR product could be clearly identified (Fig. 2D). Even when gels were overloaded, we could not find any band corresponding to the larger Bbv 1 restriction fragment (138 bp, highlighted by an arrow), which would have illustrated that a small part of RNA was in the unedited state. Figure 2D, thus, illustrates that we would have been able to detect markedly <1% of unedited GluR2 mRNA.
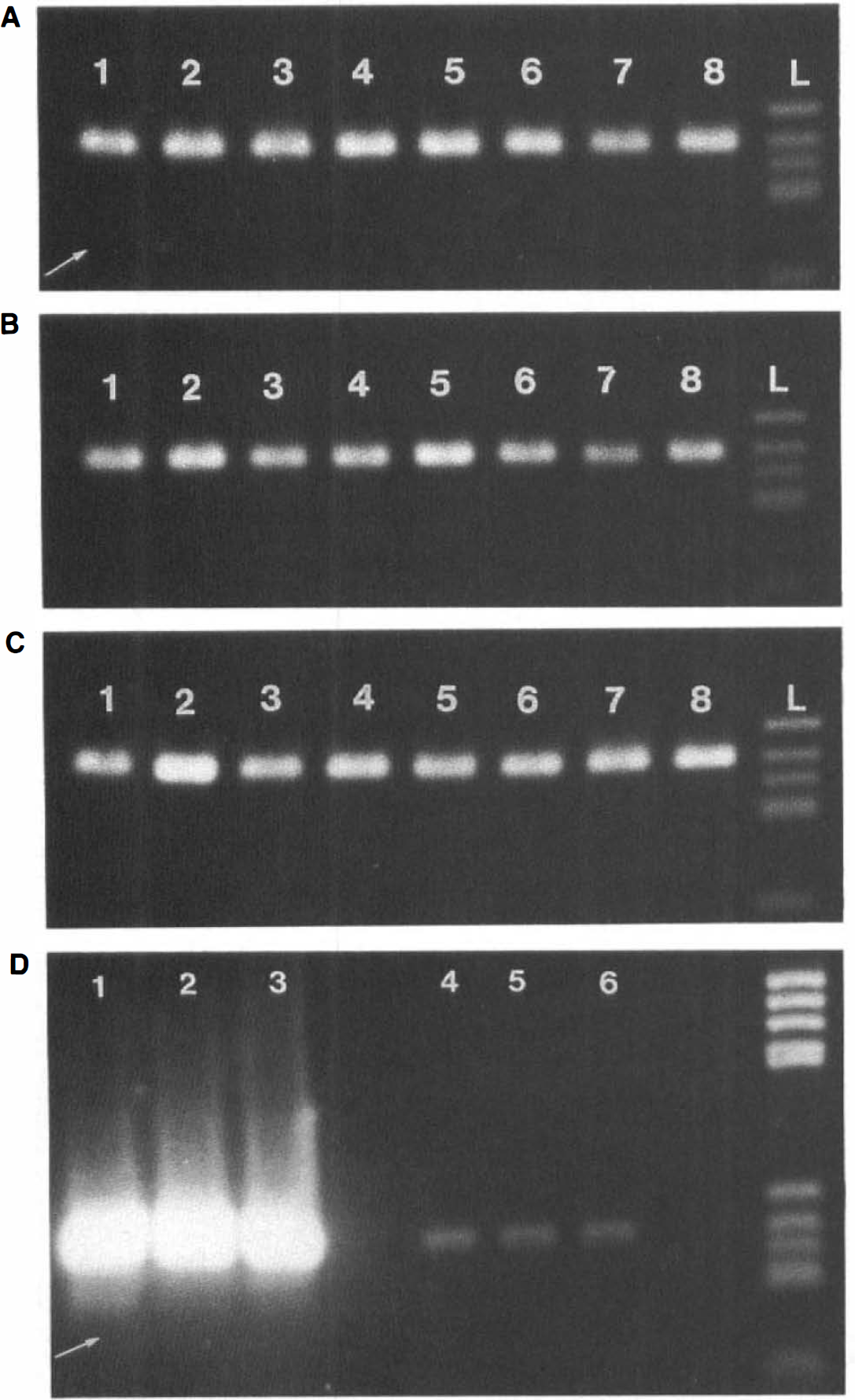
Gels illustrating ischemia-induced changes in the extent of GluR2 mRNA editing. Samples were taken from the cerebral cortex (
