
Abstract
Neuronal loss is common to many neurodegenerative diseases. Although necrosis is a common histopathologic feature observed in neuropathologic conditions, evidence is increasing that apoptosis can significantly contribute to neuronal demise. The prevalence of either type of cell death, apoptosis or necrosis, and the relevance for the progression of disease is still unclear. The debate on the occurrence and prevalence of one or the other type of death in pathologic conditions such as stroke or neurotoxic injury may in part be resolved by the proposal that different types of cell death within a tissue reflect either partial or complete execution of a common death program. Apoptosis is an active process of cell destruction, characterized morphologically by cell shrinkage, chromatin aggregation with extensive genomic fragmentation, and nuclear pyknosis. In contrast, necrosis is characterized by cell swelling, linked to rapid energy loss, and generalized disruption of ionic and internal homeostasis. This swiftly leads to membrane lysis, release of intracellular constituents that evoke a local inflammatory reaction, edema, and injury to the surrounding tissue. During the past few years, our laboratories have studied the signals and mechanisms responsible for induction or prevention of apoptosis/necrosis in neuronal injury and this is the subject of this review.
Apoptosis plays an important role in both physiologic and pathologic conditions (Kerr et al., 1972; Wyllie et al., 1980). In the development of the embryo, overproduction of cells ultimately requires that surplus elements die as part of the process that balances growth and differentiation with death. In the case of the central and peripheral nervous systems, apoptosis occurs extensively during development (Deckwerth and Johnson, 1993). However, dysregulation of apoptosis may also underlie the etiology of several diseases. For example, a decrease in the rate of apoptosis may precipitate proliferation of neoplastic cells and development of autoimmune diseases (Thompson, 1995), whereas an increased rate of apoptosis may be involved in both acute and chronic neurodegenerative diseases (Kure et al., 1991; MacManus et al. 1993; Raff et al., 1993). Studies in vitro and in vivo have recently shown that apoptosis could be a final cause of demise in the neurodegenerative processes underlying Alzheimer's disease, Parkinson's disease, and acquired immunodeficiency syndrome (AIDS) dementia, as well as neuronal cell death in the penumbra of an area of cerebral ischemia (Games et al., 1995; Linnik et al., 1993; Mitchell et al., 1994; Su et al., 1994; Petito and Roberts, 1995; Dickson, 1995; Adle-Biassette et al., 1995; An et al., 1995; An et al., 1996; Gelbard et al., 1995; Lipton, 1996).
Ca2+, MITOGEN-ACTIVATED PROTEIN KINASE SIGNALING, CASPASES, AND APOPTOSIS
Although there are probably multiple signals leading to apoptosis, recent work suggests that activation of a protease, or family of proteases, related to the nematode gene ced-3 and human interleukin-1β converting enzyme may be a common point of convergence in many cell types (Yuan et al., 1993). Activation or suppression of this protease family may be triggered by intracellular messengers such as Ca2+, but also by cyclic adenosine monophosphate and free radicals, including nitric oxide (NO) and superoxide anion. The biochemical machinery responsible for these events appears to be present in all mammalian cells, except blastomeres. Some of these proteolytic systems may be Ca2+ dependent, and direct evidence that an increase in Ca2+ after mitochondrial Ca2+ overload can mediate apoptotic death has been provided by several laboratories including ours (Juntti-Berggren et al., 1993; Nicotera et al., 1994; Nicotera et al., 1996). Thus, an increase in the intracellular Ca2+ level appears to represent a relatively common trigger for apoptosis in cells of diverse tissue origins. Proteins that mediate the effects of Ca2+ are ubiquitous. Recent evidence in neurons and other cell types has suggested that Ca2+ may trigger intracellular cascades, e.g., involving mitogen-activated protein kinase signaling pathways (Fig. 1) (Lipton, 1997; Kawasaki et al., 1997). In some cases, caspases can cleave mitogen-activated protein kinases, whereas kinases can also lead to caspase activity (Cardone et al., 1997). In addition, Ca2+ may activate directly other protease families and recent work suggests that calpains cooperate with caspases in triggering neuronal apoptosis under excitotoxic conditions (Leist et al., 1997b). Undoubtedly, a complex network of intracellular signaling cascades is involved in such apoptotic pathways. Further work to develop therapeutic interventions to ameliorate apoptosis would benefit from a detailed knowledge of potential points of control. On the other hand, the existence of alternative execution pathways may prevent the use of specifically targeted drugs.
