
Abstract
Matrix metalloproteinases (MMPs), a family of proteolytic enzymes which degrade the extracellular matrix, are implicated in blood—brain barrier disruption, which is a critical event leading to vasogenic edema. To investigate the role of reactive oxygen species (ROS) in the expression of MMPs in vasogenic edema, the authors measured gelatinase activities before and after cold injury (CI) using transgenic mice that overexpress superoxide dismutase-1. A marked induction of pro-gelatinase B (pro-MMP-9) was seen 2 hours after CI and was maximized at 12 hours in wild-type mice. The pro-MMP-9 level was significantly lower in transgenic mice 4 hours (P < 0.001) and 12 hours (P < 0.05) after CI compared to wild-type mice. The activated MMP-9 was detected from 6 to 24 hours after injury. A mild induction of pro-gelatinase A (pro-MMP-2) was seen at 6 hours and was sustained until 7 days. In contrast, the activated form of MMP-2 appeared at 24 hours, was maximized at 7 days, and was absent in transgenic mice. Western blot analysis showed that the tissue inhibitors of metalloproteinases were not modified after CI. The results suggest that ROS production after CI may contribute to the induction and/or activation of MMPs and could thereby exacerbate endothelial cell injury and the development of vasogenic edema after injury.
Matrix metalloproteinases (MMPs) are known as a family of calcium-requiring, zinc-containing proteolytic enzymes, which degrade the extracellular matrix in a variety of physiologic and pathophysiologic conditions and play a crucial role in extracellular remodeling. They are secreted as inactive zymogens and require enzymatic cleavage of the N-terminus for activation (Murphy, 1995). Among MMPs, gelatinase A (MMP-2) and gelatinase B (MMP-9) are able to digest the endothelial basal lamina, which plays a major role in maintaining the integrity of the blood-brain barrier (BBB). These MMPs are increasingly being implicated in the pathogenesis of several central nervous system diseases (Yong et al., 1998), such as cerebral ischemia (Rosenberg et al., 1996, 1998; Romanic et al., 1998; Gasche et al., 1999), multiple sclerosis (Anthony et al., 1997), seizures (Zhang et al., 1998), bacterial meningitis (Paul et al., 1998), and intracerebral hemorrhage (Rosenberg and Navratil, 1997). These studies suggest the strong relationship between MMP activation and vasogenic edema formation though BBB disruption.
Vasogenic edema is one of the most common forms of brain edema observed in clinical practice, in which the functional integrity of the endothelial cells is altered, and permeability of brain capillary endothelial cells to macromolecules is increased by disruption of the tight junctions that characterize the BBB (Chan et al., 1984). Cold injury (CI) is well known as a classic model for vasogenic brain edema and trauma, in which increased BBB permeability and the pathogenesis that follows are well-characterized (Chan et al., 1991). In previous studies, increased BBB permeability was detected within 30 minutes, lasted several hours to 3 to 4 days in the lesional and perilesional areas after CI, and was followed by formation of the infarction (Chan et al., 1987; Nag, 1996; Murakami et al., 1999). Furthermore, growing evidence suggests that oxygen radicals play an important role in the pathogenesis of lesions induced by CI (Chan et al., 1991; Oury at al., 1993).
In the present study, we sought to investigate the potential role of MMPs on CI as a suitable and well-characterized model for vasogenic edema. By zymographic measurement of gelatinase activity, we detected activation of MMP-9 with early induction of pro-MMP-9 and pro-MMP-2, which might be consistent with acute BBB disruption, and subsequent mild induction of pro-MMP-2 and its activation 7 days after CI. To further investigate the role of reactive oxygen species (ROS) on MMP induction and/or activation after CI, we examined the gelatinase activity in transgenic mice that overexpress the endogenous antioxidant enzyme copper, zinc superoxide dismutase (SOD-1) before and after CI. The results showed the inhibitory effects of SOD-1 on both MMP induction and activation, suggesting the contribution of ROS to the formation of vasogenic edema by regulating MMP expression.
MATERIALS AND METHODS
Superoxide dismutase-1 transgenic mice
Heterozygous SOD-1 transgenic mice of the SOD-1 TgHS/ SF-218-3 strain carrying human SOD-1 genes were derived from the founder stock previously described (Epstein et al., 1987). They were bred on a CD-1 mouse background. The SOD-1 transgenic mice were identified by quantitative demonstration of SOD-1 using nondenaturing gel electrophoresis followed by nitroblue tetrazolium staining (Epstein et al., 1987). There were no observable phenotypic differences between SOD-1 transgenic and wild-type normal littermates.
Cold injury and histologic assessment
SOD-1 transgenic mice and wild-type normal littermates (male, 35 to 40 g, 3 months old) were subjected to CI according to the methods previously described (Chan et al., 1991; Morita-Fujimura et al., 1999). All procedures were in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and were approved by Stanford University's Administrative Panel on Laboratory Animal Care. Experimental animals were anesthetized with chloral hydrate (350 mg/kg) and xylazine (4 mg/kg) intraperitoneally and placed in a stereotactic frame. The scalp was incised on the midline and the skull was exposed. A metal probe, 4 mm in diameter and cooled with dry ice, was applied to the surface of the skull with a 100-g weight for 30 seconds. The experimental animals were killed at 1, 2, 4, 6, 8, 12, and 24 hours and 3 and 7 days after CI.
Matrix metalloproteinase extraction
MMP extraction from brain tissue was performed as previously described, with minor modifications (Zhang and Gottschall, 1997). Approximately 20 to 50 mg of damaged brain tissue and the homologous tissue from the contralateral cortex were cut into pieces 1 to 24 hours and 3 and 7 days after CI and were homogenized by douncing 15× in Teflon-glass tissue homogenizers (Wheaton, Millville, NJ, U.S.A.) in 10 vol of cold lysis buffer (basic buffer: 50 mmol/L Tris-HCl [pH 7.6], 150 mmol/L NaCl, 5 mmol/L CaCl2, 0.05% BRIJ-35, 0.02% NaN3, containing 1% Triton X-100). Five microliters of the homogenates were stored to measure the total amount of protein. The homogenates were then centrifuged at 4°C at 12,000 × g for 5 minutes. Fifty microliters of the supernatants were stored and used for Western blot analysis of tissue inhibitor of metalloproteinase (TIMP) −1 and TIMP-2, membrane type-1 matrix metalloproteinase (MT-MMP-1), and β-actin. The supernatants were also further incubated with a 1:10 volume of gelatin sepharose 4B (Pharmacia Biotech AB, Uppsala, Sweden) for 1 hour at 4°C with constant shaking. After incubation, samples were centrifuged at 500 × g for 2 minutes. Gel pellets were washed once with basic buffer. After centrifugation at 500 × g for 2 minutes, the pellets were resuspended in elution buffer (basic buffer containing 10% dimethylsulfoxide and 20% volume of lysis buffer). Purified samples were analyzed by zymography, type IV collagenolytic activity assay, and Western blotting. Protein concentrations were determined by the bicinchoninic acid method (BCA kit, Pierce, Rockford, IL, U.S.A.).