Representative gels illustrating ischemia-induced changes in the extent of GluR5 and GluR6 mRNA editing are shown in Figs. 3A–C and 4A–C, and quantitative results are displayed in Figs. 5 and 6. In control animals, a relatively large fraction of GluR5 mRNA molecules was in the unedited state: only 57 ± 9, 39 ± 6, and 43 ± 7% of all GluR5 mRNA molecules were edited in the cerebral cortex, striatum, and hippocampus, respectively (Fig. 5). After ischemia, the extent of GluR5 mRNA editing increased slightly in all brain structures studied; however, these differences were statistically significant only in the striatum (Figs. 3B and 5). Here, the extent of editing increased significantly from 39 ± 6 to 57 ± 9% after 30 min ischemia and 4 h of recovery (p < 0.05 versus control). This significant increase in the extent of GluR5 mRNA editing was stable up to 8 h after ischemia (p < 0.05 versus control) and fell slightly when the recirculation period was extended to 24 h (Figs. 3B and 5).
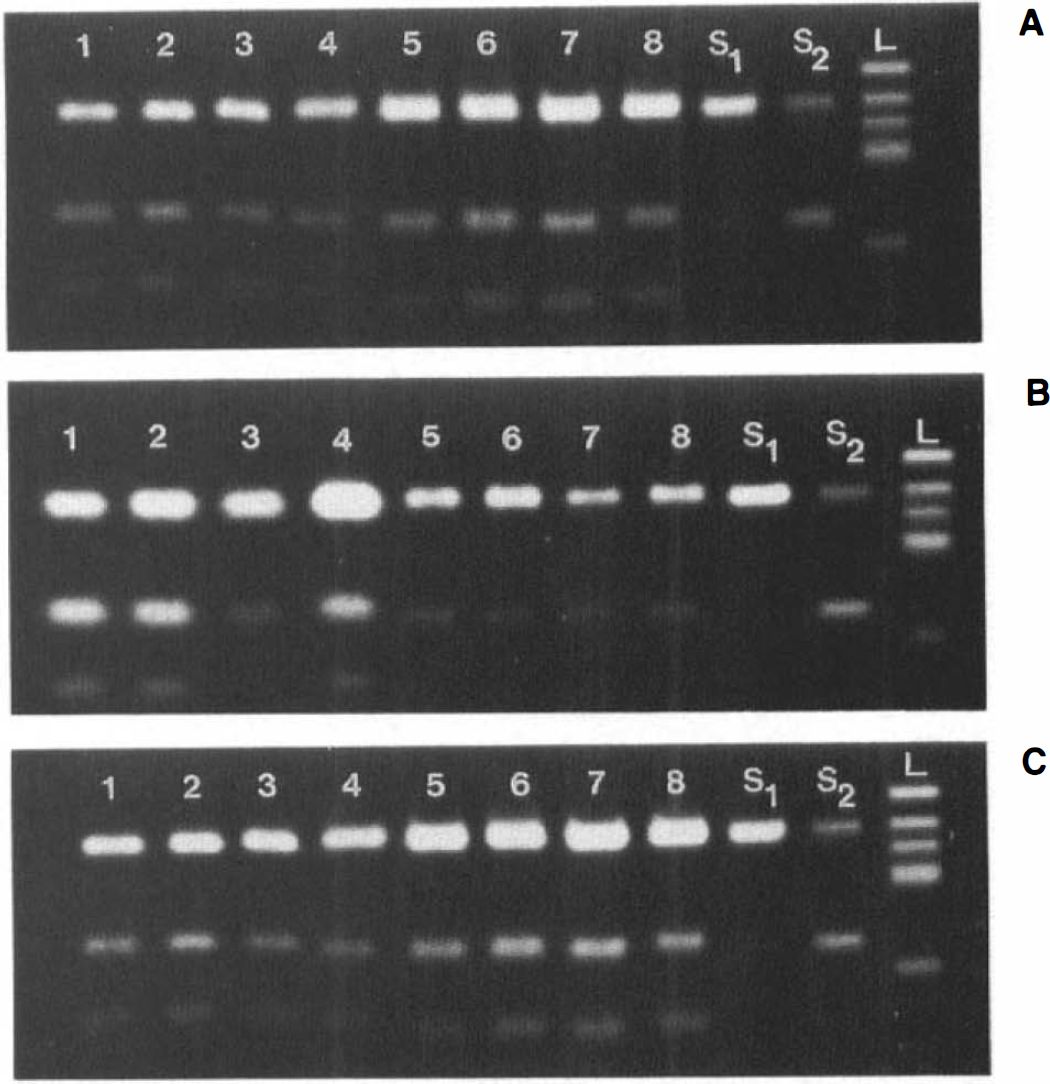
Gels illustrating analysis of ischemia-induced changes in the extent of GluR5 mRNA editing. Samples were taken from the cerebral cortex (
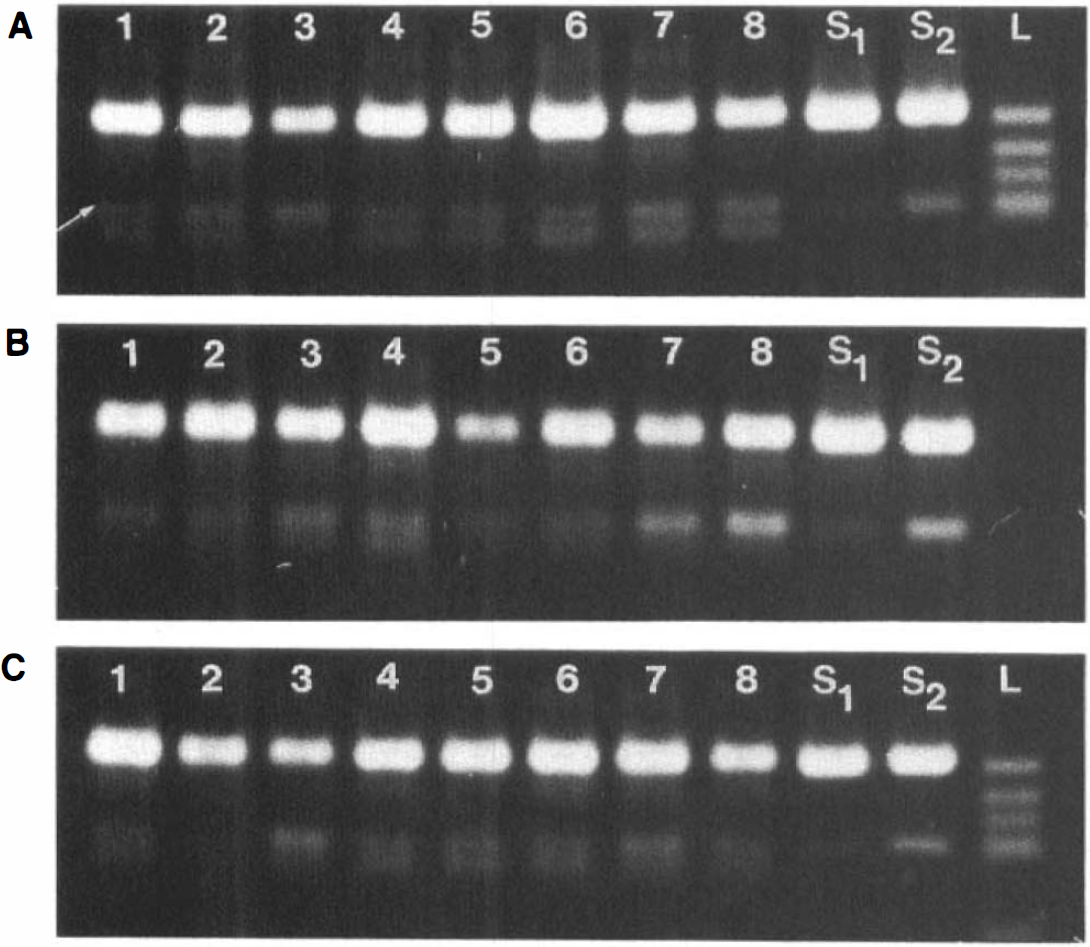
Gels illustrating ischemia-induced changes in the extent of GluR6 mRNA editing. Samples were taken from the cerebral cortex (