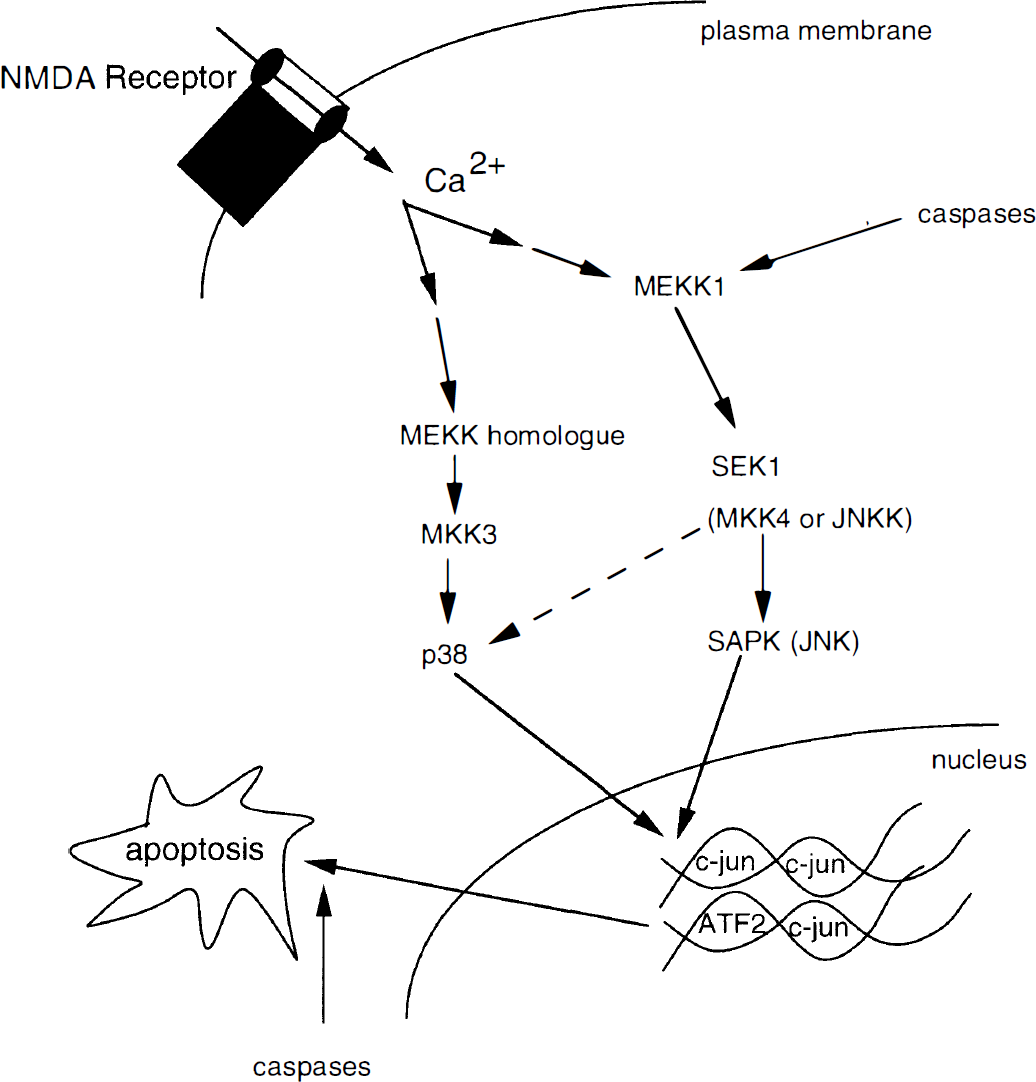
Hypothetical pathway leading from NMDA receptor activation, excessive Ca2+ influx, and intracellular signaling pathways contributing to neuronal apoptosis. After NMDA receptor activation and Ca2+ influx, subsequent activation of MAP kinases lead to transcriptional activation of c-jun. The p38 pathway may be proapoptotic, while activation of MAP kinase pathways via MEKK1 (MEK kinase 1, a MAP kinase kinase) may be antiapoptotic (Yujiri et al., 1998). MEKK1 activates the JNK (c-Jun aminoterminal kinase) pathway along with the ERK (extracellular signal-regulated protein kinase) pathway. Caspases, in addition to contributing to apoptosis downstream from MAP kinase activation, may also cleave MEKK1. When cleaved by caspases, a fragment of MEKK1 signals a proapoptotic response that further amplifies caspase activity, in contrast to the antiapoptotic effect of full-length MEKK1. NMDA, N-methyl-
REACTIVE OXYGEN INTERMEDIATES AND APOPTOSIS
The accumulation of reactive oxygen intermediates (ROI) can result in the perturbation of the cellular prooxidant—antioxidant balance, termed oxidative stress. Excessive increases in ROI have been implicated as an important mechanism of cytotoxicity in a number of biological systems, including neurons. Both apoptosis and necrosis can be induced by oxidative stress. For example, in some cell types low pro-oxidant concentrations induce apoptosis, whereas higher concentrations promote necrosis (Dypbukt et al., 1994). The involvement of free radicals in the pathogenesis of a number of neurodegenerative disorders, including Alzheimer's disease, is suggested by a number of observations. Behl et al. (1994) have reported that the β-amyloid peptide kills neural and other cells by H2O2-mediated mechanisms. Troy and Shelanski have shown that suppression of copper/zinc superoxide dismutase (Cu, Zn-SOD) activity induces apoptosis in PC 12 cells (Troy and Shelanski (1994). Analogously, antioxidants may protect cells from apoptosis (Buttke and Sandstrom, 1994). There are several possible roles for oxidation in apoptosis. Protein oxidation may be an essential influence on gene expression required for the signals leading to apoptosis. Oxidative events, e.g., may affect either directly or indirectly caspase activation, DNA binding of several transcription factors, and cytoskeletal alterations in cells undergoing apoptosis (Abate et al., 1990; Meyer et al., 1993; Troy et al., 1996).