Zymography
Expression of the proform and activated form of MMP-2 and MMP-9 was analyzed by zymography (Kleiner and Stetler-Stevenson, 1994), which was performed according to the manufacturer's instructions (Novex, San Diego, CA, U.S.A.). One fifth volume of nonreducing sample buffer (0.4 mol/L Tris, pH 6.8, 5% sodium dodecyl sulfate [SDS], 20% glycerol, 0.05% bromophenol blue) was added to the samples obtained as above and incubated for 10 minutes at room temperature. The samples were then loaded onto 10% SDS polyacrylamide gel electrophoresis gels containing 0.1% gelatin (Novex). After running the gels, they were incubated with 2.5% Triton X-100 two times for 20 minutes, equilibrated with 1 × developing buffer (Novex), and incubated for over 18 hours in the developing buffer at 37°C with gentle agitation. The gels were stained with coomassie blue (0.5% coomassie R-250, 30% methanol, 10% acetic acid) for 30 minutes and destained in washing solution (30% methanol, 10% acetic acid) as necessary. White bands on a blue background indicated zones of digestion corresponding to the presence of different pro-MMPs and activated MMPs on the basis of their molecular weight. One tenth nanogram of human pro-MMP-9 and human pro-MMP-2 (Oncogene Research, Cambridge, MA, U.S.A.) and 0.01 ng of activated MMP-9/MMP-2 (Oncogene Research) were used as the MMP standards. To quantify the protein expression, the gels were scanned using a densitometer (GS-700, Bio-Rad Laboratories, Richmond, CA, U.S.A.) and analyzed using Multi-Analyst 1.0.2 software (Bio-Rad). Recovery of pro-MMP-9 and pro-MMP-2 through the purification steps was estimated using this zymographic analysis.
Type IV collagenase activity assay
Type IV collagenase activity assay was performed using a commercially available kit (Chemicon International, Temecula, CA, U.S.A.). According to the manufacturer's protocol, 50 μL of samples were incubated with fluorescein-5-isothiocyanate -labeled type IV collagen solution, first for 20 minutes at room temperature and then for 2 hours at 42°C in a water bath. Samples were then placed on ice for 5 minutes. Three hundred microliters of cold enzyme stop reagent/extraction solution were added to all samples which were then stored for 35 minutes on ice. Residual undigested collagen was precipitated by centrifugation (10 minutes at 12000 × g). A standard curve was made using MMP-9 standard solution provided by the manufacturer. Proteolytic activity of the injured samples was measured with or without ethylene diaminetetraacetic acid (0.5 mmol/L), which is known to inhibit MMPs. Finally, fluorescence intensity of sample supernatants was measured with a fluorometer (Molecular Devices, Sunnyvale, CA, U.S.A.) at 538 nm/485 nm.
Immunohistochemistry of gelatinase B
Anesthetized animals were perfused with 10 U/mL heparin and subsequently with 4% formaldehyde in 0.1 mol/L phosphate-buffered saline (pH 7.4) 24 hours after CI. The brains were removed, postfixed for 36 hours in 4% formaldehyde, sectioned at 75 mm on a vibratome, and processed for immunohistochemistry. The sections were incubated with a blocking solution containing 20% normal goat serum (Vector, Burlingame, CA, U.S.A.) and reacted with antihuman MMP-9 polyclonal antibody (Anawa, Wangen, Switzerland) at a dilution of 1:100. Immunohistochemistry was performed using the avidin-biotin technique (Vector), and then the nuclei were counterstained with methyl green solution for 10 minutes. As a negative control, sections were incubated without primary antibodies. For histologic assessment, alternate slices from each brain section were stained with cresyl violet.
Western blot analysis
The supernatants from brain homogenates were prepared as mentioned above and used for Western blot analysis for TIMP-1, TIMP-2, MT-MMP-1, and β-actin. Equal volumes of 2 × SDS sample buffer (Novex) were added to the supernatants from brain homogenates. About 5 μg of each sample per lane were loaded onto 12% Tris-glycine gels and transferred to polyvinylidene diflouride membranes. After 1 hour of blocking, the membranes were incubated with an optimal dilution of primary antibodies against TIMP-1 (rabbit polyclonal antibody, Chemicon, no. AB770), TIMP-2 (rabbit polyclonal antibody, Chemicon, no. AB801), and MT-MMP-1 (rabbit polyclonal antibody, Chemicon, no. AB8102) in 0.5% dry milk phosphate-buffered saline/0.1% Tween 20 at 4°C overnight. After washing, the membranes were further incubated with horseradish peroxidase-conjugated antirabbit immunoglobulin G (Boehringer Mannheim, Indianapolis, IN, U.S.A.) and developed using an enhanced chemiluminescence system (ECL plus Western blotting detection reagents, Amersham International, Buckinghamshire, England). The membranes were reprobed with monoclonal anti-β-actin antibody (Sigma, St. Louis, MO, U.S.A.) and developed as above. MMP-9 Western blotting was performed as previously described (Gasche et al., 1999). The affinity-purified MMP samples were mixed with the same volume of 2 × SDS sample buffer (Novex) and boiled for 5 minutes. The samples were then loaded onto 8% Tris-glycine gels and transferred to polyvinylidene diflouride membranes. The primary antibodies were diluted 1:1000 for rabbit polyclonal antibody against human MMP-9 (Anawa, no. 5980-0907) and incubated with the membranes. The membranes were further incubated with horseradish peroxidase-conjugated antirabbit immunoglobulin G (Boehringer Mannheim) and developed as above.
Statistical analysis
Data are presented as the mean ± standard deviation. Statistical comparisons were evaluated by ANOVA followed by unpaired Student's t test using StatView software, version 4.5 (Abacus Concepts, Berkeley, CA, U.S.A.). P < 0.05 was considered to be statistically significant.