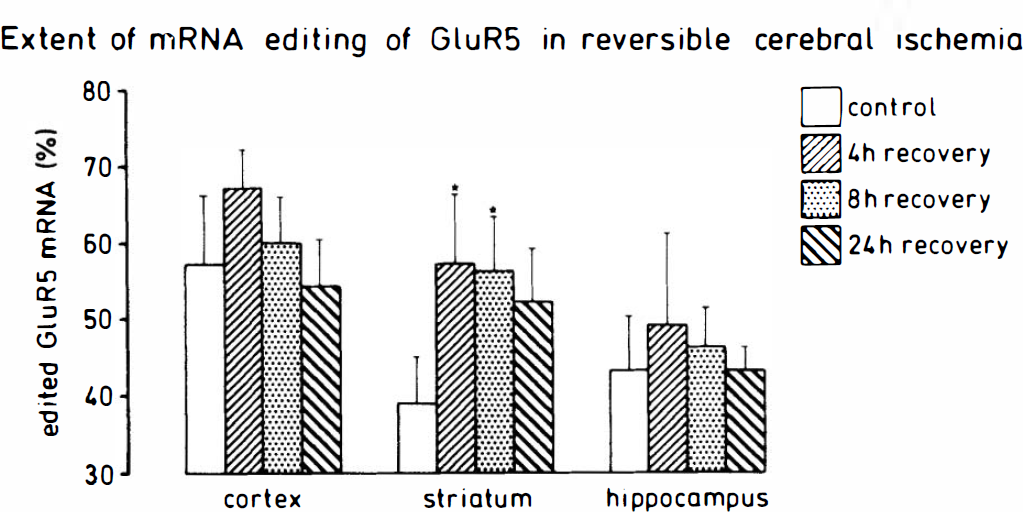
Quantification of ischemia-induced changes in the extent of GluR5 mRNA editing. The extent of RNA editing of the glutamate receptor subunit GluR5 was quantified in tissue samples after 30 min forebrain ischemia followed by 4, 8, or 24 h of recirculation. The extent of GluR5 mRNA editing was evaluated as described in Materials and Methods. Values given are means ± SD (n = 4 each group). Statistical analysis was performed using ANOVA followed by Scheffe's F-test. Statistically significant differences between experimental groups and controls are indicated by *p < 0.05 (versus control).
