
Abstract
During cerebral ischemia blood–brain barrier (BBB) disruption is a critical event leading to vasogenic edema and secondary brain injury. Gelatinases A and B are matrix metalloproteinases (MMP) able to open the BBB. The current study analyzes by zymography the early gelatinases expression and activation during permanent ischemia in mice (n = 15). ProMMP-9 expression was significantly (P < 0.001) increased in ischemic regions compared with corresponding contralateral regions after 2 hours of ischemia (mean 694.7 arbitrary units [AU], SD ± 238.4 versus mean 107.6 AU, SD ± 15.6) and remained elevated until 24 hours (mean 745,7 AU, SD ± 157.4). Moreover, activated MMP-9 was observed 4 hours after the initiation of ischemia. At the same time as the appearance of activated MMP-9, we detected by the Evan's blue extravasation method a clear increase of BBB permeability, Tissue inhibitor of metalloproteinase-1 was not modified during permanent ischemia at any time. The ProMMP-2 was significantly (P < 0.05) increased only after 24 hours of permanent ischemia (mean 213.2 AU, SD ± 60.6 versus mean 94.6 AU, SD ± 13.3), and no activated form was observed. The appearance of activated MMP-9 after 4 hours of ischemia in correlation with BBB permeability alterations suggests that MMP-9 may play an active role in early vasogenic edema development after stroke.
Blood–brain barrier (BBB) integrity protects the neuronal microenvironment (Cserr and Bundgaard, 1986). When this integrity is lost, inflammatory cells and fluid penetrate the brain, causing edema and cell death (Fishman, 1975), Impermeability of the BBB is maintained by microvascular endothelial cells through their tight junctions (Siflinger-Birnboim et al., 1987) and basal lamina. The latter is composed of type IV collagen, fibronectin, laminin, various proteoglycans, and heparan sulfates (Yurchenco and Schittny, 1990), Metalloproteinases (MMP) are proteolytic enzymes (Zn2+ -endopeptidases) secreted as zymogen, which must be cleaved to be fully active. They are involved in the remodeling of the extracellular matrix in a variety of physiologic and pathophysiologic conditions. Among MMP, gelatinase A (MMP-2) and gelatinase B (MMP-9), also known as type IV collagenases, are able to digest the endothelial basal lamina leading to the opening of the BBB (Rosenberg et al., 1992, 1996a,b, 1998). Inhibition of MMP reduces brain capillary damage in mice with experimental autoimmune encephalomyelitis (Gijbels et al., 1994). During cerebral ischemia-reperfusion, endothelial basal lamina dissolution starts as soon as 2 hours after initiation of ischemia and continues during reperfusion (Hamann et al., 1995), leading to a potentially harmful accumulation of leukocytes in tissue (Chen et al., 1992). Increased BBB permeability can be detected a few hours after the ischemic insult (Belayev et al., 1996; Kondo et al., 1997; Rosenberg et al., 1998). Expression of MMP-9 and MMP-2 has been shown to be significantly increased during stroke in human (Anthony et al., 1997) and rat models of focal ischemia (Romanic et al., 1998; Rosenberg et al., 1996b; Rosenberg et al., 1998). In the animal studies, MMP-9 increase was significant only after 12 hours after ischemia, revealing a discrepancy between MMP induction and the first signs of BBB alterations reported to occur earlier (Hamann et al., 1995; Rosenberg et al., 1998). Recent experimental data show that inhibition of MMP can reduce the degree of brain infarction produced by permanent focal ischemia (Romanic et al., 1998) and can decrease the BBB opening after 3 hours of reperfusion following 2 hours of focal ischemia (Rosenberg et al., 1998). These results imply that MMP play a critical role in brain injury after stroke. But so far no study has reported the early induction and activation of latent MMP-9 or MMP-2 after ischemia. Our study evaluates the effect of ischemia on the early expression and activation of gelatinases in permanent middle cerebral artery occlusion (pMCAO) using a recently described in vivo method of MMP extraction (Zhang and Gottschall, 1997). We also determined if MMP expression was correlated with BBB permeability alterations in mice after focal stroke.
MATERIALS AND METHODS
Focal cerebral ischemia in mice
Adult male CD-1 mice (35 to 40 g) were subjected to permanent focal ischemia by intraluminal middle cerebral artery blockade with a nylon suture, as described by others (Yang et al., 1994). The mice (n = 15) were anesthetized with 2.0% isoflurane in 30% oxygen and 70% nitrous oxide using a face mask. Central temperature was maintained at 37°C with a homeothermic blanket. Cannulation of a femoral artery allowed the monitoring of blood pressure and arterial blood gases, with samples for analysis being taken immediately after cannulation and 10 minutes after occlusion. Following the midline skin incision, the left external carotid artery was exposed, and its branches were electrocoagulated. An 11.0-mm 5-0 surgical monofilament nylon suture, blunted at the end, was introduced into the left internal carotid artery through the external carotid artery stump and tightly fixed at the appropriate position at the end of surgery.