RESULTS
Early induction and activation of pro-gelatinase B is followed by delayed activation of gelatinase A after cold injury in mice
The expressions of pro- and activated MMPs were first examined by gelatin zymography (Fig. 1). In order to detect pro- and activated MMP-9 and MMP-2 with better sensitivity, the gelatinases were purified up to 217.5 ± 35.0-fold (n = 5) and concentrated 5 times from brain homogenates with 85% to 90% of MMP recovery, using their selective affinity to gelatin sepharose after centrifugation, as described in Materials and Methods (Fig. 1A). Pro-MMP-9 was increased 2 hours after injury with a molecular weight of 96 kDa and was maximized at 12 hours in wild-type mice (Fig. 1B, lanes 1 to 8). Activated MMP-9 with a molecular weight of 88 kDa appeared 4 hours after injury and was also maximized at 12 hours (Fig. 1B, lanes 3 to 6), whereas it was not detected in the control (Fig. 1B, lane 1) and contralateral hemispheres (data not shown). A slight induction of pro-MMP-2 with a molecular weight of 72 kDa was observed from 6 hours and was further increased at 3 days (Fig. 1B). The MMP-2 activated form was first observed from 24 hours after injury with a molecular weight of 64 kDa and was maximized 7 days after CI (Fig. 1B, lanes 6 to 8). Figure 2 demonstrates the quantitative analysis of zymographic measurements of pro-MMP-9 (Fig. 2, upper graph) and pro-MMP-2 (Fig. 2, lower graph). Induction of pro-MMP-9 became significant at 12 hours and 3 days when pro-MMP-9 was 100.9-fold and 73.6-fold, respectively, compared with the control (Fig. 2, upper graph). Induction of pro-MMP-2 was significant 6 hours, 12 hours, 3 days, and 7 days after CI. Three days after CI, pro-MMP-2 induction was maximized and was 3.80-fold compared with the control (Fig. 2, lower graph), which was milder than pro-MMP-9. In order to further evaluate the actual proteolytic activity of the extracted samples used for zymographic analysis, we performed an enzymatic activity assay using fluorescein-5-isothiocyanate-labeled type IV collagen as substrates of MMP-9 and