
Abstract
Oxidative stress generated during stroke is a critical event leading to blood–brain barrier (BBB) disruption with secondary vasogenic edema and hemorrhagic transformation of infarcted brain tissue, restricting the benefit of thrombolytic reperfusion. In this study, the authors demonstrate that ischemia-reperfusion–induced BBB disruption in mice deficient in copper/zinc-superoxide dismutase (SOD1) was reduced by 88% (P < 0.0001) and 73% (P < 0.01), respectively, after 3 and 7 hours of reperfusion occurring after 1 hour of ischemia by the inhibition of matrix metalloproteinases. Accordingly, the authors show that local metalloproteinase-generated proteolytic imbalance is more intense in ischemic regions of SOD1 mice than in wild-type litter mates. Moreover, active in situ proteolysis is, for the first time, demonstrated in ischemic leaking capillaries that produce reactive oxygen species. By showing that oxidative stress mediates BBB disruption through metalloproteinase activation in experimental ischemic stroke, this study provides a new target for future therapeutic strategies to prevent BBB disruption and potentially reperfusion-triggered intracerebral hemorrhage.
Blood–brain barrier (BBB) integrity protects the neuronal microenvironment (Cserr and Bungaard, 1986). When this barrier integrity is lost, inflammatory cells and fluid penetrate the brain, causing edema and cell death (Fishman, 1975). Impermeability of the BBB is maintained by microvascular endothelial cells through their tight junctions and basal lamina (Siflinger-Birnboim et al., 1987). The latter is composed of type IV collagen, fibronectin, laminin, and various proteoglycans (Yurchenco and Schittny, 1990). Matrix metalloproteinases (MMPs) are proteolytic enzymes (Zn2+-endopeptidases) secreted as zymogen and cleaved to their fully active form in the interstitial space. Among the MMPs, gelatinase A (MMP-2) and gelatinase B (MMP-9), also known as type IV collagenases, are able to digest the endothelial basal lamina leading to the opening of the BBB (Rosenberg et al., 1996, 1998; Romanic et al., 1998; Gasche et al., 1999) and leukocyte transmigration (Lou et al., 1999). During cerebral ischemia, endothelial basal lamina dissolution starts as soon as 2 hours after the onset of ischemia, continues during reperfusion (Hamann et al., 1995), and is rapidly followed by an increase in BBB permeability (Kondo et al., 1997; Belayev et al., 1998; Rosenberg et al., 1998; Gasche et al., 1999). The MMPs play an active role in secondary brain injury after focal ischemia (Romanic et al., 1998; Rosenberg et al., 1998; Gasche et al., 1999), but the mechanisms implicated in the activation of MMP-generated proteolysis during stroke have not been elucidated. Reactive oxygen species (ROS) are implicated in BBB disruption during stroke, as illustrated by the fact that mice lacking copper/zinc-superoxide dismutase (SOD1) are highly susceptible to focal cerebral ischemia–reperfusion, with exacerbated vasogenic edema and a higher mortality than wild-type (WT) animals (Kondo et al., 1997). Accordingly, ROS inhibition prevents tissue plasminogen activator–triggered cerebral hemorrhage after stroke (Asahi et al., 2000). In this context, we hypothesize that MMPs, in particular, gelatinases, are predominantly activated in SOD1-deficient animals during stroke and that MMP inhibition could prevent BBB disruption in those animals. In the current study, we show that MMP-9 and MMP-2 are, indeed, predominantly activated in SOD1−/− animals after focal cerebral ischemia–reperfusion and that MMP inhibition prevents BBB disruption in these animals. In addition, using an in situ method to detect the actual proteolytic unbalance related to MMP activation, we show that a strong proteolysis is present in brain capillary walls concomitantly with an increased production of superoxide anions.
MATERIALS AND METHODS
SOD1−/− mice and transient focal cerebral ischemia
We conducted the procedures involving animals and their care in conformity with Stanford University's Administrative Panel on Laboratory Animal Care. We used a strain of SOD1 knockout mice, designated 129/CD1-Sod1 <tm1 Cep> (Cephalon, West Chester, PA, U.S.A.) (Reaume et al., 1996) and extensively crossed with animals with a CD-1 background (10 generations). We did not detect any phenotypic difference between the knockout and WT mice (Kondo et al., 1997). Homozygous animals (SOD1−/−) had a 95% decrease in SOD1 activity (Kondo et al., 1997). SOD1−/− adult males and their litter mates (35 to 40 g) were subjected to focal ischemia for 1 hour by intraluminal middle cerebral artery occlusion as described (Yang et al., 1994). The temperature was maintained at 37 ± 0.5°C. We monitored blood pressure, arterial blood gases, and regional cerebral blood flow (rCBF).