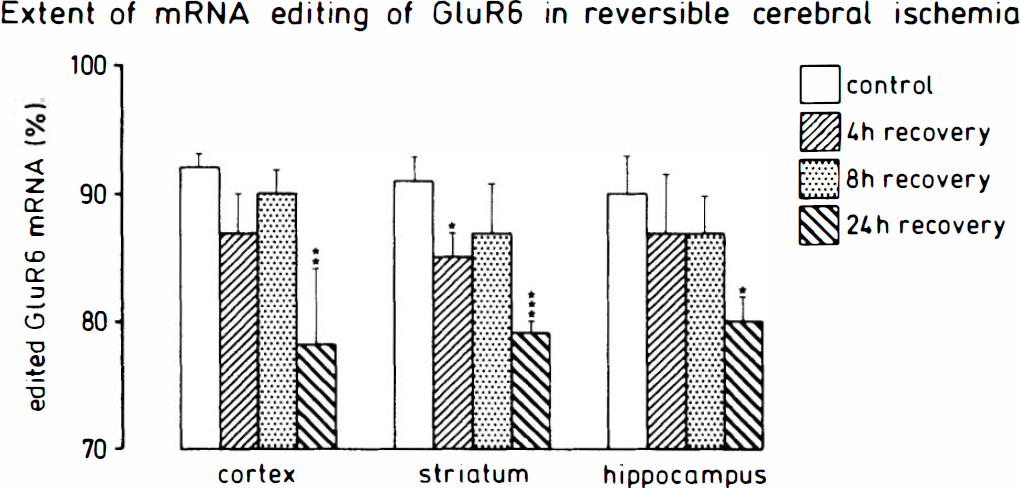
Quantification of ischemia-induced changes in the extent of GluR6 mRNA editing. The extent of RNA editing of the glutamate receptor subunit GluR6 was quantified in tissue samples after 30 min forebrain ischemia followed by 4, 8, or 24 h of recirculation. The extent of GluR6 mRNA editing was evaluated as described in Materials and Methods. Values given are means ± SD (n = 4 each group). Statistical analysis was performed using ANOVA followed by Scheffe's F-test. Statistically significant differences between experimental groups and controls are indicated by *p < 0.05, **p < 0.01, and ***p < 0.001 (versus control).
In our studies of GluR6 mRNA editing, we observed an additional faint band in some of the samples after PCR amplification. This band was, however, clearly separated on the electrophoresis gel from the larger restriction fragment produced after Bbv 1 incubation (Fig. 4), and, therefore, did not obscure the results. The extent of GluR6 mRNA editing was significantly higher than the extent of GluR5 editing (Figs. 4A-C and 6): 92 ± 1, 91 ± 2, and 90 ± 3% of all GluR6 mRNA molecules were in the edited state in the cortex, striatum, and hippocampus, respectively, of control animals. After 30 min ischemia and 4–24 h of recovery, the extent of GluR6 mRNA editing decreased significantly in all brain structures studied (Figs. 4A–C and 6). However, the time course of this reduction in the extent of GluR6 mRNA editing varied in different brain structures. In the striatum, a significant decrease was already apparent after 4 h of recovery (decrease from 91 ± 2 to 85 ± 2%, p < 0.05 versus controls), and the level dropped further when the recirculation time was extended to 24 h (to 79 ± 1%, p < 0.001 versus control). In the cerebral cortex and hippocampus, in contrast, no significant reduction in the extent of GluR6 mRNA editing could be observed before 24 h of recovery (Fig. 6) (p < 0.01 and p < 0.05 versus controls in the cortex and hippocampus, respectively). At this time period, the level of GluR6 mRNA editing was reduced equally in all three brain structures.
DISCUSSION
In the present series of experiments, ischemia-induced changes in the extent of mRNA editing of the AMPA/kainate receptor subunits GluR2, GluR5, and GluR6 were quantified using a recently developed analytical approach (Paschen et al., 1994b). This system is based on isolation of total RNA, reverse transcription into cDNA, subunit-specific PCR, and evaluation of PCR products by restriction digest with Bbv 1, followed by electrophoresis and image analysis of the photographed gels. This approach is relatively simple and allows the running of several samples in parallel. The only possible limitation is the dynamic OD range of the film, i.e., quantification of editing in samples in which the extent of editing is extremely high or low. Even this limitation may be overcome in the future by HPLC quantification of the fractions of PCR product originating from edited and unedited mRNA by HPLC. With the analysis technique used in the present series of experiments, small fractions corresponding to <1% unedited RNA can still be clearly visualized, as indicated in Fig. 2D.