REACTIVE NITROGEN INTERMEDIATES AS PHYSIOLOGIC MESSENGER OR PATHOLOGIC MEDIATOR OF APOPTOSIS
Reactive nitrogen intermediates (RNI) are generated by the progressive oxidation of the terminal guanidine residue of
NO has a dual role as physiologic messenger and as a contributor to lethal processes. Classically, NO mediates endothelium-dependent relaxation, takes part in neurotransmission, and is a key player in the cellular immune response (Dawson et al., 1992; Nathan, 1992). Multiple reactions occur between oxygen, superoxide, and transition metals with the following products: N2O3 [equivalent to (NO2−)(NO+)], peroxynitrite (OONO−), and metal-NO adducts, respectively. These reactions determine the biological activity of the NO group in its various redox-related forms. Other reactions involving the transfer of NO+ equivalents (with one less electron than NO) result in nitrosative reactions at nucleophilic centers with critical cysteine sulfhydryls, producing S-nitrosothiol formation. This reaction, termed S-nitrosylation, occurs preferentially at specific consensus motifs of amino acid residues centered around a critical cysteine sulfhydryl which reacts with the NO group, and serves to regulate protein function akin to phosphorylation of critical serine, threonine, or tyrosine residues (Lipton et al., 1993; Stamler et al. 1997). Accordingly, thiol- and transition metal-containing proteins serve as major target sites for NO-related species (Stamler, 1994). NO-target interaction achieves both cyclic guanosine monophosphate (cGMP)–dependent and cGMP-independent transducing mechanisms. cGMP-independent NO-induced responses account for the antimicrobial, the cytostatic, and in many cases the cytotoxic capacity of NO. Excess production of NO has been shown to underlie, at least in part, glutamate-induced neuronal toxicity in cultures of cortical and striatal neurons (Dawson et al., 1991). Finally, radicals generated by the interaction of NO with oxygen species can induce DNA damage (Noronha-Dutra et al., 1993). Our own studies have recently focused on the cytotoxicity of NO in conjunction with reactive oxygen species in cortical neurons and cerebellar granule cells.