Matrix metalloproteinase inhibition
The SOD1-deficient and WT mice were randomly assigned to receive the MMP inhibitor in a blinded fashion, p-aminobenzoyl-gly-pro- d -leu- d -ala-hydoxamate (AHA) (60 mg/kg in phosphate-buffered saline intraperitoneally), or a placebo 30 minutes before surgery. Because no data regarding inhibitory activity and brain penetration of AHA were available, we carried out a pilot study to assess its antigelatinolytic activity. We performed a kinetic study, in vitro, of MMP-9– and MMP-2–induced fluorescein release from a gelatin fluorescein conjugate (Molecular Probes, Eugene, OR, U.S.A.) in the presence of different concentrations of the inhibitor. Twenty microliters of gelatin fluorescein conjugate (100 μg/mL) were incubated with p-aminophenylmercuric acetate–activated MMP-9/MMP-2 (1 μg/mL; Chemicon, Temecula, CA, U.S.A.) and 0, 10, 100, and 1000 nmol/L of AHA. Quantification of fluorescein release was performed with a fluorometer. The inhibitory activity and brain penetration of AHA in vivo was assessed by fluoromicroscopic detection of in situ gelatinolytic activity in the ischemic regions of five mice.
Matrix metalloproteinase extraction and gelatin zymography
Extraction of MMP from the brains was performed as described (Zhang and Gottschall, 1997; Gasche et al., 1999). Supernatants obtained from homogenized ischemic and contralateral corresponding brain tissues were recovered and incubated for 60 minutes with gelatin-Sepharose 4B (Pharmacia Biotech, Uppsala, Sweden). After incubation and centrifugation, samples were resuspended for 30 minutes in 150 μL of elution buffer containing dimethyl sulfoxide. The MMP activity of the eluates was measured by gelatin zymography using a published method (Kleiner and Stetler-Stevenson, 1994). The same protein quantity from each sample was loaded onto 10% zymogram gels (Novex, San Diego, CA, U.S.A.). Ten microliters of mouse pro–MMP-9 and activated MMP-9 (6 ng/mL; Chemicon) and human pro–MMP-9 and activated MMP-2 standards (15 ng/mL; Oncogene Research, Cambridge, MA, U.S.A.) were loaded into each gel to allow comparison between different gels.
In situ gelatinolytic activity assay and immunofluorescence
In situ gelatinolytic activity was assessed using a commercially available kit (Molecular Probes). After 1 hour of ischemia or 1 hour of ischemia followed by 3 hours of reperfusion, the animals were killed, and the brains were frozen. Brain sections, 20 μm thick, were incubated for 2 hours at 37°C in a solution of gelatin fluorescein conjugate (40 μg/mL) provided by the manufacturer. This procedure was performed carefully without any washing. The in situ gelatinolysis was revealed by the appearance of fluorescent brain constituents. Astrocytes and endothelial cells were identified by immunofluorescence staining of glial fibrillary acidic protein and factor VIII, respectively. Sections were incubated with the primary antibody (polyclonal rabbit anti-cow glial fibrillary acidic protein, 1:500; polyclonal rabbit anti-human factor VIII, 1:100; Dako, Carpinteria, CA, U.S.A.) and the secondary antibody (polyclonal swine anti-rabbit TRITC, 1:60; Dako). Neurons were stained with a mouse anti–neuron-specific nuclear protein (NeuN) monoclonal antibody (1:100; Chemicon) using the Dako ARK kit (Dako). Staining was revealed with Texas Red–labeled streptavidin. Irrelevant corresponding first antibodies or serum were used for negative controls. Nuclear counterstaining was achieved with 2 μg/mL Hoechst 33258 (Molecular Probes). Sections of the same anatomic levels were examined microscopically.
Detection of superoxide anion production
The production of superoxide anions was investigated using the hydroethidine (HEt) oxidation method. Hydroethidine is diffusible into the brain and selectively oxidized to ethidium, which is not able to pass the BBB (Murakami et al., 1998) and fluoresces in red. The HEt oxidation is carried out by superoxide anions but not other free radicals (Bindokas et al., 1989; Benov et al., 1998). An intravenous injection of HEt (6.25 mg/kg in 0.3 mol/L dimethyl sulfoxide–phosphate-buffered saline) was performed 30 minutes before the animals were killed. Determination of ethidium production in the brain parenchyma was performed after 4 hours of ischemia–reperfusion. After killing, brains were frozen and qualitative studies were performed by fluoromicroscopy on 20-μM brain sections.
Evans blue extravasation
Immediately after reperfusion, 2.5 mL/kg of 2% Evans blue in normal saline were injected into the right jugular vein. Animals were killed 3 and 7 hours after ischemia. For quantitative measurements, brain hemispheres were homogenized in 300 μL of N,N-dimethyl-formamide (Sigma, St. Louis, MO, U.S.A.), incubated for 18 hours at 55°C, and centrifuged. Evans blue extravasation was quantified in the supernatants by spectrophotometry. For qualitative examinations, brains were snap-frozen, sectioned into 20-μm slices, and examined by fluoromicroscopy.
Statistical analysis
Results had a normal distribution and were expressed as means plus/minus the standard deviation. Comparisons among multiple groups were performed using an analysis of variance (Fisher's protected least-significant difference post-hoc test), whereas comparisons between two groups were achieved using the Student's t-test. A P value of < 0.05 was considered statistically significant.
RESULTS
Physiologic parameters
During the surgical procedure, we recorded the mean arterial blood pressure, arterial blood gases, and rCBF in WT, placebo-treated SOD1−/− and AHA-treated (ICN Biomedicals, Aurora, OH, U.S.A.) SOD1−/− animals. Mean arterial blood pressure, pH, Pa o2 and Pa co2 remained in physiologic ranges and did not significantly vary among the three groups. Despite suture removal, a significant restoration of rCBF (>70%) could not be achieved in 40% of the operated animals. These animals were excluded from the study. Their number was evenly distributed among the different groups. Among the animals studied, residual rCBF and reperfusion rCBF were not different among the three groups.