MMP-2. We detected increased activity in digestion of type IV collagen at 12 hours (95.8 ± 32.6 mU/mL, mean ± SD, n = 3) and 7 days (53.9 ± 61.2 mU/mL, n = 3) compared with the normal control (1.5 ± 0.5 mU/mL, n = 3). These activities were inhibited by adding 0.5 mmol/L ethylene diaminetetraacetic acid, confirming the involvement of MMPs in the activities observed (data not shown). Because no activated form of MMP-9 was observed at 7 days, collagenase activity detected at 7 days was considered to mainly reflect MMP-2 activity.
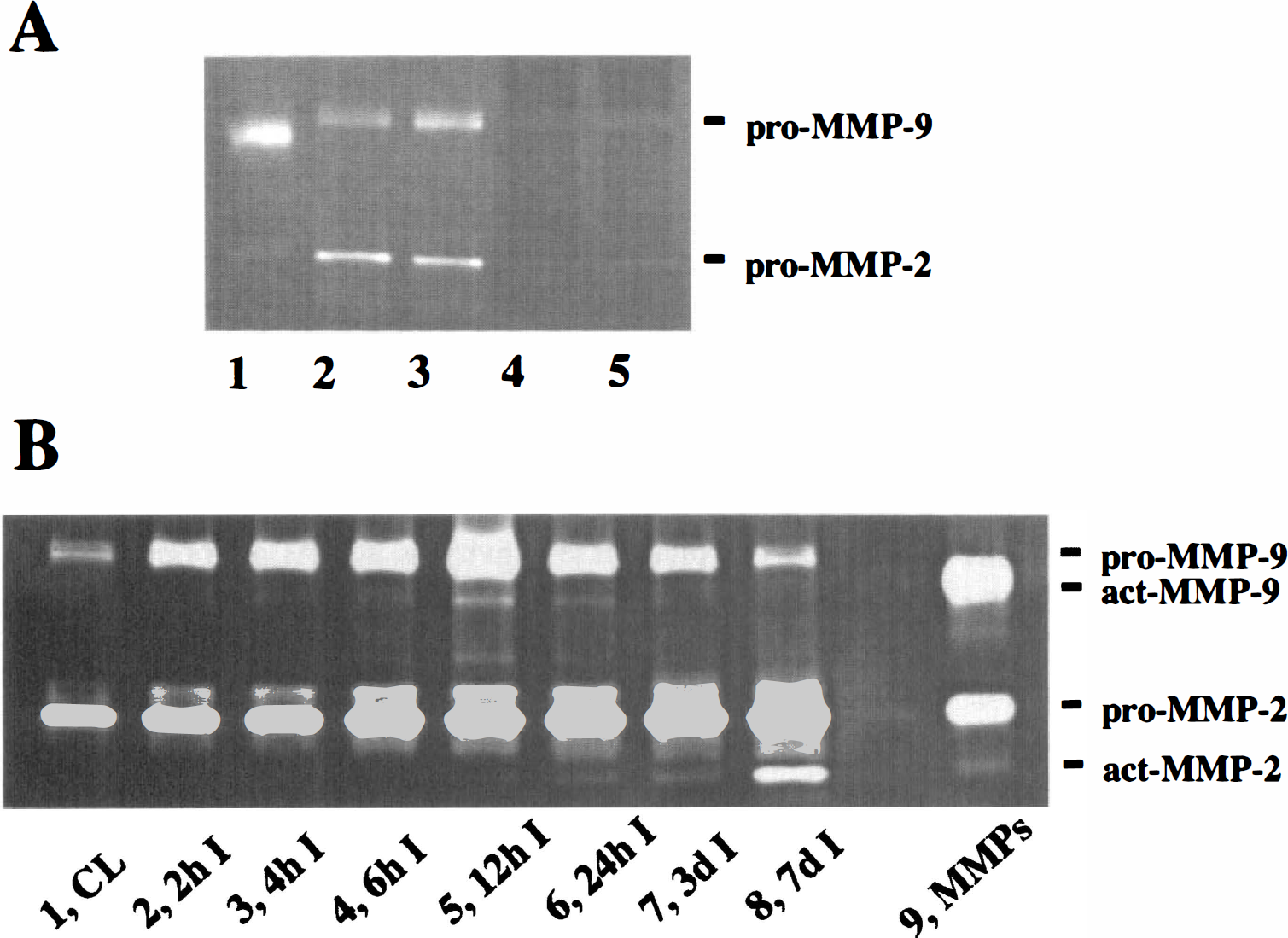
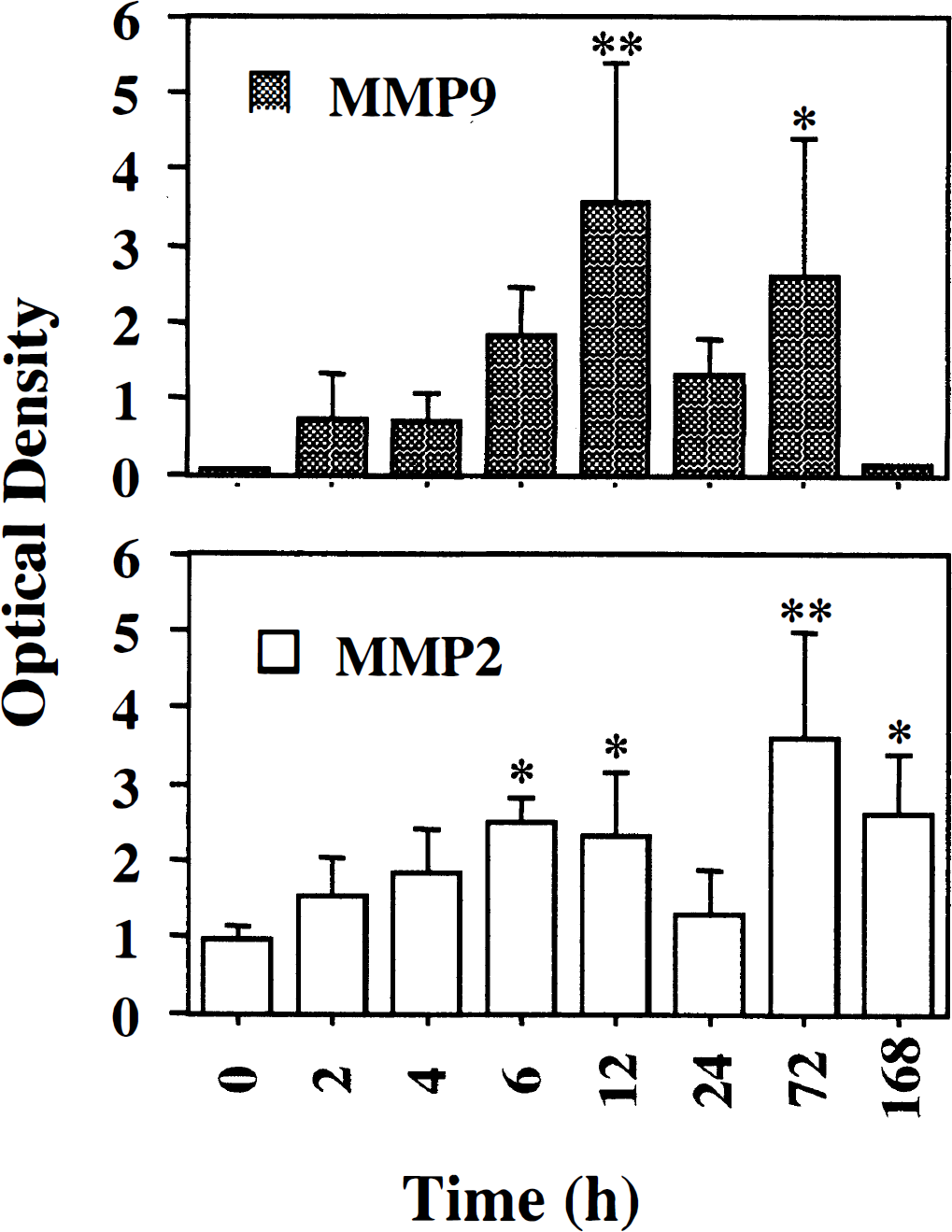
Quantitative analysis of gelatin zymography of mice brain extracts after cold injury (CI). Graphs show quantitative analysis of zymograms from three independent studies. Optical density values among three zymograms were adjusted using standard human pro-gelatinase B (MMP-9) and pro-gelatinase A (MMP-2) (Oncogene Research). Statistical analysis showed a significant increase at 12 and 72 hours in pro-MMP-9 expression and at 6, 12, 72, 168 hours in pro-MMP-9 expression (*P < 0.05, **P < 0.001, n = 3, versus normal control; upper panel). A mild increase in pro-MMP-2 reached significance 3 days after CI (lower panel).
Immunohistochemical analysis demonstrates perilesional upregulation of gelatinase B after cold injury
To examine the anatomic distribution of induced MMP-9, we performed immunohistochemical staining 24 hours after CI (Fig. 3). A strong MMP-9 immunoreactivity was observed mainly in the perilesional area (Fig. 3A, area 1, and 3C), but not in the core of the lesion or control brain (Fig. 3A, areas k and m, and 3B). There was no immunoreactivity in the specimens that were treated without a primary antibody (data not shown). In our previous study, we confirmed this antibody's immunoreactivity to mouse pro-MMP-9 and activated MMP-9 by Western blotting (Gasche et al., 1999). In the present study, Western blot analysis of MMP-9 further confirmed the same immunoreactivity of the antibody against mouse MMP-9 induced after brain edema (data not shown).