Metalloproteinase extraction
Metalloproteinase extraction from brains was performed according to a previously described method (Zhang and Gottschall, 1997). All procedures were performed on ice. Brain tissues were homogenized with 2-mL Teflon glass homogenizers in a lysis buffer (50 mmol/L Tris-HCl pH 7.6, 150 mmol/L NaCl, 5 mmol/L CaCl2, 0.05% BRIJ-35, 0.02% NaN3, 1% Triton X-100). Ten-microliter aliquots of the homogenates were saved for total protein measurements (BCA kit, Pierce, Rockford, IL, U.S.A.). Homogenates were centrifuged at 12,000 × g for 5 minutes. The supernatants were recovered and incubated for 60 minutes with gelatin-sepharose 4B (Pharmacia Biotech AB, Uppsala, Sweden) with constant shaking. After incubation, samples were centrifuged at 500 × g for 2 minutes. Pellets were washed with a buffer containing 50 mmol/L Tris-HCl pH 7.6, 150 mmol/L NaCl, 5 mmol/L CaCl2, 0.05% BRIJ-35, and 0.02% NaN3. After a second centrifugation, the pellets were resuspended in 150 μL of elution buffer (50 mmol/L Tris-HCl pH 7.6, 150 mmol/L NaCl, 5 mmol/L CaCl2, 0.05% BRIJ-35, 0.02% NaN3, dimethylsulfoxide 10%) for 30 minutes. The MMP activity of the eluates was measured by gelatin zymography.
Standard zymography
Zymographies were performed using a published method (Kleiner and Stetler-Stevenson, 1994) with minor modifications. Ten microliters of nonreducing sample buffer (0.4 mol/L Tris pH 6.8, 5% sodium dodecyl sulfate [SDS], 20% glycerol, 0.05% bromophenol blue) were added to each sample. Samples then were loaded onto 8% SDS-polyacrylamide gel electrophoresis (PAGE) gels in which 0.15% porcine skin gelatin (Sigma, St. Louis, MO, U.S.A.) was copolymerized. Gels were run at 40 rA for 3 hours. After migration, gels were incubated with 2.5% Triton-X 100 twice for 1 hour at room temperature, washed for 10 minutes in Tris HCl-NaCl-CaCl2 buffer (TNCA) (50 mmol/L Tris-HCl pH 7.5, 200 mmol/L NaCl, 5 mmol/L CaCl2) and further incubated for 16 hours in TNCA in a water bath at 37°C. Gels were stained for 90 minutes in coomassie blue (1% coomassie brilliant blue, 30% methanol, 10% acetic acid) and destained in 30% methanol/10% acetic acid, four times for 5, 15, 30, and 60 minutes, respectively. White bands on a blue background indicated zones of digestion corresponding to the presence of different MMP, identified on the basis of their molecular weight. Twenty microliters of MMP-9 and MMP-2 standards (Oncogene Research, Cambridge, MA, U.S.A.) were loaded into each gel to allow comparison between the different gels. Bands were scanned using a densitometer (GS-700, Bio-Rad Laboratories, Richmond, CA, U.S.A.), and quantitation was performed using Multi-Analyst 1.0.2 software (Bio-Rad).
Type IV collagenase activity assay
Type IV collagenase activity assay was performed using a commercially available kit (Chemicon International, Temecula, CA, U.S.A.). According to the manufacturer protocol, samples were incubated with TNCA and fluorescein isocyanate–labeled type IV collagen solution, first for 20 minutes at room temperature and then for 2 hours at 42°C in a water bath. Samples then were placed on ice for 5 minutes. Three hundred microliters of cold enzyme stop reagent/extraction solution were added to all samples, which were further stored for 35 minutes on ice. Residual undigested collagen was precipitated by centrifugation (10 minutes at 12,200 × g). A standard curve was performed using MMP-9 standard solution provided by the manufacturer. Proteolytic activity of ischemic samples was measured in presence or absence of ethylenediamine tetraacetic acid (EDTA, 0.5 mmol/L), which is known to inhibit MMP. Finally, fluorescence intensity of sample supernatants was measured with a fluorimeter (Molecular Devices, Sunnyvale, CA, U.S.A.) at 520 nm/495 nm.
Reverse zymography
Reverse zymographies were performed using a published method (Oliver et al., 1997) with a few modifications. Ten microliters of nonreducing sample buffer (0.4 mol/L Tris pH 6.8, 5% SDS, 20% glycerol, 0.05% bromophenol blue) were added to the samples. Samples then were loaded onto 15% SDS-PAGE gels in which 2.2 mg/mL porcine skin gelatin (Sigma), and 150 ng/mL of proMMP-9 were copolymerized. Gels were run at 40 mA for 3 hours. After migration, gels were incubated with 2.5% Triton-X 100 twice for 1 hour at room temperature, washed for 10 minutes in TNCA (50 mmol/L Tris pH 7.5, 200 mmol/L NaCl, 5 mmol/L CaCl2), and further incubated for 16 hours in TNCA in a water bath at 37°C. Gels were stained for 30 minutes in coomassie blue (1% coomassie brilliant blue, 30% methanol, 10% acetic acid) and destained in 30% methanol/10% acetic acid four times for 5, 15, 45, and 60 minutes, respectively. Tissue inhibitor of metalloproteinase-1 (TIMP-1; Oncogen Research) was identified as a 28-kd blue band. Twenty microliters of TIMP-1 were loaded as standard. Bands were scanned using a densitometer (GS-700; Bio-Rad).
Immunohistochemistry
Anesthetized animals were perfused with 10 U/mL heparin and subsequently with 4% formaldehyde in 0.1 mol/L phosphate-buffered saline (PBS) (pH 7.4) at 4 and 24 hours after permanent ischemia, respectively. Brains were removed, post-fixed for 36 hours in 4% formaldehyde, sectioned at 50 μm on a vibratome, and processed for immunohistochemical study. The sections were incubated with blocking solution containing 20% normal goat serum (Vector, Burlingame, CA, U.S.A.) and reacted with rabbit anti-human MMP-9 polyclonal antibody (Anawa, Wangen, Switzerland) at a dilution of 1:50. Immunohistochemical study was performed using the avidin–biotin technique (Vector), and then the nuclei were counterstained with methyl green solution for 10 minutes. As a negative control, sections were incubated without primary antibodies. For histologic assessment, alternate slices from each brain section were stained with cresyl violet.