NO-INDUCED NEURONAL CELL DEATH: CONTRIBUTION OF APOPTOSIS
Excessive activation of excitatory amino acid receptors has been implicated as a mechanism for neurotoxicity in both acute and chronic neurologic diseases, ranging from stroke and head trauma to Alzheimer's disease and amyotrophic lateral sclerosis to Hungtington's disease and AIDS dementia (Rothman and Olney, 1987; Choi, 1988; Meldrum and Garthwaite, 1990; Coyle and Puttfarken, 1993; Lipton and Rosenberg, 1994). This form of neuronal cell death has been termed excitotoxicity. The underlying process responsible for neuronal cell death after overactivation of glutamate receptor subtypes — of which the N-methyl-
The interaction of glutamate with excitatory amino acids receptors initiates a cascade of events involving excessive Ca2+ entry and activation of several enzymes, including phospholipases, proteases, and NOS (Dawson et al., 1991; Choi, 1988; Manev et al., 1989; Meldrum and Garthwaite, 1990; Coyle and Puttfarken, 1993; Lipton and Rosenberg, 1994). Phospholipase A2 activation leads to the generation of arachidonic acid and other metabolites as well as to the formation of oxygen free radicals. This can lead to oxidative stress and, together with the activation of Ca2+ -dependent catabolic processes (Nicotera et al., 1992), can lead to either necrosis or apoptosis depending on the intensity of the insult (Bonfoco et al., 1995; Ankarcrona et al., 1995). In addition, it has been shown that NO — generated by activation of NOS — can react with O2− to form OONO− (peroxynitrite) (Beckman et al., 1990). This RNI, or one of its degradation products, is neurotoxic (Lipton et al., 1993). In fact, NO alone, even at high levels, is not toxic to cortical neurons, but only becomes neurotoxic after reaction with O2− to form ONOO− (Lipton et al., 1993).
We have investigated whether apoptosis or necrosis can be induced in cerebrocortical neurons in culture by overstimulation of glutamate receptors, with consequent influx of excessive Ca2+, and downstream production of NO and O2−. We used high and low concentrations of glutamate agonists (such as NMDA), NO-donors [such as 3-morpholinosydnonimide (SIN-1) and S-nitroso-cysteine (SNOC)], or peroxynitrite (OONO−). We found that exposure of cortical cultures to relatively short durations or low concentrations of NMDA, SNOC, SIN-1, or peroxynitrite induced delayed neuronal cell death characterized by apoptotic features. In contrast, intense exposure to high concentrations of NMDA or peroxynitrite induced relatively rapid necrotic cell death in neurons (Bonfoco et al., 1995). Superoxide dismutase (SOD) and catalase attenuated neuronal cell death, most likely by reducing the formation of peroxynitrite because they were only effective if peroxynitrite had not yet formed. These findings suggest that the intensity of the original insult may determine the ensuing pathway to either necrotic or apoptotic neuronal cell death. The nature of the original insult as well as the decision to enter the necrotic versus the apoptotic pathway might have therapeutic implications in terms of the possible effectiveness of SOD/catalase or NMDA-antagonists, as well as the necessary timing of such interventions.
GLUTAMATE ELICITS NECROSIS OR APOPTOSIS DEPENDING ON MITOCHONDRIAL FUNCTION AND ENERGY SUPPLY
Our laboratories have also been investigating the mechanisms underlying the decision of neurons to undergo apoptosis or necrosis after glutamate exposure (Ankarcrona et al., 1995). We found that glutamate can induce either early necrosis or delayed apoptosis in cerebellar granule cell cultures, similar to the aforementioned results on cerebrocortical neurons. During and shortly after exposure to glutamate, a subpopulation of cerebellar granule cells died by necrosis. In these neurons, mitochondrial membrane potential collapsed, nuclei swelled, and intracellular debris were scattered in the incubation medium. Neurons surviving the early necrotic phase recovered mitochondrial potential and energy levels. Later, they underwent apoptosis, as shown by the formation of apoptotic nuclei and chromatin degradation into high and then low molecular weight fragments. These results suggest that energy generation and mitochondrial function are critical factors in the decision on the mode of execution of neuronal death. During exposure to glutamate in our experiments, both mitochondrial membrane potential and ATP levels decreased in many neurons. In those with irreversibly dissipated mitochondrial membrane potentials, necrosis rapidly ensued. The initial surviving neuronal population recovered both mitochondrial membrane potential and energy levels, and subsequently underwent delayed apoptosis. In fact, before DNA fragmentation and apoptotic nuclei were evident, mitochondrial membrane potential, metabolism, and energy charge had been restored to near normal levels. At that point, repeated challenge with glutamate resulted in repeated collapse of mitochondrial membrane potential, rapidly followed by necrosis of a second neuronal subpopulation. According to one hypothesis, loss of energy and onset of necrosis simply prevent the activation of the default apoptotic cell death program. This postulate is supported by the observation that treatment of cerebellar granule cells with the combination of a relatively low concentration of glutamate (100 µmol/L) plus the irreversible mitochondrial uncoupler carbonyl cyanide m-chlorophenylhydrazone (CCCP, 10 µmol/L) resulted in necrosis of 60% of the neuronal population rather than in the delayed apoptosis observed with this low concentration of glutamate alone. Evidence that nonneuronal cells triggered to undergo apoptosis are instead forced to die by necrosis when energy levels are rapidly compromised, has been recently provided (Leist et al., 1997a).