Gelatinase expression and activity in wild-type versus SOD1−/− mice
Latent (pro-MMP) and active (MMP) forms of MMP-9 and MMP-2 were determined first by zymography after gelatin affinity–based extraction. Immediately after ischemia (1 hour), no active form of MMP-9 or MMP-2 was observed (Fig. 1). After 1 hour of ischemia followed by 3 hours of reperfusion, pro–MMP-9 was dramatically increased in the SOD1−/− mice (n = 10) and moderately increased in the WT animals (n = 10) (Fig. 1). This increase was statistically significant when compared with the contralateral hemispheres (P = 0.0001); the difference between pro–MMP-9 values in the ischemic hemispheres of the SOD1−/− mice compared with those of the WT animals also was significant (P = 0.0012). At 3 hours of reperfusion, the active form of MMP-9 was observed in both groups but was significantly higher in the SOD1−/− mice (P = 0.0001, Fig. 1). On the other hand, whereas no active form of MMP-2 was observed at 3 hours of reperfusion in the WT mice, a faint band corresponding to the active form of MMP-2 could be observed in the ischemic hemisphere of the SOD1−/− mice. No significant increase in the pro-form of MMP-2 was observed in either group at 3 hours of reperfusion. Because zymography does not accurately represent the actual proteolytic unbalance occurring in the ischemic brain regions, we performed an in situ gelatinolytic assay on brain slices obtained from the SOD1−/−(n = 5 per group) and WT animals (n = 5 per group) at the same time points as regular zymographies. In the nonischemic corresponding regions of the contralateral hemispheres, no in situ gelatinolytic activity was detected (Figs. 2A and 2B). Although no activated form of MMP-9 or MMP-2 was detected by zymography at 1 hour of ischemia, the in situ gelatinolytic activity clearly was identifiable in the ischemic cortex and caudate putamen of the SOD1−/− and WT mice (Figs. 2C and 2D). At 3 hours of reperfusion, the in situ activity was more pronounced than at 1 hour of ischemia, and overall, the SOD1−/− mice showed a higher activity in the ischemic areas than the WT animals (Figs. 2E and 2F). To confirm the validity of our in situ gelatinolytic activity assay, we also performed an in situ collagenolytic (type IV collagen) assay, which confirmed the results of the former method (Fig. 2, insets).
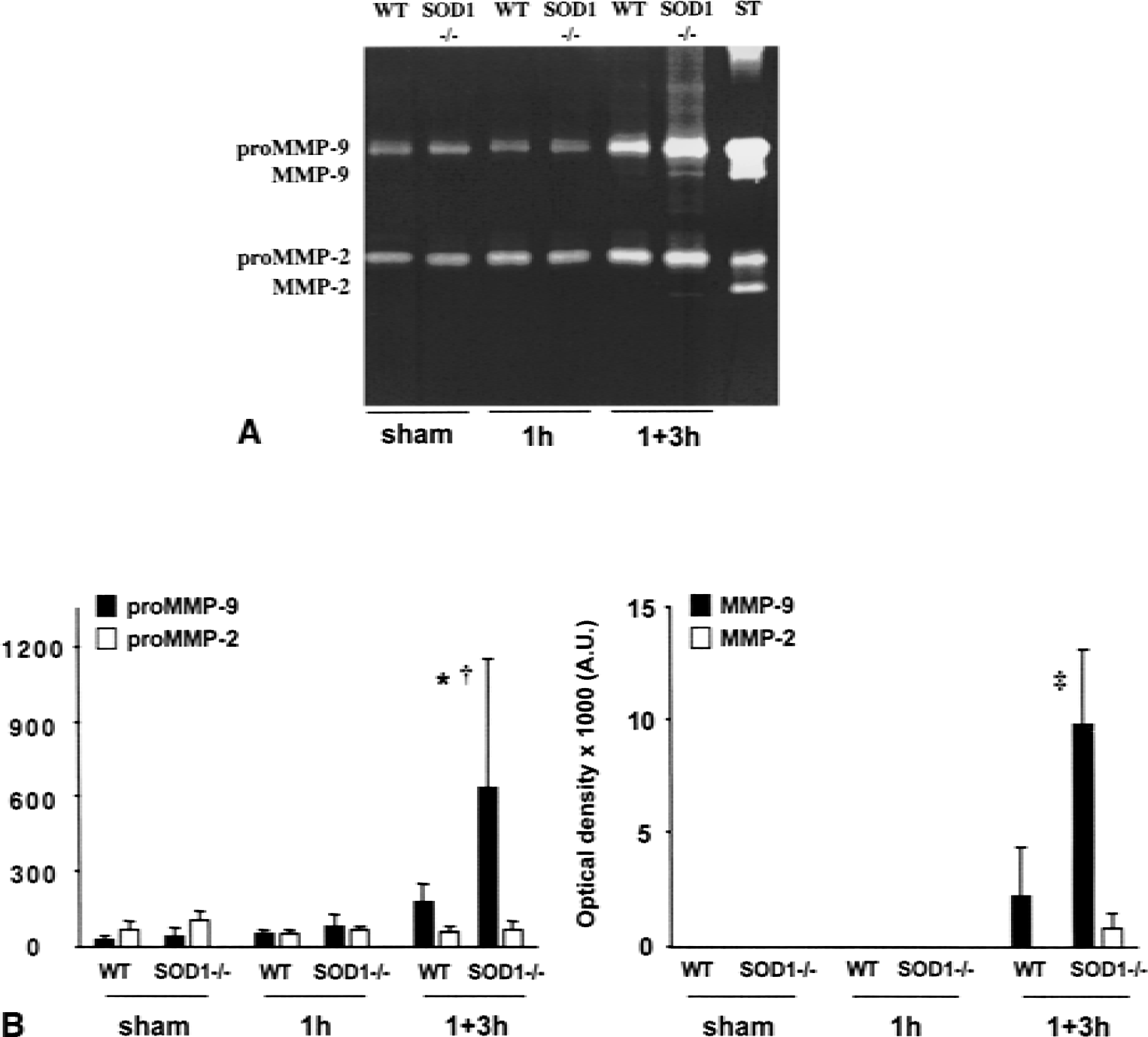
Zymographic analysis in wild-type (WT) and deficient in copper/zinc-superoxide dismutase (SOD1−/−) mice after ischemia–reperfusion.