Western blot
Extracted samples, as well as the MMP-9 standard, were loaded onto SDS-polyacrylamide gel under nonreducing conditions. After migration, proteins were electrotransferred onto a polyvinylidene difluoride membrane (Novex, San Diego, CA, U.S.A.). The membrane was blocked overnight in 5% dry milk (Bio-Rad) PBS/0.1% Tween 20 at 4°C. After washing in PBS/0.1% Tween 20, membranes were incubated for 90 minutes at 37°C with rabbit anti-human MMP-9 poly clonal antibody (1/500 in PBS/0.1% Tween 20; Anawa). Membranes were washed three times and incubated for 60 minutes at 37°C with peroxidase-labeled sheep anti-rabbit immunoglobulin G (Boehringer Mannheim, Indianapolis, IN, U.S.A.) used at a dilution of 1/12,500 in PBS/0.1% Tween 20. After extensive washing, bands were revealed with an enhanced chemiluminescence reagent (ECL plus Western Blotting Analysis System, Amersham Life Science, Arlington Heights, IL, U.S.A.) for 1 minute and autoradiographed on Hyperfilm ECL (Amersham) for varying times.
Evans blue extravasation
Five minutes before pMCAO, 0.1 mL 4% of Evans blue in saline was injected in the jugular vein. Animals were perfused with 10 U/mL heparin and subsequently with 4% formaldehyde in 0.1 mol/L PBS (pH 7.4) at 2, 4, and 6 hours after permanent ischemia, respectively. Brains were removed, postfixed for 24 hours in 4% formaldehyde, and sectioned on a vibratome. Sections were dried at room temperature in a dark box, stained with 2.5 × 0.1 mg/mL Hoechst 33258 (Molecular Probes, Eugene, OR, U.S.A.) for nuclear staining and mounted. The BBB breakdown was assessed by fluoromicroscopic detection of Evans blue extravasation.
Statistical analysis
Results of MMP activities are expressed as mean ± SD. Comparisons between multiple groups were performed using an analysis of variance (Bonferroni/Dunn t test), whereas comparisons between two groups were achieved using the Student's t test. P < 0.05 was considered statistically significant.
RESULTS
Physiologic data and cerebral infarction
Physiologic parameters showed no significant differences in MABP, body temperature, and arterial blood gas analyses between each time point. The preischemic physiologic variables (mean ± SD) were as follows; MABP (mm Hg) 73 ± 5.6; body temperature (°C) 36.5 ± 0.1; Pa
Gelatinases and TIMP-1 expression
Latent (proMMP) and activated forms (MMP) were measured using the technique of zymography, in which proMMP-9 and MMP-9 appeared as light bands on a blue background with molecular weights of 96 and 88 kd, respectively, whereas proMMP-2 was identified at 72 kd. As our cleaved form of MMP-9 was higher than usually reported (84 kd), we incubated several extracted samples from nonischemic regions (showing no band at 88 kd) with 1 mmol/L p-aminophenylmercuric acetate known to activate MMP. The zymography performed on these artificially activated samples and the corresponding samples from ischemic regions (genuinely showing the 88-kd band) showed similar bands at 88 kd in treated and nontreated samples (data not shown), allowing us to assume that the cleaved form observed in ischemic samples was the regular cleaved form of MMP-9.
Low levels of proMMP-9 and proMMP-2 were detected in the contralateral corresponding brain region of mice subjected to pMCAO and in normal brains (Fig. 1A). No modification of MMP levels was observed over time in the contralateral control regions. During pMCAO, a strong increase of the proMMP-9 level was observed in the ischemic regions as soon as 2 hours after occlusion (Fig. 1A). This increase of proMMP-9 already was maximal at 2 hours and persisted at similar levels after 4 and 24 hours of ischemia, respectively (Fig. 1A). Although no band corresponding to the activated form of MMP-9 was observed in either normal brains or in contralateral regions, activation of MMP-9 was first observed at 4 hours of ischemia and increased at 24 hours (Fig. 1A). On the other hand, proMMP-2 was mildly induced by permanent ischemia; indeed, only 24 hours of ischemia significantly increased the enzyme level (Fig. 1A). No activated form of MMP-2 could be found at any time point. Mean values of MMP activities are summarized in Table 1.