Until recently, the role of mitochondria in apoptosis had been a matter of debate. The observation that apoptosis occurs in respiration-deficient cells had initially cast doubts on the role of mitochondrial energy production in apoptotic cell death (Jacobson et al., 1993; Newmeyer et al., 1994). In view of more recent findings (Ankarcrona et al., 1995; Leist et al., 1997a), it is more likely, that mitochondrial energy generation is needed to retain some of the apoptosis execution subroutines. To examine the events that determine the mode of execution of cell death (apoptosis or necrosis) after energy deprivation, cells can be exposed to a single insult and then individual parts of the death program can be blocked by manipulating the intracellular ATP level. With this approach it has been possible to show that when ATP levels are reduced, typical apoptotic stimuli instead cause necrosis (Leist et al., 1997a). If the intracellular ATP concentration is markedly reduced during a critical time window, activation of downstream caspases and almost all typical apoptotic changes are prevented. Stimulated cells die, nonetheless, but this demise has necrotic features. These findings provide direct evidence that the complete apoptotic program involves energy-requiring steps, at least in the cells types that we have studied, including neurons. More recent work suggests that one of the ATP-requiring steps is the formation of a protein complex between Apaf-1, pro-caspases, and cytochrome c released from damaged mitochondria. This complex is needed for the activation of execution caspases, including caspase-3 (Fig. 2) [for review see Green and Kroemer (1998)].
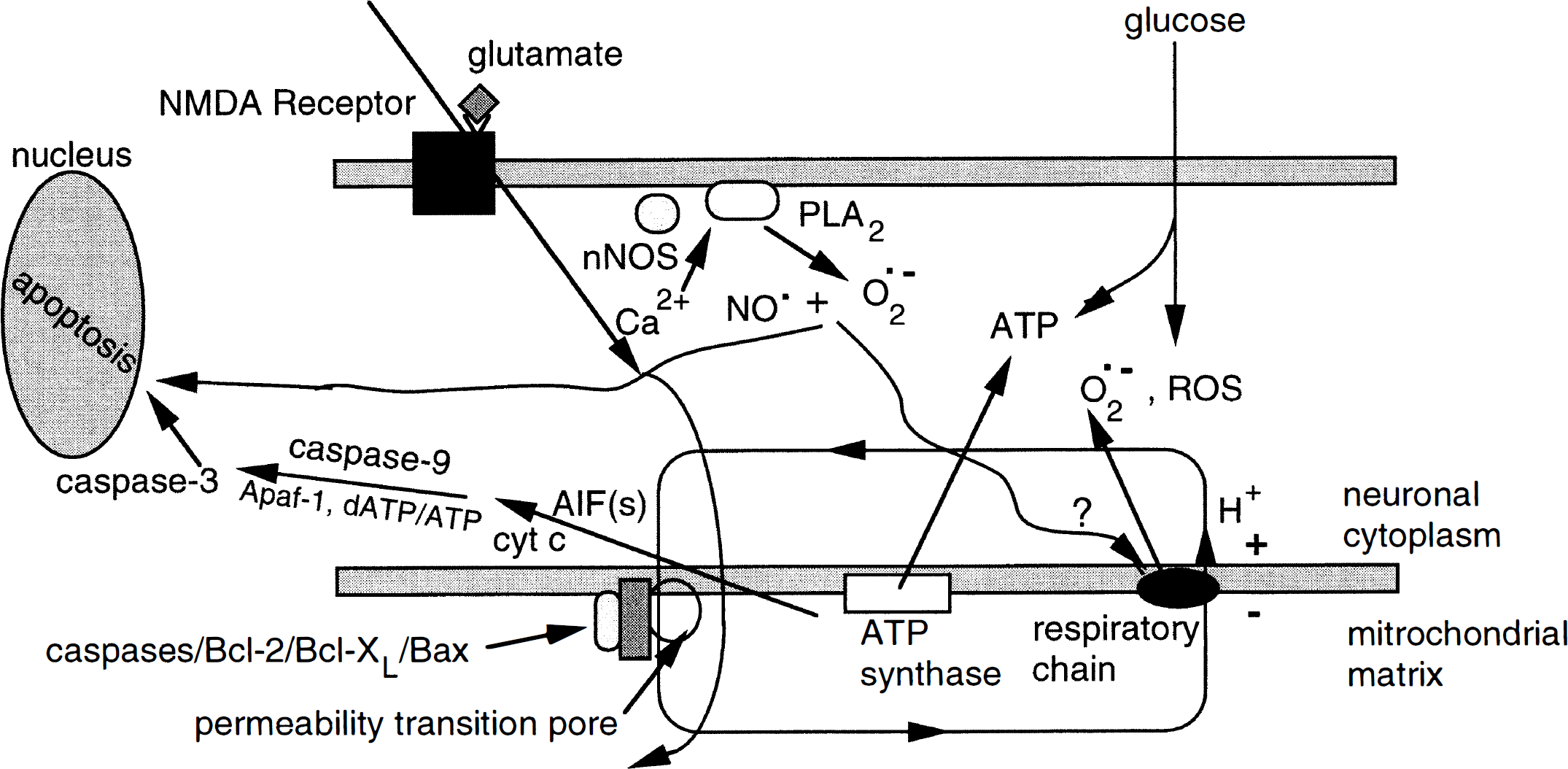
Model incorporating several features of apoptotic injury in neurons. After a relatively mild insult by glutamate at the NMDA receptor (initiating excessive Ca2+ influx and generating NO and O2− to form ONOO−), the mitochondrial membrane potential transiently depolarizes and a drop in ATP transiently results, but this not a sufficient compromise in energy