After having established this analytical system, the supplier of Bbv 1 (New England Biolabs) provided recombinant enzyme, with which we were not able to produce sharp electrophoresis bands unless the restriction digest was heated. However, after heating to 94°C, we obtained faint extra bands with higher molecular weights than those obtained with the uncut PCR product (Paschen and Djuricic, 1994, 1995). We then inserted a heating step of 60°C into the protocol and were, thus, able to eliminate these extra bands. This change in the protocol probably explains why we observed a slightly lower extent of GluR5 mRNA editing than we had in previous studies (Paschen and Djuricic, 1994, 1995).
The present series of experiments was designed to investigate whether transient forebrain ischemia induces any change in the extent of mRNA editing of GluR2, GluR5, or GluR6. It is noteworthy that mRNA editing changed in different ways in each of the AMPA/kainate receptor subunits. mRNA editing of GluR2 was not changed by ischemia; this mRNA species remained completely edited in all the brain structures studied. Editing of GluR5 was significantly increased in the striatum, whereas GluR6 editing was significantly reduced in all the three brain structures investigated. Since editing of these different subunits was changed so diversely, the underlying mechanisms and possible consequences for the neurons affected probably also differ. In the following discussion, the three subunits will, therefore, be treated separately.
In all the samples tested, GluR2 mRNA was completely edited, indicating that this editing reaction seems to be very stable in most physiological and pathological states of the brain. In fact, in the rat brain, the GluR2 subunit has been found to be completely edited in the physiological state in all brain structures studied to date, and in both embryonic and the adult tissues (Paschen et al., 1994a). In this respect, the GluR2 subunit differs considerably from the GluR5 and GluR6 subunits, the editing of which is sharply upregulated during embryonal development (Bernard and Khrestchatisky, 1994; Paschen et al., 1994a). We also noted a similar stability in the extent of GluR2 mRNA editing in the human brain both in the physiological state and in patients suffering from Huntington's disease (Paschen et al., 1994b). In contrast to this high extent of GluR2 mRNA editing, Nutt and Kamboj (1994) observed a significantly lower degree of GluR2 editing in the substantia nigra (72%) and corpus striatum (89%) of the normal human brain.
After cerebral ischemia, the extent of GluR5 mRNA editing was increased significantly in the striatum (by ∼46% 4 h after ischemia), but not significantly in the cortex or hippocampus. In the experimental model used in the present study, neuronal cell damage was produced in the lateral striatum, and this pathological process is already manifested morphologically 3–6 h after ischemia (Pulsinelli et al., 1982). We did not distinguish between lateral and medial striatum before analysis, so we do not yet know whether the extent of editing was increased in either portion of the striatum or in both structures. In principle, an increase in the fraction of edited GluR5 mRNA molecules could be caused by an increased rate either of GluR5 mRNA editing or degradation of unedited GluR5 mRNA. We have two plausible explanations for the observed increase in GluR5 mRNA editing observed in the striatum after ischemia. The first is that this is an adaptive response, namely, an increase in the fraction of edited GluR5 mRNA, taking place in those neurons that are not damaged by transient ischemia. If this interpretation is correct, then the upregulation of GluR5 mRNA editing could be the result of reparative mechanisms that do not affect vulnerability directly. The second is that neurons exhibiting a large fraction of unedited GluR5 are particularly vulnerable to cerebral ischemia. If this assumption is correct, then the observed increase in the extent of editing would result from a breakdown of unedited GluR5 mRNA in neurons in the striatum irreversibly damaged after ischemia. It is interesting to note that in the hippocampal CA1 subfield—a brain region known to be most vulnerable to transient cerebral ischemia—GluR5 mRNA is completely unedited, as indicated by single-cell PCR experiments (Mackler and Eberwine, 1993). These observations corroborate our view that a high level of unedited GluR5 might result in a higher sensitivity of neurons to transient cerebral ischemia. The exact mechanism underlying the observed increase in GluR5 mRNA editing could be established in subsequent experiments by analyzing the extent of GluR5 editing in single neurons of both the medial and lateral striatum after transient forebrain ischemia.