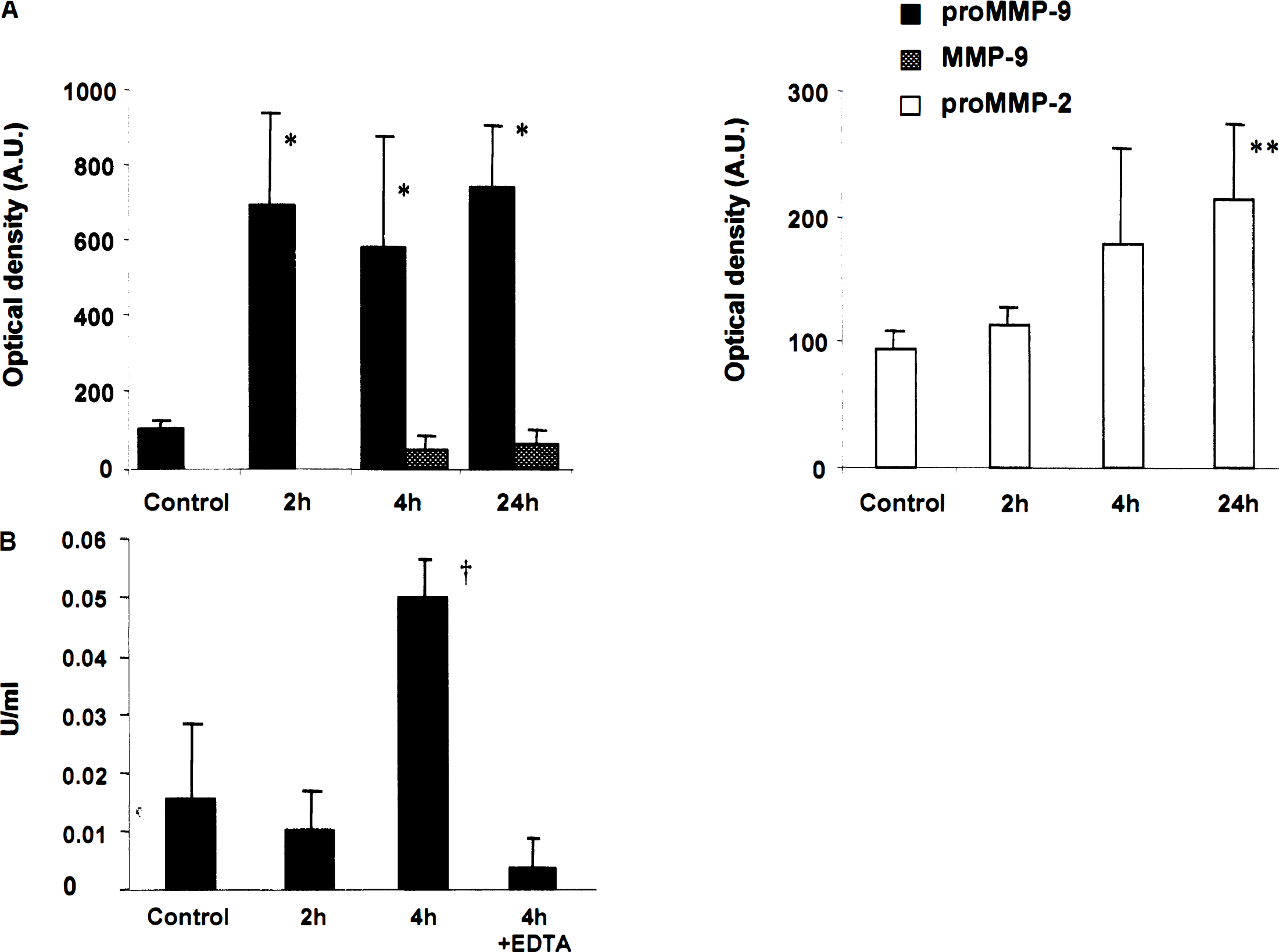
MMP-9 and MMP-2 activities in brain after permanent middle cerebral artery occlusion in mice
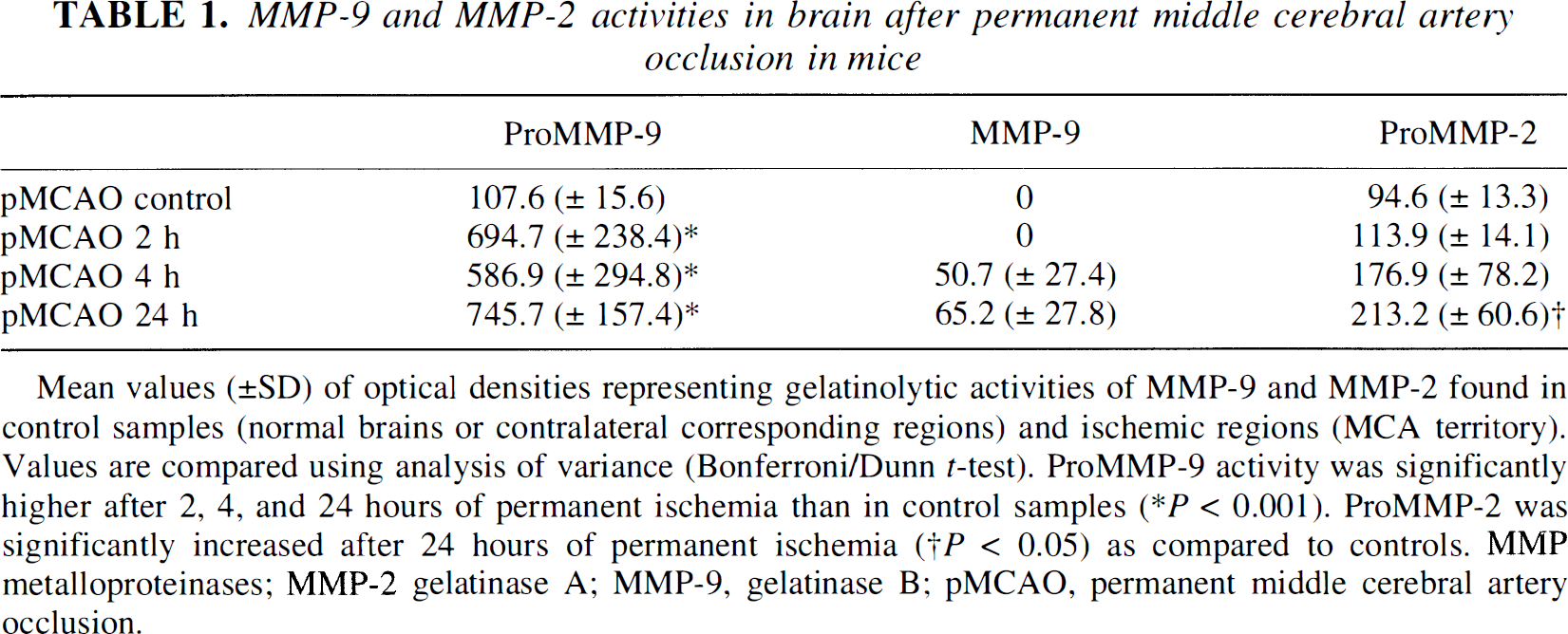
Mean values (±SD) of optical densities representing gelatinolytic activities of MMP-9 and MMP-2 found in control samples (normal brains or contralateral corresponding regions) and ischemic regions (MCA territory). Values are compared using analysis of variance (Bonferroni/Dunn t-test). ProMMP-9 activity was significantly higher after 2, 4, and 24 hours of permanent ischemia than in control samples (* P < 0.001). ProMMP-2 was significantly increased after 24 hours of permanent ischemia († P < 0.05) as compared to controls. MMP metalloproteinases; MMP-2 gelatinase A; MMP-9, gelatinase B; pMCAO, permanent middle cerebral artery occlusion.