to severely disrupt the pumps and therefore does not result in necrosis. Rather, mitochondrial permeability transition occurs and a slight swelling of the mitochondria ensues. Caspases, Bcl-2, Bcl-XL, and Bax, located at the mitochondrial inner membrane, may affect permeability transition (Marzo et al., 1998). As previously shown in other cell types, it is hypothesized in neurons that as ATP synthesis recovers, apoptotic-inducing factors (AIF) are released from mitochondria. These AIF include cytochrome c (cyt c) which, in conjunction with apoptotic protease activating factor-1 (Apaf-1) and dATP/ATP, triggers activation of caspase-9 and in turn caspase-3, leading to apoptosis. NMDA, N-methyl-
Thus, mitochondria can have an active role in cell death by releasing proteins that are required for caspase activation (i.e., cytochrome c). An initial loss of mitochondrial membrane potential may be an important signal for apoptosis as long as the ATP level is sufficient to allow caspase-3 activation (Green and Kroemer, 1998). Alternatively, in some systems, selective damage of the outer mitochondrial membrane without measurable loss of membrane potential may cause release of intermembrane proteins (Green and Kroemer, 1998). In this case, mitochondria would still be capable of producing ATP. In neurons, a decrease in mitochondrial membrane potential — as observed in our studies during the initial phases of glutamate injury — may lead to the initial release of signaling molecules from the mitochondria. In the presence of sufficient ATP levels (i.e., after mild insults) this could trigger execution of apoptosis via caspases-9/3. In ATP-depleted neurons (i.e., after more intense or prolonged insults), execution of cell death may result from a loss of ionic homeostasis, with resultant swelling and osmotic lysis, producing a typically necrotic morphology. Alternatively, a morphology intermediate between necrotic and apoptotic may develop, depending on exactly when the programmed cell death pathway was interrupted by the extreme loss of energy.