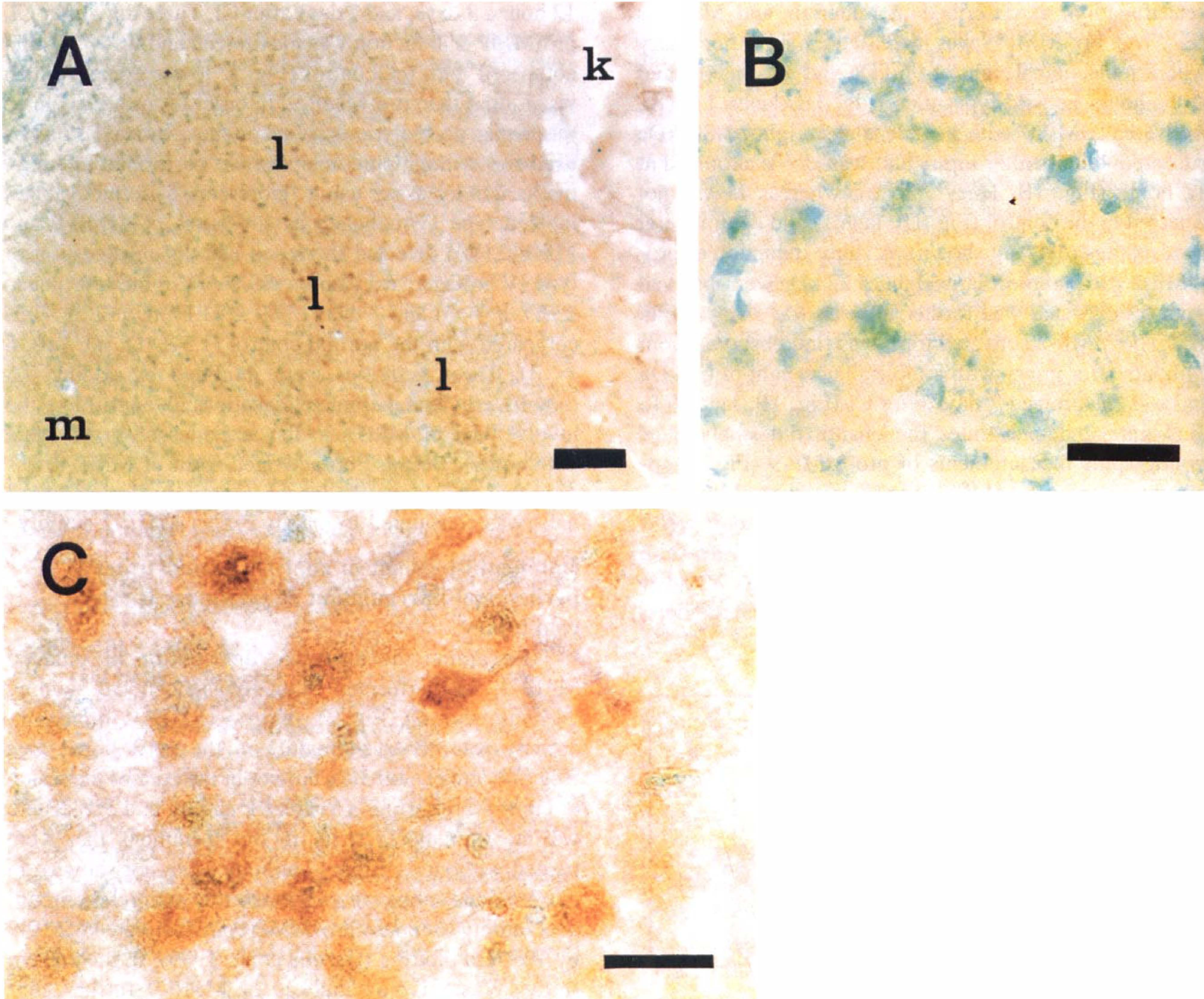
Immunohistochemical analysis of gelatinase B (MMP-9) demonstrates the anatomic distribution of MMP-9 induction 24 hours after cold injury (CI).
Western blot analysis demonstrates the small contribution of tissue inhibitors of metalloproteinase and membrane type-1 matrix metalloproteinase to changes in matrix metalloproteinases activation after cold injury
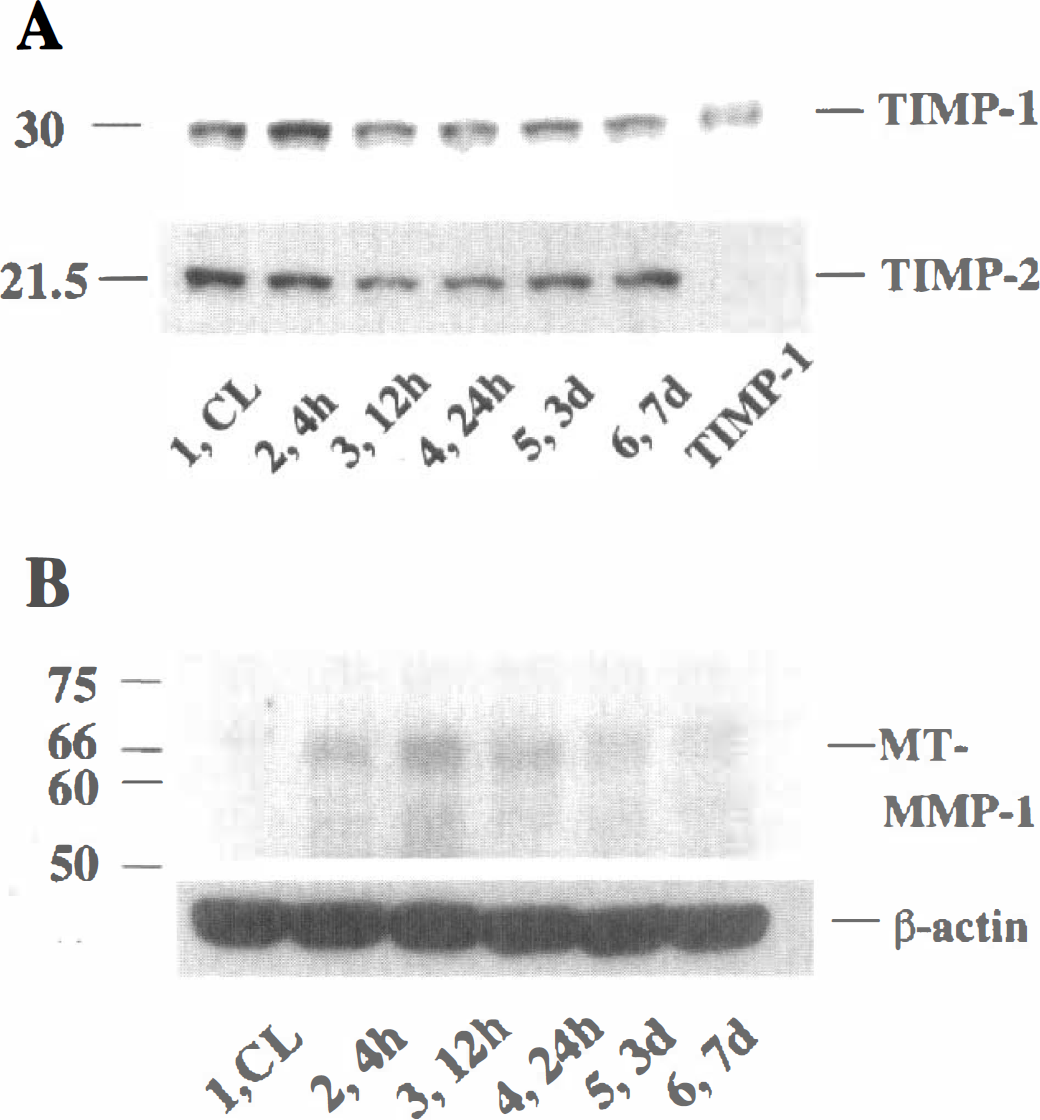
To examine whether MMP activity is modulated by TIMP-1 and TIMP-2, protein expression of TIMP-1 and TIMP-2 in brain homogenates after CI was analyzed using Western blot analysis. A characteristic 30-kDa band of TIMP-1 in the normal control (Fig. 4A, upper panel, lane 1) was observed and was not modified by the injury (Fig. 4A, upper panel, lanes 2 to 6). TIMP-2 appeared as a characteristic band of 21.5 kDa in the normal control (Fig. 4A, lower panel, lane 1) and was not modified by the injury (Fig. 4A, lower panel, lanes 2 to 6). To further investigate whether the delayed activation of MMP-2 is directly regulated by the expression of MT-MMP-1, which is known to activate MMP-2 by processing proMMP-2, its expression was also examined by Western blot analysis (Fig. 4B, upper panel). MT-MMP-1, which appeared as a characteristic band of 66 kDa in the normal control, was slightly induced from 4 hours, was maximized at 12 hours, and gradually decreased until 7 days after CI (Fig. 4B, upper panel).
