
Abstract
The authors show that the inhibitor of the succinate dehydrogenase, 3-nitroproprionic acid (3-NPA), which in high doses and with chronic administration is a neurotoxin, can induce profound tolerance to focal cerebral ischemia in the rat when administered in a single dose (20 mg/kg) 3 days before ischemia. Infarcts were approximately 70% and 35% smaller in the 3-NPA preconditioned groups of permanent and transient focal cerebral ischemia, respectively. This regimen of 3-NPA preconditioning neither induced necrosis, apoptosis, or any other histologically detectable damage to the brain, nor did it affect behavior of the animals. 3-NPA led to an immediate (1-hour) and long-lasting (3-day) decrease in succinate dehydrogenase activity (30% reduction) throughout the brain, whereas only a short metabolic impairment occurred (ATP decrease of 35% within 30 minutes, recovery within 2 hours). The authors found that 3-NPA induces a burst of reactive oxygen species and the free radical scavenger dimethylthiourea, when administered shortly before the 3-NPA stimulus, completely blocked preconditioning. Inhibition of protein synthesis with cycloheximide given at the time of 3-NPA administration completely inhibited preconditioning. The authors were unsuccessful in showing upregulation of mRNA for the manganese superoxide dismutase, and did not detect increased activities of the copper-zinc and manganese superoxide dismutases, prototypical oxygen free radicals scavenging enzymes, after 3-NPA preconditioning. The authors conclude that it is possible to pharmacologically precondition the brain against focal cerebral ischemia, a strategy that may in principal have clinical relevance. The data show the relevance of protein synthesis for tolerance, and suggests that oxygen free radicals may be critical signals in preconditioning.
Keywords
In many organs a short ischemic interval, in itself not sufficient to cause tissue damage, induces tolerance to more severe and prolonged reductions in organ perfusion: ischemic preconditioning. Clinical applications of ischemic preconditioning have already successfully been tested in cardiology. Ischemic preconditioning also exists in the brain, but no clinically applicable protocols have emerged and mechanisms have remained enigmatic. In addition to ischemia itself, other stimuli may afford tolerance to cerebral ischemia, e.g., repeated spreading depressions, cytokines, inflammatory stimuli such as lipopolysaccharide, or inhibition of the succinate dehydrogenase (SDH) (Prass et al., 1998). Ischemic tolerance is induced in many cell types by a number of different stimuli, however, tolerance may be triggered and executed by common mechanisms (Hochachka et al., 1996). A review of the literature suggests that the generation of oxygen free radicals (OFR) may represent such a common trigger (Schumann et al., 1999).
3-Nitropropionic acid (3-NPA), an irreversible inhibitor of the SDH (Hassel and Sonnewald, 1995) located in the tricarboxylic acid cycle and in complex II (SDH, cytochrome b) in the oxidative phosphorylation, induces OFR formation (Beal et al., 1995). It can generate tolerance to hypoxia in in vitro models of cerebral ischemia (Riepe et al., 1997), as well as against global cerebral ischemia in vivo (Nakase et al., 1997; Sugino et al., 1999). We here describe for the first time that a single dose of 3-NPA given intraperitoneally induces profound tolerance to permanent or transient cerebral ischemia in the rat, without producing tissue damage. It is shown that 3-NPA, while transiently decreasing SDH activity and energy rich phosphates, leads to a burst of OFR, and that the inhibition of OFR formation or protein synthesis completely blocks preconditioning. We speculate that a pharmacologic mode of preconditioning, as shown here, may have clinical relevance and that 3-NPA induced tolerance represents an attractive model to investigate endogenous protective mechanisms of the brain.
METHODS
All 161 animals used in this study were treated in accordance with the Guidelines for the use of Animals in Neuroscience Research of the Society for Neuroscience. NPA- and drug-pretreatment was performed by intraperitoneal injection (pH 7.4). There where no major differences in anesthesia, recording of physiologic data, and methods used for brain extraction between groups. All surgical and treatment procedures were performed in one series by the same surgeon in a randomized fashion.
Focal cerebral ischemia
In male Wistar rats (250 to 300 g) the right ipsilateral common carotid artery (CCA) and the middle cerebral artery (MCA) were permanently or transiently (90 minutes of ischemia) occluded (CCA/MCAO) under halothane anesthesia as described by Brint et al. (1988). In addition to the right CCA the left CCA was temporarily occluded for I hour (90 minutes in the reperfusion group). Animals were killed 4 days after CCA/MCAO.