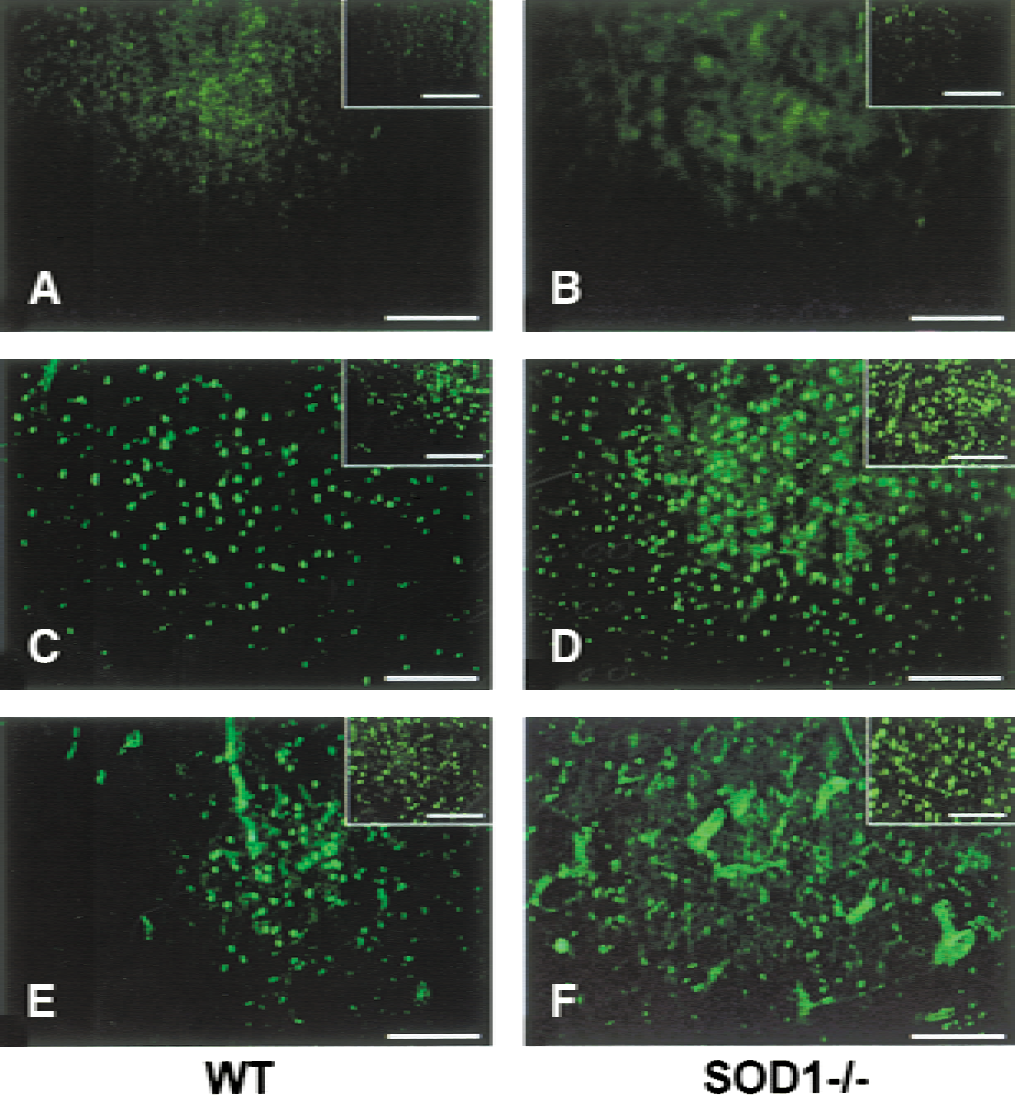
Gelatinolysis in brain regions of wild-type (WT) and deficient in copper/zinc-superoxide dismutase (SOD1−/−) mice after middle cerebral artery occlusion. Representative microphotographs showing the gelatinolytic activity (green fluorescence) observed in ischemic brain regions after 1 hour of ischemia
Blood–brain barrier permeability evaluation in wild-type and SOD1−/− mice
According to our previous study (Kondo et al., 1997), BBB permeability determined by the Evans blue method was significantly higher in the ischemic hemisphere of the SOD1−/− mice than the WT mice. Evans blue leakage was 4.4-fold higher in the SOD1−/− mice after 3 hours of reperfusion and twofold higher after 7 hours of reperfusion. Moreover, a spatial correlation was observed between capillaries showing gelatinolytic activity and Evans blue extravasation (Fig. 3). To demonstrate the role played by MMPs in the exacerbation of BBB disruption found in SOD1−/− animals, we compared the level of BBB leakage after ischemia–reperfusion in the SOD1−/− animals