Ca2+, MITOCHONDRIA, ROI, Bcl-2, AND CASPASES
Ca2+ influx triggered by excessive activation of NMDA receptors results not only in mitochondrial depolarization, but also in Ca2+ overload of mitochondria and subsequent ROI production (Nicholls and Akerman, 1982; Dykens, 1994; Ankarcrona et al., 1995; Dugan et al., 1995; White and Reynolds, 1996; Schinder et al., 1996). Ca2+ influx also initiates a cascade of biochemical processes in the cell cytosol including the activation of several enzymes such as phospholipases, proteases, and nNOS, as elaborated above. These events subsequently lead to the formation of additional ROI as well as other free radicals including NO. NO and ROI have previously been implicated in excitotoxic neuronal death (Beckman et al., 1990; Dawson et al., 1991; Lipton et al., 1993; Coyle and Puttfarken, 1993; Bonfoco et al., 1995). More recent data (Schulz et al., 1996; Tenneti et al., 1998) suggest that caspases act upstream from ROI accumulation. Potential sources of ROI under excitotoxic conditions in neurons include mitochondria (Beal, 1992). Calcium-dependent depolarization of mitochondria can contribute to the accumulation of ROI (Dykens, 1994). The relative contribution of these different sources of ROI during excitotoxicity is not clear at the present time. The subsequent steps leading to apoptosis are also not yet well worked out in neurons, but model systems based on other tissues suggest that mitochondrial permeability transition occurs with release of apoptotic-inducing factors and cytochrome c, which contribute to nuclear damage and apoptosis (Fig. 2) (Golstein, 1997). Moreover, caspases, Bcl-2, and Bcl-XL, most likely located at the mitochondrial inner membrane, affect permeability transition in some cell types (Yang et al., 1997; Kluck et al., 1997).
PROTEASE FAMILIES AND EXECUTION OF NEURONAL APOPTOSIS
Execution of cell death may involve distinct proteolytic families. Each protease family may become predominant depending on different stimuli or intensity of insult. Moreover, different proteases may interact in autocatalytic loops. For example, there is evidence that inhibitors of Ca2+ activated proteases, or calpains, are as effective as caspase inhibitors in preventing neuronal apoptosis following some forms of excitotoxicity (Leist et al., 1998). Calpains may process pro-caspases, whereas active caspases may cleave Ca2+– transport proteins or endogenous calpain inhibitors, such as calpastatin. Caspase activation seems to be the predominant execution pathway in physiologic cell death and after moderate insults. Accordingly, brain development is severely disturbed in animals with targeted deletions of the apoptosis executing caspases-3 and −9 (Hakem et al., 1998; Kuida et al., 1998). However, thymocytes from caspase-9 (–/–) embryos are still sensitive to apoptosis elicited by ultraviolet radiation or anti-CD95 antibodies (Hakem et al., 1998), which suggests a differential requirement for this execution caspase in different types of apoptotic cell death. In adult neurons dying after a severe insult, caspase-3/9-dependent pathways can be inhibited, but other proteolytic systems can still execute cell death (Leist et al., 1997b).
These considerations may be relevant when designing therapeutic approaches. Observations in stroke models suggest that apoptosis occurs mainly in the borderzone region of perfusion (the penumbra), while necrosis dominates in the more severely stressed areas of the ischemic core. Intervention a few hours after the ischemic insult is aimed at reducing spread of the lesion and inhibiting delayed cell death in the borderzone area. Assuming that the degree of injury dictates the activation of distinct pathways for the execution of cell death, it is apparent that caspase inhibitors, e.g., may be most effective in areas where the intensity of the excitotoxic insult is mild but ineffective in regions where the stress is more intense. Thus, strategies that combine agents to reduce the overall intensity of the insult and the overall lesion size (i.e., NMDA-antagonists or selective nNOS inhibitors) with agents that block execution of apoptosis (caspase inhibitors) may prove more successful than individual agents (Schulz et al., 1998). Similar considerations may hold in more chronic neurodegenerative diseases as the intensity of the insult varies with time in a particular brain region.
Caspases may act at several regulatory points in the cascade leading to apoptosis. It is possible that a particular caspase family member might be transiently activated either upstream or downstream of mitochondrial events (Susin et al., 1997). Development and use of very specific inhibitors for individual caspase family members will be required to study the detailed role of these enzymes more extensively. Our recent results suggest that caspases are critical mediators of excitotoxin-induced neuronal apoptosis. ROI, lipid peroxidation, and chromatin condensation appear to be downstream events to caspase activation while Ca2+ influx and mitochondrial depolarization are upstream in the neuronal signaling pathways that contribute to apoptosis (Leist et al., 1997b; Tenneti et al., 1998).