To further evaluate the actual proteolytic activity of our extracted samples, we performed a type IV collagenase activity assay, which showed a significant increase of collagenolytic activity in samples taken from brains of mice subjected to 4 hours of ischemia (Fig. 1B). This activity was inhibited by EDTA, confirming the involvement of MMP. Since no active MMP-2 was observed on our zymographies, we can assume that the digestion of type IV collagen revealed in the assay resulted from MMP-9.
We finally evaluated the balance between the expression of MMP-9 and its natural inhibitor, TIMP-1, which was measured using the technique of reverse zymography. The inhibitor was spontaneously expressed in contralateral regions, and no modification of TIMP-1 could be found after permanent ischemia (Fig. 2).
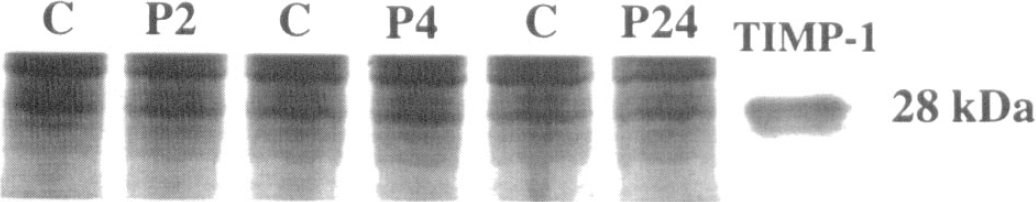
Reverse zymographies were performed to identify tissue inhibitor of metailoproteinase-1 (TIMP-1, 28 kd). Representative examples from contralateral and ischemic regions show that TIMP-1 is not regulated by ischemia.
Regional MMP-9 expression also was analyzed by immunohistochemical study (Fig. 3). Whereas MMP-9 was weakly but diffusely (cortex and caudate putamen) detectable in control samples (Fig. 3A), a strong immunoreactivity was observed during pMCAO at 4 and 24 hours of ischemia, respectively, which had a regional predominance at the inner boundary of the ischemic cortex (Fig. 3B and Fig. 3D). There was a mild expression of MMP-9 at the middle cerebral artery territory cortex (Fig. 3C) and no expression at the caudate putamen (Fig. 3E). There was no immunoreactivity in the control specimens, which were treated without primary antibody (Fig. 3F). Table 2 summarizes the regional distribution of (pro)MMP-9 observed by immunohistochemical study. By Western blot analysis (Fig. 4), proMMP-9 immunoreactivity was evident as a characteristic band of molecular mass 96 kd from both the nonischemic control and from the sample after 4 and 24 hours MCAO, respectively. On the contrary, the activated form of MMP-9 was not recognized in nonischemic controls but was significantly increased after 4 and 24 hours of ischemia, respectively. These data confirm the reactivity of the antibody to mice MMP-9 used in this study, and that the activated form of MMP-9 increased after focal cerebral ischemia.