treated preventively with AHA or a placebo. In preliminary studies, we assessed AHA inhibitory activity in vitro using a quantitative gelatinolytic activity assay, which confirmed the dose-dependent inhibition of activated gelatinases (Fig. 4A). The BBB penetration and in vivo inhibitory activity of AHA were confirmed by the absence of residual gelatinolytic activity in ischemic regions (4 hours after surgery) of five mice treated with AHA (Fig. 4B). To determine the efficiency of AHA in preventing BBB disruption in SOD1−/− animals, eight animals per group and time point were studied by blinded observation (total 32). The MMP inhibition significantly reduced BBB leakage by 88% 3 hours (P ≤ 0.0001) and by 73% 7 hours (P ≤ 0.01) after ischemia (Fig. 4C). As expected, MMP inhibition also diminished BBB leakage in wild type animals to 67% of control values (P ≤ 0.01) after 3 hours and 65% of control values (P ≤ 0.01) after 7 hours of reperfusion.
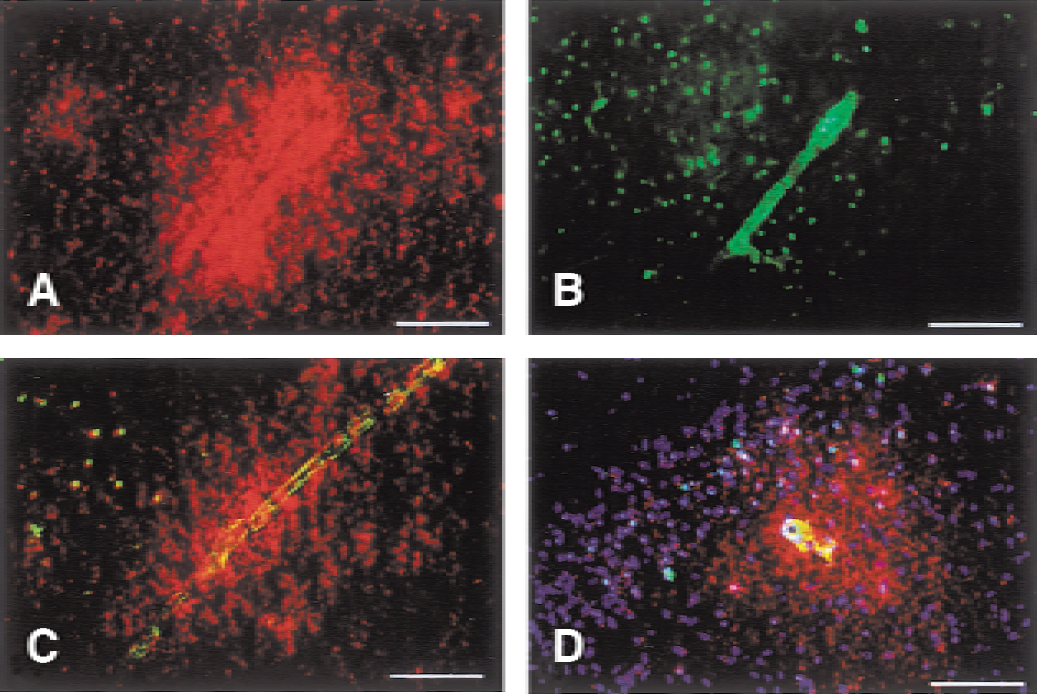
Co-localization of Evans blue leakage and vascular gelatinolytic activity.