Induction and activation of matrix metalloproteinases were reduced by superoxide dismutase-1 overexpression in mice
To further investigate the molecular mechanisms of MMPs after CI, we examined the effect of SOD-1 on MMP induction and activation. We used SOD-1 transgenic mice that overexpress SOD-1 2.9-fold in the brain cortex compared with nontransgenic mice, as previously described (Chan et al., 1991). Figure 5 demonstrates expression of pro-MMP-9 in both wild-type and transgenic mice. There were no differences in pro-MMP-9 basal expression between the transgenic and wild-types (Fig. 5A, lanes 1 and 2). Pro-MMP-9 induction in the injured area was apparently reduced in SOD-1 transgenic mice compared with that in wild-type mice from 4 hours after CI (Fig. 5A). A quantitative analysis showed significant reduction of pro-MMP-9; this expression in transgenic mice was approximately 28.8% (n = 7, P =0.002) and 47.5% (n = 6, P =0.05) of wild-type mice at 4 hours and 12 hours, respectively (Fig. 5B, 4 and 12), whereas no difference was seen in pro-MMP-9 expression in control animals (Fig. 5B). The effect of SOD-1 on MMP-2 induction and activation 7 days after CI is shown in Fig. 6. Induction of pro-MMP-2 was also significantly reduced by SOD-1 overexpression (Figs. 6A and 6B, pro-MMP-2, n = 3, P < 0.05). Furthermore, activation of MMP-2 observed 7 days after injury in wild-type mice was not detected in SOD-1-overexpressing transgenic mice (Figs. 6A and 6B, MMP-2, n = 3, P < 0.05). No difference was seen in pro-MMP-9 expression in control animals (Fig. 6B, CL).
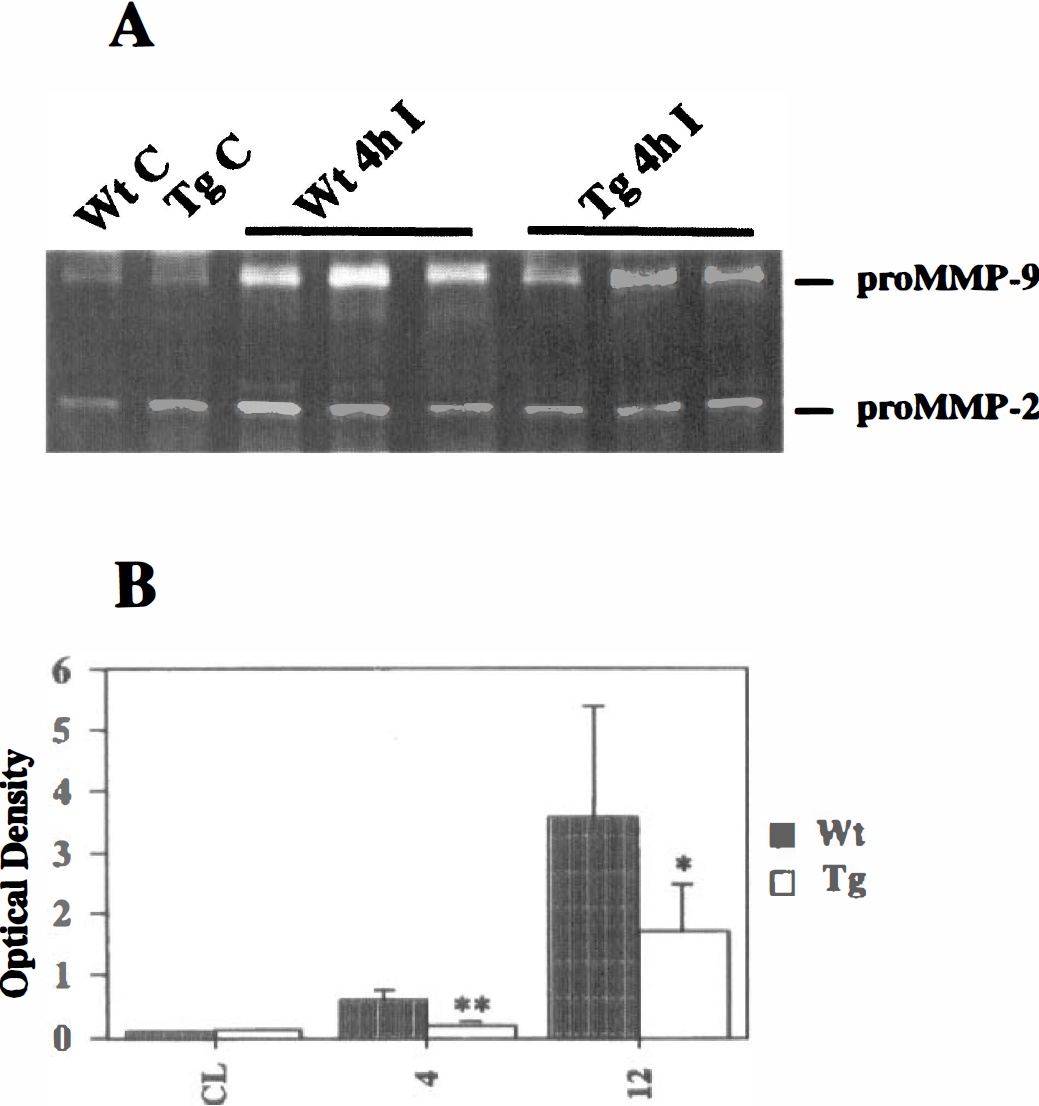
Induction of pro-gelatinase B (MMP-9) was reduced in mice that overexpressed superoxide dismutase-1 (SOD-1) compared with wild-type (Wt) animals.