NO AND INHIBITION OF CELL DEATH
In addition to the contribution of NO to neuronal cell death described above, it is important to outline the mechanisms whereby NO-related species can also be neuroprotective. Recent work has shown that S-nitrosylation of critical cysteine sulfhydryl groups of the NMDA receptor, of p21ras during MAP kinase signaling, and in the active site of caspase enzymes can regulate protein activity. Curtailing excessive activity of the NMDA receptor by S-nitrosylation is neuroprotective (Lipton et al., 1993; Lipton and Stamler, 1994). Similarly, downstream from NMDA receptor activation, S-nitrosylation of a critical cysteine residue in p21ras or of the cysteine residue in the active site of all known caspase enzymes decreases their activity and also affords protection to neurons from NMDA receptor-mediated apoptotic events (Tenneti et al., 1997; Melino et al., 1997; Yun et al., 1998). Thus, depending on its redox state, the NO group can contribute to excitotoxicity (via formation of peroxynitrite in conjunction with superoxide anion) or provide neuroprotection (by downregulating the activity of both the NMDA receptor and its downstream activation of p21ras and caspases).
Besides directly inhibiting p21ras and caspases, NO may interfere with the execution of apoptosis at different steps, without necessarily affecting the rate of cell death [i.e., NO can change the mode of demise from apoptosis to necrosis (Melino et al., 1997)]. For example, NO can inhibit caspase activation by mechanisms in addition to S-nitrosylation (Kim et al., 1997), e.g., by NO-dependent formation of cGMP that can interfere with cell death signaling upstream from caspase activation (Mannick et al., 1994; Kim et al., 1997; Hebesteit et al., 1998). Moreover, because a well documented action of NO is inhibition of the mitochondrial respiratory chain, it seems conceivable that the resulting ATP-depletion might be relevant to the effects of NO on cell death. In fact, recent results have suggested that NO prevents caspase activation by inhibiting mitochondrial respiration, and thereby decreasing intracellular ATP levels (Leist et al., 1999). The prevention of cell death in this system is only ephemeral. Cell demise is delayed, and doomed cells die eventually by necrosis. When non-mitochondrial, glycolytic ATP generation was supported via glucose supplementation to the culture medium, death reverted to its apoptotic from.
In vivo, halting the apoptotic program may have two possible implications: (1) cells protected by NO via stopping the apoptotic execution cascade would have time to recover from a transient or mild insult, and thus survive; (2) cells exposed to a lethal, normally apoptotic insult would eventually lyse without being removed by phagocytosis. Thus, depending on the situation, endogenous mediators, such as NO, either may prevent cell demise entirely or convert an apoptotic insult into a necrotic one. In the latter case, the release of factors from dead cells and the ensuing inflammation would further aggravate tissue damage.
CRITERIA FOR DISTINGUISHING APOPTOSIS FROM NECROSIS
A variety of techniques, ranging from morphological to biochemical to molecular, have been developed to distinguish apoptotic from necrotic cell death. Importantly, more than one criterion must be used to make this distinction, and, as alluded to above, certain events can produce an overlap in the appearance of the two forms of cell death, e.g., if tissue macrophages are insufficient to phagocytose apoptotic cells, secondary necrosis can occur. Techniques for determining the mode of cell death and a list of caveats limiting the usefulness of this distinction were recently reviewed (Bonfoco et al., 1997).
SUMMARY
In conclusion, the type of cell death encountered in neurons exposed to excitotoxins, with subsequent influx of excessive Ca2+ and generation of ROI and RNI, depends on the intensity of the exposure and involves two temporally distinct phases of necrosis and apoptosis. Similar events could possibly occur in both acute and chronic neurodegenerative disorders resulting from a variety of insults. Necrosis — associated with extreme energy depletion — may simply reflect the failure of neurons to perform the default apoptotic cell death program used to efficiently dispose of aged or otherwise unwanted cells. In contrast, the maintenance of balanced energy production may be a decisive factor in determining the degree, type, and progression of neuronal injury to apoptosis caused by excitotoxins and free radicals.