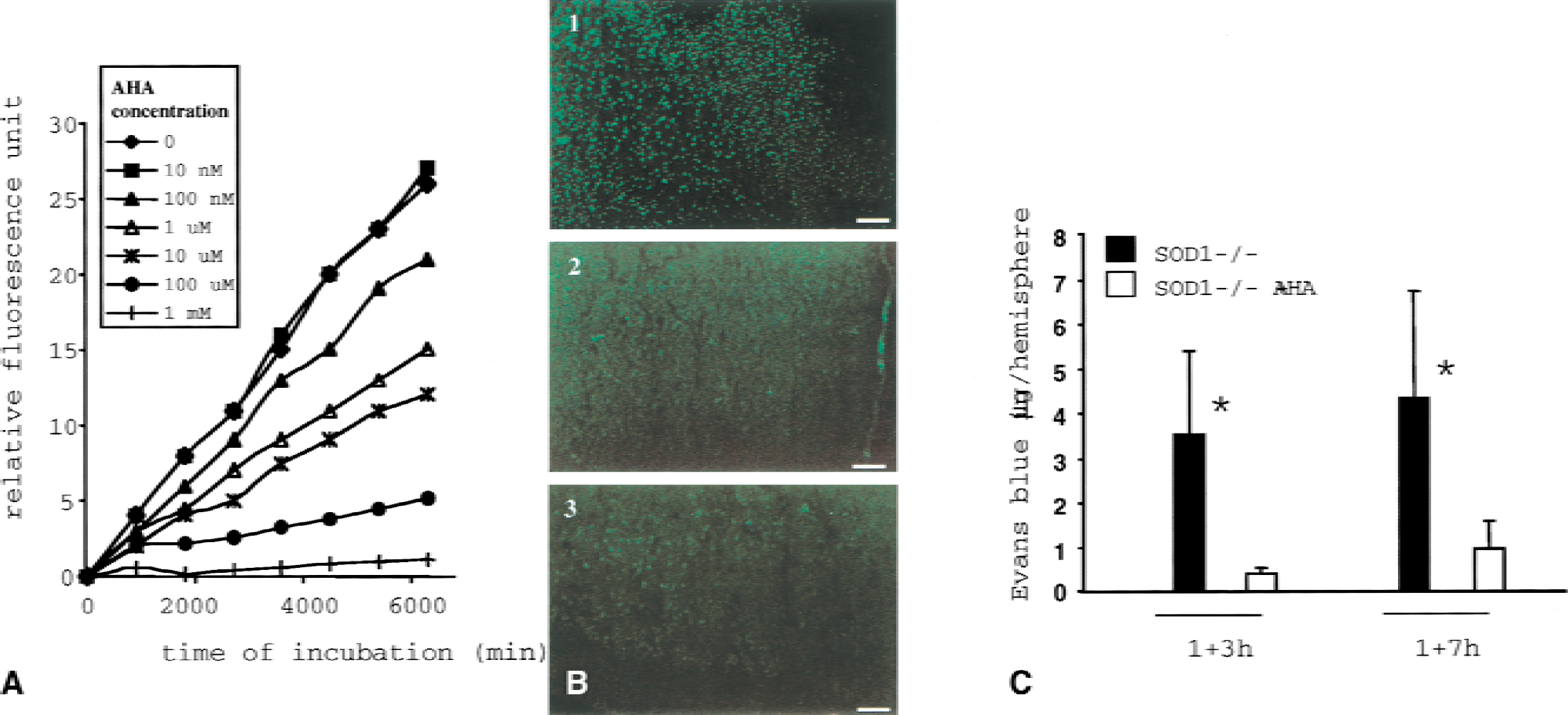
Evaluation of p-aminobenzoyl-gly-pro- d -leu- d -ala-hydoxamate (AHA) inhibitory activity on matrix metalloproteinases (MMPs) in vitro and in vivo.
In situ co-localization of gelatinolytic activity and superoxide anion production
Gelatinolysis was observed clearly in capillary walls and in cells distributed throughout the ischemic cortex and caudate putamen (Fig. 2). To determine the cellular origin of the proteolytic activity, we performed a labeling for factor VIII, glial fibrillary acidic protein, and NeuN, together with the in situ gelatinolytic activity assay. Factor VIII staining revealed a close relationship between the endothelial cells and the in situ gelatinolytic activity (Fig. 5A). Whereas a co-localization between capillary gelatinolysis and astrocytic processes at the capillary wall level often was observed, no gelatinolytic activity could be seen in the cellular body of astrocytes (Fig. 5B). In some cells, the gelatinolytic activity was superimposed on the nucleus (Fig. 5C); most of the cells showing this characteristic activity appeared to be NeuN positive (Fig. 5D).