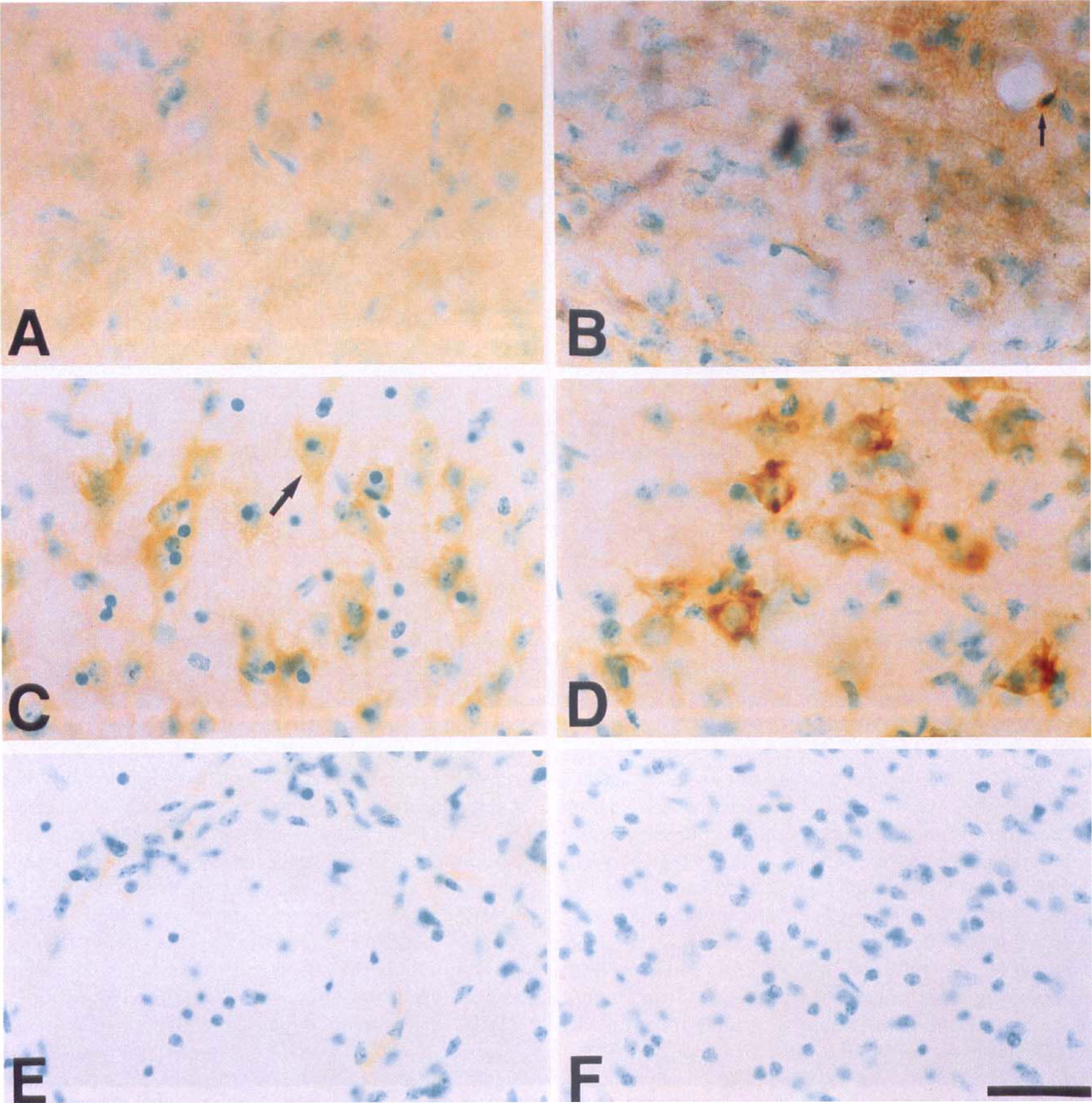
Immunohistochemical analysis of MMP-9. High magnification photomicrographs (630×; bar = 30 μm) illustrating immunoreactivity with the anti-human MMP-9 antibody in the different ischemic regions after 4 and 24 hours of permanent ischemia, respectively, as summarized in Table 2.
Immunohistochemical expression of (pro)MMP-9 in brain tissue after permanent middle cerebral artery occlusion in mice

Summary of (pro)MMP-9 expression detected by immunohistochemistry. Analysis of enzyme distribution in brain ischemic and nonischemic regions of mice subjected to pMCAO. Control, contralateral corresponding region; pMCAO 4 h, 4 hours of permanent ischemia; pMCAO 24 h, 24 hours of permanent ischemia; –, no immunoreactivity to (pro)MMP-9; –(+), very mild immunoreactivity to (pro)MMP-9; +, mild immunoreactivity to (pro)MMP-9; ++, strong immunoreactivity to (pro)MMP-9; +++, very strong immunoreactivity to (pro)MMP-9. MCA, middle cerebral artery; MMP, metalloproteinase; MMP-9, gelatinase B; pMCAO, permanent middle cerebral artery occlusion.
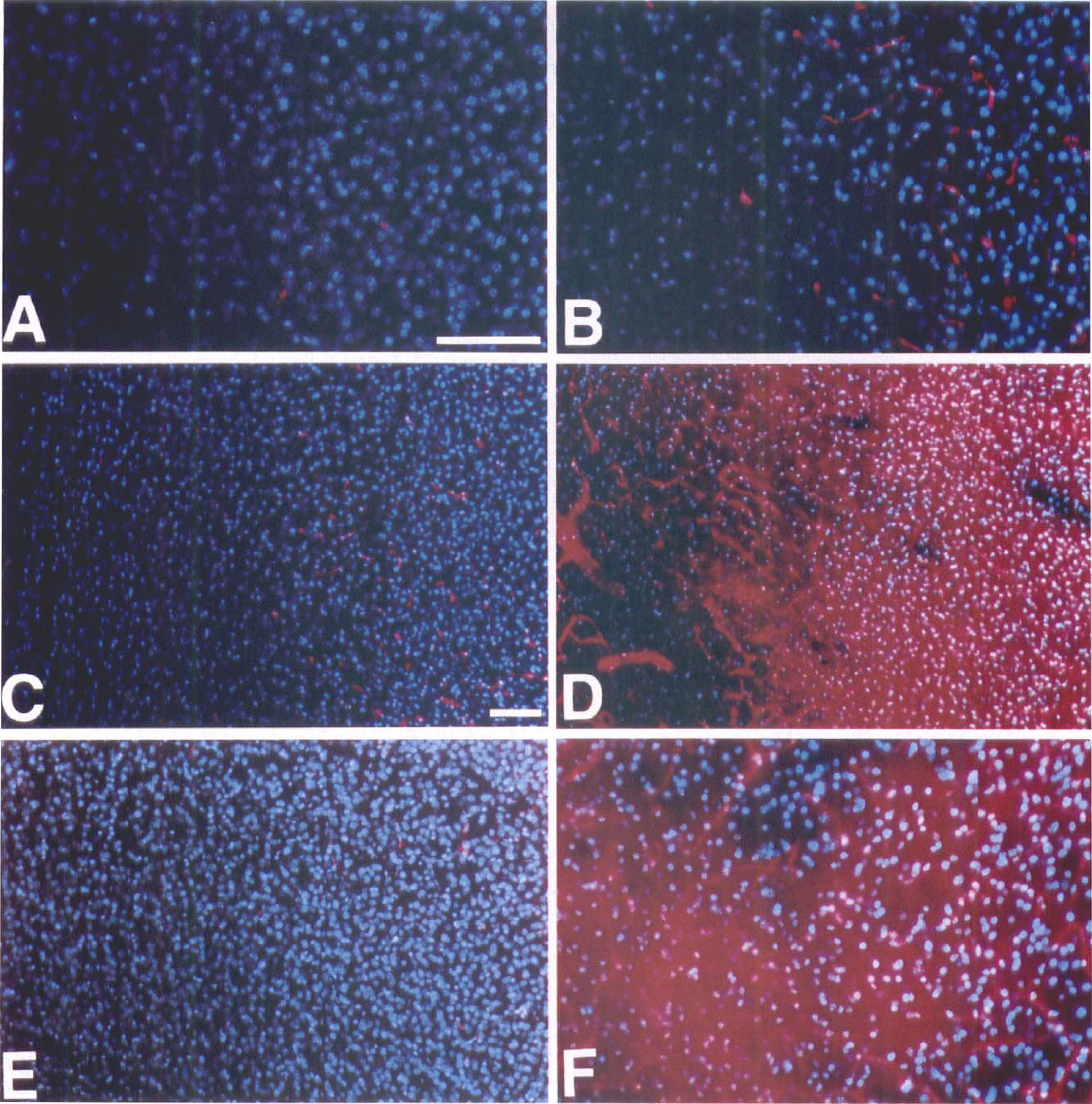
Vasogenic edema detection
Blood–brain barrier permeability was evaluated in ischemic regions and in contralateral regions at 2, 4, and 6 hours of ischemia, respectively. Evans blue leakage was first detected in ischemic regions after 4 hours of occlusion (Fig. 5D); this extravasation was confirmed at 6 hours (Fig. 5F). Evans blue extravasation was detected in the ischemic cortex and in the ischemic caudateputamen. In ischemic regions after 2 hours of ischemia and in contralateral regions at each time point of ischemia, the fluorescent dye was detected only in the vessels as shown in Figs. 5A to 5C and 5E.
DISCUSSION
This study shows for the first time that 2 hours of permanent focal ischemia in mice induce a significant (P < 0.001) and strong increase of proMMP-9 expression. On the other hand, the activated form of MMP-9, which plays the actual pathophysiologic role in BBB alteration, is induced 4 hours after the onset of ischemia. Previous studies performed using a model of pMCAO in rats have shown that proMMP-9 and, possibly, activated MMP-9 are significantly increased only after 12 hours of ischemia (Romanic et al., 1998; Rosenberg et al., 1998; Rosenberg et al., 1996b). In those studies, no proMMP-9 was observed in control samples, whereas proMMP-2 was constitutively expressed. Similar results were obtained after transient MCAO (tMCAO) in rats (Rosenberg et al., 1998). In our study, not only proMMP-2 but also proMMP-9 were detected in normal mice brains, and their levels were similar to those detected in the contralateral (control) regions sampled from mice subjected to pMCAO. The apparent discrepancies between our results and those of previous studies can be explained by technical differences. We used a recently described in vivo method of MMP purification (Zhang and Gottschall, 1997), which allowed us to substantially increase the threshold of MMP-9 detection, as suggested by the fact that we already detected the proform of MMP-9 in control samples. Nevertheless, since previous studies were performed on spontaneously hypertensive rats, we cannot exclude that the differences observed are related to the animal species.
The appearance of the activated form of MMP-9 after a few hours of ischemia in the current study was accompanied by a clear alteration of BBB permeability (Fig. 5). These results of BBB alteration are consistent with recent data showing that vasogenic edema can be detected after 4 hours of focal ischemia–reperfusion in rats (Rosenberg et al., 1998; Belayev et al., 1996) and 2 to 8 hours after tMCAO in mice (Kondo et al., 1997). Nevertheless, the increase of MMP-9 during permanent ischemia, together with the appearance of a clear leakage of the BBB observed in our model, occur earlier than in a previous report, showing that BBB impermeability was preserved after 6 hours of ischemia, whereas a clear breakdown of the BBB (measured by labelled aminoisobutyric acid) was observed after an equal period of ischemia–reperfusion (Yang and Betz, 1994). Although the aggravation of edema by reperfusion of damaged vessels cannot be questioned, caution should be used when comparing BBB permeability modifications induced by pMCAO and tMCAO using intravenous tracers. Indeed, evaluation of BBB leakage during pMCAO with tracers reaching poorly nonperfused regions could underestimate the actual BBB damage. In our study, to solve this problem, we injected the Evans blue dye shortly before pMCAO. This technical procedure allowed us to detect BBB permeability alterations as early as 4 hours after the onset of ischemia, On the other hand, the examination of Evans blue extravasation by fluoromicroscopic study increased the sensitivity of the method, as illustrated by the fact that extravasation was hardly detectable by standard microscopic examination (data not shown).