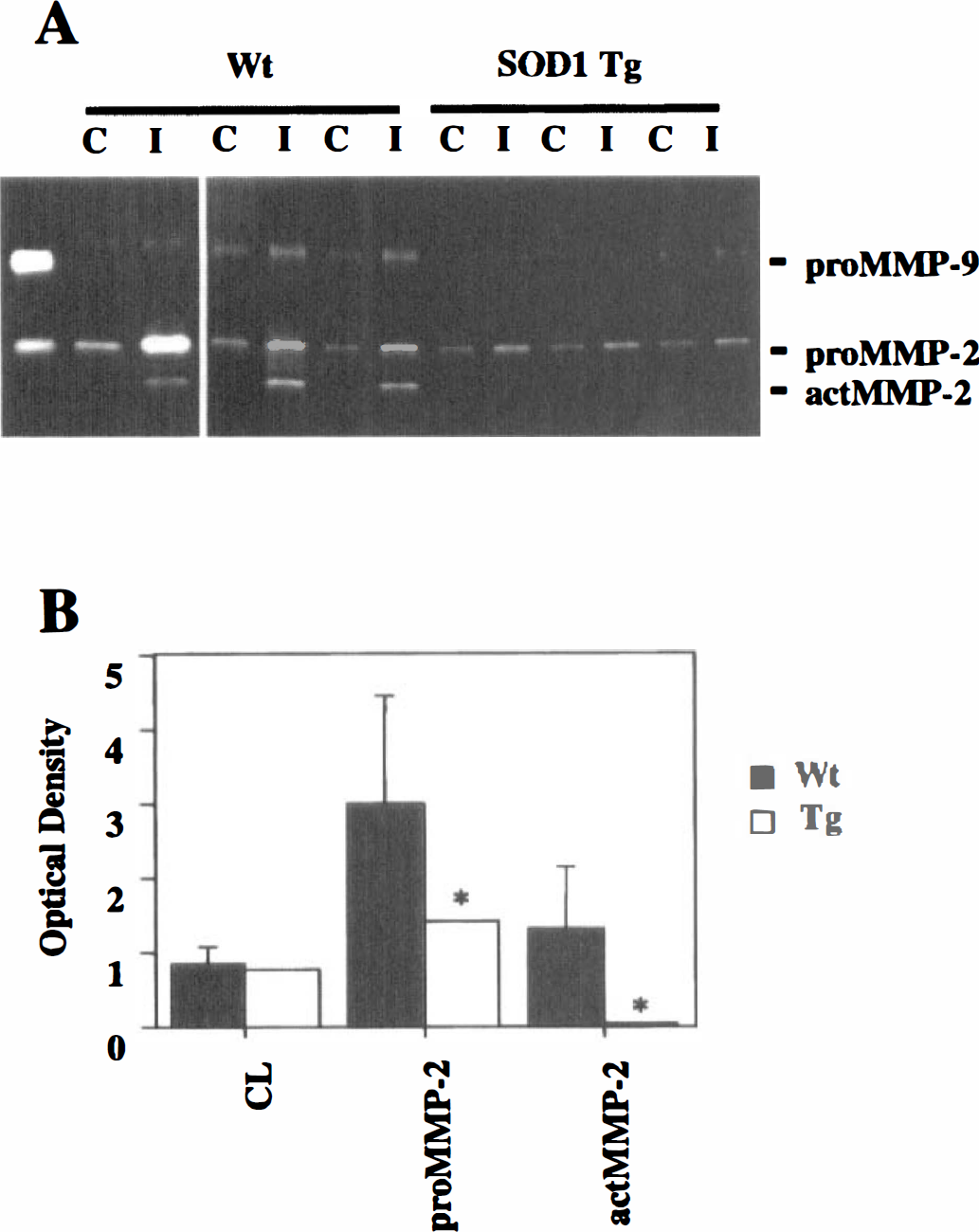
Activation and induction of gelatinase A (MMP-2) was reduced in mice that overexpressed superoxide dismutase-1 (SOD-1) compared with wild-type animals.
DISCUSSION
The present study provides the first evidence that MMP-9 and MMP-2 was induced independently after CI, which is known as a model for vasogenic brain edema and trauma (Chan et al., 1991) and that this induction was prevented by the overexpression of the antioxidant enzyme SOD-1. We used gelatin affinity gel to concentrate and purify MMP-9 and MMP-2 (Zhang and Gottschall, 1997), which enabled us to detect pro- and activated MMPs with better sensitivity (Fig. 1A). Early induction of pro-MMPs was maximized at 12 hours and was more prominent in pro-MMP-9 (100.9-fold compared with normal control) than in pro-MMP-2 (2.5-fold compared with normal control), whereas activation of MMP-9 was observed at 2 hours until 24 hours, with its peak at 12 hours (Figs. 1 and 2). A secondary mild induction of pro-MMP-2 was maximized at 3 days and lasted until 7 days, with a strong, clear band of activated MMP-2 (Figs. 1 and 2).
To investigate the contribution of ROS to the molecular mechanisms of MMP induction and activation after CI, we examined the effect of SOD-1 overexpression using transgenic mice, which was reported to cause a significant reduction in vasogenic edema after CI (Chan et al., 1991). The early induction of pro-MMP-9 at 4 and 12 hours after injury was significantly reduced by SOD-1 overexpression (Fig. 5), and the activation of MMP-2 at 7 days was apparently inhibited by SOD-1 overexpression (Fig. 6). These results suggest that SOD-1 prevents the early induction of MMP-9 and could thereby reduce the subsequent vasogenic edema formation and infarction after CI (Chan et al., 1991). The mechanism of this reduction of MMP-2 activation at 7 days by SOD-1 is not clear. It is conceivable that the lesser amount of brain damage did require less MMP-2 activation in transgenic mice compared with wild-type animals. We do not rule out the possibility that MMP-2 in transgenic mice was activated earlier than the time point at which we observed activation in the wild-type animals. Although further examination is necessary to clarify this point, our data suggest the multiple involvement of ROS in the mechanism of MMP induction and activation. In fact, it has been suggested that ROS regulate the activity of MMPs in vitro (Rajagopalan et al., 1996).