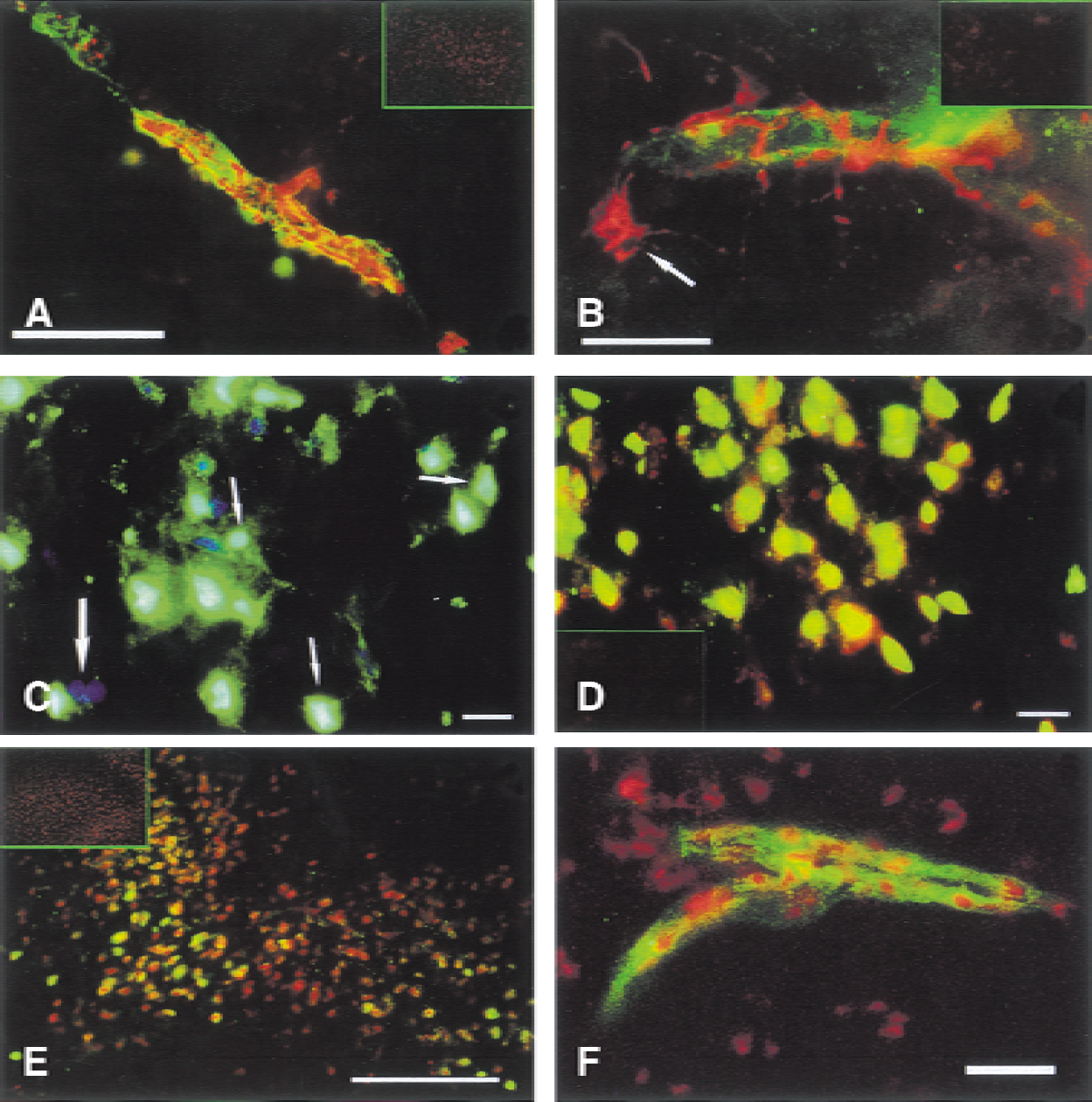
Cellular localization of gelatinolytic activity and its co-localization with ethidium. Representative microphotographs showing the co-localization of factor VIII, glial fibrillary acidic protein (GFAP), NeuN, and oxidized hydroethidine (Het) with the gelatinolytic activity observed in the ischemic brain regions of wild-type mice.
Because SOD deficiency prevents normal superoxide anion removal from the cells, we hypothesized that the gelatinolytic activity would preferentially localize where superoxide anions were produced. Indeed, a clear co-localization of gelatinolytic activity and oxidized HEt was observed in capillaries, possibly endothelial cells, and in nonvascular cells located in the ischemic cortex (Figs. 5E and 5F).
DISCUSSION
It is well established that the oxidative unbalance during focal cerebral ischemia is a major contributor to BBB disruption and secondary brain damage (Chan, 1994; Chan et al., 1996; Kondo et al., 1997). Conversely, although reperfusion by thrombolytic agents (tissue plasminogen activators) is the only effective stroke therapy (National Institute of Neurological Disorders and Stroke rt-PA Stroke Study Group, 1995), its value is limited by the risk of intracerebral hemorrhage (Clark et al., 2000) from loss of vascular integrity. Although it has been suggested that ROS are capable of directly injuring the endothelium (Wei et al., 1985; Zweier et al., 1988), the exact mechanism by which ROS affect brain capillary permeability in vivo is not well understood. We demonstrate that MMPs participate in oxidative stress-related BBB disruption during cerebral ischemia–reperfusion. Although the nonselective nature of the MMP inhibitor used in the current study prevents us from pointing out the precise MMP that actually is responsible for the BBB leakage in SOD1−/− mice, our results suggest that MMP-9, MMP-2, or both might be implicated in this pathophysiologic process. Indeed, the active forms of both enzymes were produced predominantly in SOD1−/− mice at the time of the massive BBB breakdown. However, zymographic detection of activated forms of MMP-9 and MMP-2 does not reveal the actual in situ activity of the enzymes. During the zymographic procedure, MMPs undergo an artificial nonproteolytic activation and are separated from their natural tissue inhibitors of MMP (TIMPs), which tightly regulate their activity in vivo. To address this issue, we performed a recently described in situ gelatinolytic activity assay (Oh et al., 1999), which confirmed that the proteolytic unbalance suggested by the zymographies was predominant in the SOD1−/− animals. Nevertheless, while we observed activated gelatinases only after 4 hours of ischemia–reperfusion on the zymographies, the in situ gelatinolytic and collagenolytic assays were already positive after 1 hour of ischemia in the ischemic regions of the SOD1−/− as well as the WT mice. Because the gelatinolytic activity was inhibited in vivo and ex vivo by different MMP inhibitors (Fig. 4B, panels 2 and 3), the enzymes responsible for this activity certainly were MMPs. Several hypotheses can be made to explain this chronologic discrepancy between the results given by regular zymographies and in situ gelatinolytic and collagenolytic assays. On one hand, gelatinolytic and collagenolytic activity assays may be more sensitive than regular zymographies, in particular because the latter were run with samples obtained from brain homogenates after a procedure involving several centrifugations, possibly leading to MMP loss. Conversely, the early proteolysis observed with the in situ assays could result from the activation of other MMPs. Gelatin is not a specific substrate of MMP-9 and MMP-2 but also is digested by other MMPs such as MMP-1, MMP-3, MMP-7, MMP-8, and MMP-13 (Sang and Douglas, 1996). Because, in our study, the type IV collagenolytic assay showed results similar to the gelatinolytic assay, possible candidates able to digest gelatin and type IV collagen are MMP-3 and MMP-7 (Sang and Douglas, 1996). Another possible cause of the temporal discrepancy between in situ and zymographic gelatinolytic activity detection could involve pro-MMP-9 or pro–MMP-2 themselves in a process of nonproteolytic conformational activation of the zymogen. The classic mechanism of MMP activation