Western blot analysis of MMP-9. Analysis was performed with a rabbit polyclonal antibody against human MMP-9. Results confirm cross-reactivity with mouse MMP-9. Representative examples are shown. Control (c) samples were obtained from normal brain or contralateral regions. The numbers 4 and 24 represent MMP-9 from ischemic regions after 4 and 24 hours, respectively, of permanent ischemia. The antibody recognized the proform as well as the activated form of MMP-9.
Photomicrographs (magnification:
The etiology of the early BBB disruption in ischemia still is not clear, although endothelial dysfunction or destruction and factors like bradykinin and histamine have been implicated (Wahl et al., 1988). The current study brings more evidence that activated MMP might play an active role in the early and prolonged alteration of the BBB. The confirmation of this hypothesis could imply new therapeutic possibilities using natural or synthetic inhibitors of MMP.
The exact mechanisms involved in MMP expression and activation during ischemia are not well defined. Early inducibility of proMMP-9 by ischemia observed in this study is not surprising according to the gene structure, which comprises an AP-1 site in its promoter region. Early genes such as c-fos and c-jun, which are able to interact with the AP-1 site and induce later gene expression, have been shown to be expressed at the mRNA level as soon as 30 minutes after ischemia (Welsh et al., 1992). Reactive oxygen species (ROS) burst brought with reoxygenation in previously ischemic regions has been invoked to explain BBB breakdown occurring at the time of reperfusion (Traystman et al., 1991). But oxygen free radicals also are significantly induced during ischemia before effective reperfusion. Sustained ROS production has indeed been detected during pMCAO (Peters et al., 1998). Moreover, studies conducted in our laboratory have demonstrated the key pathophysiologic role played by ROS during brain ischemia (Chan et al., 1996; Kondo et al., 1997; Murakami et al., 1997, 1998). The extent of hemispheric enlargement and infarct volume after pMCAO is markedly increased in manganese superoxide dismutase knockout mice compared with wild-type animals (Murakami et al., 1998), suggesting that ROS play a major role after pMCAO and tMCAO. In our study, MMP-9 expression analyzed by immunohistochemical study during pMCAO was clearly preponderant in regions still supplied with some perfusion (inner boundary of ischemic cortex), although it was absent in the ischemic core, suggesting a possible role of oxygen free radicals in MMP-9 gene induction. Since in vitro MMP activation was reported to be closely related to ROS (Weiss et al., 1985) and in vivo Evans blue extravasation in copper superoxide dismutase knockout mice was shown to occur as early as 2 hours after focal ischemia (Kondo et al., 1997), it would be of great interest to evaluate if MMP-9 induction/activation is affected in superoxide dismutase-deficient mice. These studies are currently being undertaken in our laboratory.
Pro-gelatinase B also appears to be under the control of proinflammatory cytokines. Indeed, elevated levels of proMMP-9 have been detected in the CSF of patients with multiple sclerosis (Gijbels et al., 1992). Moreover, both MMP protein activity and mRNA levels are increased in rats with experimental autoimmune encephalomyelitis (Pagenstecher et al., 1998). In vitro, proMMP-9 can be induced by tumor necrosis factor-alpha in various cell types such as astrocytes (Gottschall et al., 1995), microglial cells (Gottschall and Deb, 1996) and fibroblasts (Hanemaaijer et al., 1993). Because of its earliness, induction of proMMP-9 at 2 hours observed in our model of focal ischemia probably cannot be attributed to tumor necrosis factor, which is produced mainly by leukocytes (monocytes) known to infiltrate infarcted areas later during reperfusion. However, it is interesting that tumor necrosis factor mRNA induction can be detected in neurons as early as 3 hours after the initiation of permanent ischemia (Liu et al., 1994).
The mechanism of MMP activation consists in the cleavage of a propeptide. The result of this cleavage is a shorter protein, which is enzymatically active although the proform is not. The zymographic detection of activated (cleaved) forms of MMP, although indicative, does not reveal the actual proteolytic activity of the enzymes. The presence of active forms of type IV collagenases (MMP-9 or MMP-2) in our samples obtained from brains after 4 hours of ischemia could be confirmed by demonstrating a significant increase of type IV collagenolytic activity (inhibited by EDTA) in those samples. These results, together with our zymographic data showing no cleaved form of MMP-2, strongly suggest that the collagenolytic activity observed results from the activated form of MMP-9. Activation of MMP is a complex process that is tightly regulated by their natural inhibitors, TIMP. During the zymographic procedure, MMP are separated from their respective TIMP. To partially solve this problem and better define the balance existing between MMP-9 and its natural inhibitor TIMP-1, we measured the activity of the latter by reverse zymography under the same experimental conditions. Contrary to MMP-9, TIMP-1 was not induced during the first 24 hours by permanent ischemia. These results confirm immunohistologic findings published by others (Romanic et al., 1998) showing a mild and diffuse expression of TIMP-1 in ischemic regions and in nonischemic contralateral controls. Nevertheless, a recent study showed a significant increase of TIMP-1 mRNA after permanent and transient ischemia (Wang et al., 1998). This discrepancy between mRNA and protein expression suggests a possible posttranscriptional regulation of the inhibitor.
In conclusion, our study shows for the first time that proMMP-9 expression is significantly induced as soon as 2 hours after the onset of ischemia and that permanent ischemia triggers early proMMP-9 activation. The timing of proMMP-9 induction and activation correlated with the timing of BBB breakdown. These results are completely in accordance with those showing that the inhibition of MMP diminishes the early opening of BBB after focal ischemia (Rosenberg et al., 1998).
Footnotes
Abbreviations used
Acknowledgment
The authors thank Cheryl Christensen for editorial assistance.