Previous studies using the same model have provided evidence of the early opening of the BBB within 30 minutes (Murakami et al., 1999), which lasts for 24 hours (Chan et al., 1987, 1991). This was demonstrated by extravasation of a serum albumin tracer, Evans blue, whereas water content in the injured hemisphere was maximized at 24 hours followed by formation of infarction. In our study, induction of MMP-9 appeared as early as 2 hours (19.8-fold compared with normal control) after injury and was maximized at 12 hours with mild induction of MMP-2 (Fig. 2), which preceded the peak of water content increase (Murakami et al., 1999). Furthermore, significant attenuation of MMP-9 expression 4 and 12 hours after CI in SOD-1 transgenic mice in the present study consistently resulted in the marked decrease of lesion volume in transgenic mice 24 hours after CI (Chan et al., 1991; Murakami et al., 1999). These results together suggest the possibility that the early induction and activation of MMPs may contribute to edema formation and lesion volume after CI. However, we do not completely rule out the possibility that other factors such as mechanical damage of endothelial cells by CI also played a role in BBB permeability as shown by Evans blue extravasation at the early time points.
Recently, MMP-9 was implicated in BBB disruption in a variety of central nervous system disorders (Rosenberg et al., 1996, 1998; Paul et al., 1998; Romanic et al., 1998; Zhang et al., 1998). We have demonstrated the early induction and activation of MMP-9 after focal cerebral ischemia, in which the early appearance of activated MMP-9 was consistent with the BBB opening (Gasche et al., 1999). In fact, it was reported that the MMP inhibitor BB-1101 reduced the BBB opening after focal ischemia (Rosenberg et al., 1998), which may also support the idea that activation of MMP-9 contributes to BBB dysfunction. The early appearance of activated MMP-9 shown in the present study may also suggest the role of MMP-9 in vasogenic edema formation after CI. Further study using MMP inhibitors and/or MMP knockout mice would be necessary to address the role of MMPs in edema formation after CI.
In contrast to MMP-9, little is known about MMP-2 induction (Clark et al., 1997) and, especially, MMP-2 activation by cleavage after brain insult as in this study (Figs. 1 and 6). In the present study, a mild and biphasic pro-MMP-2 was induced: the early induction which was accompanied by a strong MMP-9 induction and the secondary induction which lasted 7 days after the injury. Furthermore, we observed characteristic activation of MMP-2 7 days after injury, when tissue remodeling is assumed to be taking place after the peak of the infarction. Therefore, this MMP-2 activity is assumed to contribute to tissue remodeling of the lesion and is unlikely to be correlated with the BBB opening and edema formation. Moreover, the activated nature of 64 kDa of MMP-2 observed by zymographic analysis was supported by the presence of the digestion of type IV collagen (Results). MMP-2 induction and MT-MMP-1 colocalization in cerebral aneurysms was recently reported, suggesting that activation of MMP-2 may contribute to vascular extracellular matrix remodeling in cerebral aneurysms (Bruno et al., 1998). MT-MMP-1, which is activated by processing of its 66-kDa proform to become active 60-kDa MT-MMP-1, is known to cleave pro-MMP-2 to become a biologically active form (Takino et al., 1995). Therefore, we sought to clarify the contribution of MT-MMP-1 to this MMP-2 activation observed 7 days after CI. Western blot analysis did not show a significant change in MT-MMP-1 expression 7 days after CI (Fig. 4B). A slight induction of MT-MMP-1 was observed much earlier (4 to 24 hours) than the activation of pro-MMP-2 (Fig. 4B). Although the mechanisms of this MMP-2 activation remain unclear, it will be interesting to determine in future studies the anatomic distribution of MT-MMP-1 and MMP-2 by immunohistochemistry to evaluate the role of MT-MMP-1 in MMP-2 activation. Additionally, we cannot exclude the possibility of other unknown factors activating MMP-2 by cleavage. Further studies may be required to clarify this point.
Activation of MMPs is a complex process, which is tightly regulated by their natural inhibitors, TIMPs. The detection by zymography of activated MMPs, although indicative, does not reveal the actual proteolytic activity of the enzyme in vivo. Indeed, during the zymographic procedure, MMPs are separated from their respective TIMPs. In order to partially solve this problem and better define the balance between MMP-9 and its natural inhibitors TIMP-1 and TIMP-2, we detected the level of their expression by Western blotting under the same experimental conditions (Fig. 4A). Unlike MMP-9 and MMP-2, TIMP-1 and TIMP-2 were not induced after CI. A recent study showed a significant increase of TIMP-1 mRNA after focal ischemia (Wang et al., 1998), whereas its protein expression was not induced (Romanic et al., 1998; Gasche et al., 1999). Although, the mRNA expression after CI was not examined, it is possible that there is a discrepancy between mRNA and protein expression because of posttranscriptional regulation of the inhibitor.
The exact mechanisms involved in the independent induction and activation of MMP-9 and MMP-2 are not well defined. As for the MMP-9 cell population, it could be induced in a variety of cells in the central nervous system, not only endothelial cells (Nguyen et al., 1998) but also astrocytes (Gottschall and Yu, 1995), oligodendrocytes (Uhm et al., 1998), microglia (Gottschall et al., 1995), and neurons (Backstrom et al., 1996). In the present study, immunohistochemistry suggested the MMP-9 expression in the heterogeneous cell populations including neurons and astrocytes (Fig. 3C). A double staining of MMP immunohistochemistry with various cell markers will provide important information to confirm this issue. Because MMP-9 has an activator protein-1 (AP-1) binding site in its promoter region, it is conceivable that the early induction of pro-MMP-9 is associated with the alteration of AP-1 DNA-protein binding activity after CI. In fact, our preliminary study demonstrated that there was an early increase in AP-1 binding activity after CI, which was observed by electrophoretic mobility shift assays (unpublished data). A significant increase in AP-1 binding activity was also reported in several studies of brain insults, such as traumatic brain injury (Yang et al., 1994) or ischemia (Salminen et al., 1995), followed by an early induction of genes, such as c-fos and c-jun, which interact with the AP-1 site (An et al., 1993). Those early genes are also sensitive to ROS (Pinkus et al., 1996). Therefore, it is also conceivable that overexpression of SOD-1 in transgenic mice may contribute to the reduction of AP-1 binding activity, thereby preventing MMP-9 induction in the present study. Further examination of AP-1 binding activity after CI in transgenic mice will address this critical issue.
In conclusion, we have demonstrated the first evidence that MMP-9 and MMP-2 are induced independently after CI and that the early induction of MMP-9 is regulated by SOD-1, whereas that of MMP-2 is not. These results suggest that SOD-1 prevents the early induction of MMP-9, thereby reducing the subsequent vasogenic edema formation and infarction after CI.
Footnotes
Acknowledgments
The authors are grateful to Charles J. Epstein, MD, Department of Pediatrics, University of California, San Francisco, School of Medicine, for his continuous collaboration by providing breeding pairs of SOD-1 transgenic mice. The authors also thank Cheryl Christensen for editorial assistance and Liza Reola, Bernard Calagui, and Jane O. Kim for technical assistance.