consists of the disruption of the interaction between a zinc molecule on the active site and a cysteine in the pro-domain, leading to the proteolytic cleavage of the zymogen and the production of the mature active form of the enzyme (Van Wart and Birkedal-Hansen, 1990). Notice that the complexity of this mechanism has prevented its demonstration in vivo. The existence of chemicals such as p-chloromercuribenzoate (Stricklin et al., 1983) or sodium dodecyl sulfate, which are able to generate active zymogen, and the early description of factors in conditioned cell culture medium activating human pro–MMP-1 with no modification of the enzyme molecular weight (Tyree et al., 1981), suggest that alternative, nonproteolytic mechanisms of MMP activation could exist in biologic systems. One wonders whether such MMP activation mechanisms could not implicate ROS. In vitro, ROS have been shown to activate the latent forms of pro–MMP-2 and pro–MMP-9, as well as neutrophil collagenase (Weiss et al., 1985; Rajagopalan et al., 1996), but the issue of a possible nonproteolytic activation of these enzymes has not been addressed. Here, we show the involvement of ROS in MMP activation in vivo, but the exact molecular mechanism involved in this activation remains unknown. Our results also show that MMP-9 gene expression is triggered by ROS. This finding is consistent with the presence of AP-1, NF-κB, and TRE sites in the promoter region of MMP-9 (Sen and Packer, 1996; Christman et al., 2000) and a previous study of our laboratory showing that gelatinases expression is diminished in SOD1-overexpressing mice compared with WT animals after cold injury (Morita-Fujimura et al., 2000).
Among MMPs, gelatinases are expressed by neurons (Pagenstecher et al., 1998), astrocytes (Gottschall and Yu, 1995), microglial cells (Gottschall et al., 1995), endothelial cells (Romanic et al., 1998), and oligodendrocytes (Oh et al., 1999) under various physiologic and pathophysiologic conditions. Only rare studies address the question of the actual proteolytic activity generated by MMPs in brain pathologies like ischemia–reperfusion (Romanic et al., 1998). In the current study, we examine the regional and cellular distribution of active gelatinolysis. This gelatinolytic activity was distributed throughout the ischemic middle cerebral artery territory in the capillary walls, also in neurons. We found a close spatial relationship between endothelial cells, astrocytic processes, and active MMPs, whereas no gelatinolytic activity could be observed in the cellular bodies of astrocytes. Strikingly, in the ischemic cortex neuronal nuclei showed a clear gelatinolytic activity. To further substantiate the relationship between ROS and activated MMPs we examined the localization of oxidized HEt and found it predominantly in ischemic capillaries and nonvascular cells with strong gelatinolytic activity. Endothelial cells subjected in vitro to hypoxia–reoxygenation produce high levels of superoxide anions (Zweier et al., 1988), which consequently mediate complex cell injury pathways and redox-sensitive gene expression (Griendling et al., 2000; Irani, 2000). Our results suggest that superoxide anion produced by endothelial cells might participate in MMP activation and secondary alteration of capillary permeability. Recent studies have shown that TIMP-1–independent activated MMP-9 was produced in endothelial cells, implying a more intricate intracellular regulation of the enzyme than previously thought (Nguyen et al., 1998).
In conclusion, the current study demonstrates the importance of ROS in MMP activation and that MMP participates in early BBB disruption triggered by oxidative stress generated during cerebral ischemia–reperfusion. We also show that active MMPs co-localize in ischemic brain regions with ROS at the level of the capillary walls and astrocytic processes. Our results allow a better understanding of the complex mechanisms implicated in BBB disruption during cerebral ischemia–reperfusion and provide a new target for future therapeutic strategies destined to prevent BBB disruption and possibly reperfusion-triggered intracerebral hemorrhage.
Footnotes
Acknowledgments:
The authors thank Dr. Richard Scott of Cephalon for the SOD1−/− mice, Jane O. Kim for breeding the animals, and Cheryl Christensen for editorial assistance.