In all three brain structures studied, the extent of GluR6 mRNA editing decreased significantly after ischemia. In the striatum, this change was already apparent only 4 h after ischemia, whereas in the cortex and hippocampus, no significant change occurred until 24 h after ischemia. We have two separate plausible explanations for the decrease in GluR6 mRNA editing observed in the present study. The first is that this decrease results from disturbances in the editing reaction. To date, the mechanisms of the editing reaction have still not been established. Evidence has been presented that this reaction is catalyzed by a nuclear adenosine deaminase specific for double-stranded RNA (Higuchi et al., 1994; Rueter et al., 1995; Yang et al., 1995). An enzyme reaction such as this could be disturbed after ischemia, either directly by an allosteric inhibition of enzyme activity or indirectly through a decrease in overall protein synthesis (Bodsch et al., 1985; Thilmann et al., 1986). An alternative explanation would be that GluR6 mRNA editing is reduced as a result of glial proliferation after ischemia. With regard to regional differences in the extent of GluR6 mRNA editing in control brains, the low extent of GluR6 mRNA editing in the white matter, which consists predominantly of glial cells, contrasted sharply with that in gray matter structures (Paschen et al., 1994b). Since glial proliferation is already evident shortly after 30 min transient forebrain ischemia in the rat, as evidenced by the increase in glial fibrillary acidic protein synthesis (Kiessling et al., 1986), it would be possible for postischemic glial proliferation to cause the observed decrease in the fraction of edited GluR6 mRNA. However, we have not yet established which of the cells in the white matter exhibit low GluR6 mRNA editing and whether these particular cells proliferate after ischemia in the gray matter, where the low rates of editing were observed.
Results of this study corroborate the view that editing in each of the AMPA/kainate receptor subunits GluR2, GluR5, and GluR6 is regulated differently after transient forebrain ischemia. Whereas editing is unchanged for GluR2, it is significantly increased in the striatum for GluR5 and significantly decreased for GluR6 in all the brain structures studied. The observed changes in the extent of editing were not dramatic: The fraction of unedited GluR5 subunit (the higher the fraction of unedited subunit, the higher the calcium conductance through the respective ion channel) was decreased in the striatum from 61 to 43%, while that of GluR6 was increased from ∼10 to 20% in all brain structures. However, any interpretation of the results should take into account the fact that the tissue samples analyzed contained both vulnerable and nonvulnerable neurons and that, most probably, changes were even more prominent within surviving or damaged neurons. Thus, further elucidation the role of mRNA editing of kainate receptor subunits GluR5 and GluR6 in the manifestation of ischemic cell damage would require cellular analysis, e.g., in situ hybridization techniques under conditions in which the extent of editing could be visualized on a regional level.
In conclusion, transient forebrain ischemia induces changes in the extent of mRNA editing of the kainate receptor subunits GluR5 and GluR6. Editing of GluR5 is significantly increased in the striatum but not in the cortex or hippocampus. Editing of GluR6 mRNA, in contrast, is reduced in all brain structures studied so far. Thus, the effect of ischemia on the editing reaction appears to be different for GluR5 and GluR6. Furthermore, brain structures, such as the cortex, striatum, and hippocampus respond to dissimilar ways to transient ischemia.
Footnotes
Acknowledgment:
The excellent technical assistance of Cordula Strecker is gratefully acknowledged. This work was supported by the Deutsche Forschungsgemeinschaft, Grant Pa 266/7-1. GluR5 and GluR6 plasmids were provided courtesy of Drs. Steve Heinemann, Martin Köhler, and Peter H. Seeburg.