Physiologic monitoring
In all animals mean arterial blood pressure was monitored continuously throughout anesthesia through a tail artery catheter, while rectal temperature was measured and maintained at 38° ± 0.5°C with a heating pad. In some of the animals temperature of the left temporal muscle as a measure of brain temperature was monitored with a needle thermometer. After surgery rectal temperature was measured daily in all animals. In six animals temperature during the 3-NPA pretreatment period was determined (time points after 3-NPA application: 30 minutes and then 1, 2, 4, 6, and 24 hours and daily up to 7 days). During surgery blood gas analysis (arterial oxygen and carbon dioxide tensions, pH, base excess) was performed every 30 minutes. Blood glucose was measured directly before MCAO (arterial) and on the first day after surgery (tail vein) in all. In six animals venous blood glucose was measured before, 1 hour, and 24 hours after 3-NPA injection. Hematocrit was determined 24 hours after MCAO in every animal.
In some experiments, laser Doppler flowmetry (LDF, Perimed Periflux Master 4001 with PF 403 probe, Jarfälla, Sweden) was used to quantify the consequences of CCA/MCAO on regional cerebral blood flow (rCBF) in the MCA territory. The skull bone was thinned to translucency and the LDF probe was placed 2 mm lateral and 3 mm caudal to the bregma over a region assumed to represent the evolving infarct core.
Histology
To assess possible brain damage by 3-NPA, three animals were killed 3 days after 3-NPA injection (20 mg/kg). After transcardial perfusion, brains were removed from the skull, fixed, and prepared for standard paraffin embedding. Serial coronal sections (20 μm) throughout the entire brain were cut using a microtome (HM330, Microm, Heidelberg, Germany). After hematoxilin/eosin or Nissl staining of adjacent sections, slides were examined using light microscopy.
DNA gel electrophoresis
We performed gel electrophoresis of brain extracts 3 days after 3-NPA injection by a commercial DNA isolation kit and a radioactive Phosphor labeling with the aid of a Phosphorimager (Molecular Dynamics, CA) on three animals. For detailed methods see Bruer et al. (1997).
Quantitative infarct volume analysis
Four days after focal cerebral ischemia the animals were reanesthetized with halothane and decapitated. The brains were rapidly removed and frozen in 2-Methylbutan (Carl Roth GmbH, Karlsruhe, Germany) at −30° C. Coronal brain sections (40 μm) were cut serially at 400 μm intervals with a cryotome (HM 500 OM, Microm, Heidelberg, Germany) at −25° C, dried over night and stained with vanadium acidic fuchsin (Victorov and Barskov, 1993). Infarct volumes were calculated from 30 to 35 coronal sections by an observer blinded to the treatment groups using an image analysis system. Because the brains were obtained 4 or 7 days after the CCA/MCAO, time points when ischemic brain edema has subsided, no correction for brain swelling was necessary (Lin et al., 1993).
ATP measurements
Thirty minutes, 1 hour, and 2 hours after intraperitoneal injection of 3-NPA brains were harvested by in vivo freezing of the whole skull in liquid nitrogen. Subsequently cortical tissue samples (approximately 200 μL) were removed from parietal brain regions in a −20 C° environment. Adenosine phosphates were determined according to Werner (1987) by reversed-phase high performance liquid chromatography (Shimadzu, Kyoto, Japan). Concentrations of adenosine phosphates in probes were calculated as nMol/mg protein and normalized to controls. Three animals were used at each time point and values were determined five times in each animal.
Succinate dehydrogenase activity measurements
One hour and then 1, 3, 5, and 7 days after 3-NPA injection animals (two per time point) were decapitated in deep anesthesia. The brains were rapidly removed, quickly frozen in 2-Methylbutan at −30°C, stored at −80°C until coronal sections (40-μm thick) were cut serially at 400-μm intervals at −20°C. For imaging of SDH activity on brain sections we used a histoblot technique. Sections were mounted on nitrocellulose membranes (Biometra, Goettingen, Germany) and fixed by drying for 30 minutes at 4°C.
The nitrocellulose histoblots with the immobilized brain slices were stained in 50 mmol/L Tris/HC1 (pH = 7.4), 10 mmol/L succinate, and 0.5 mmol/L tetranitroblue tetrazolium for 10 minutes at room temperature. Afterward slices were fixed with 1 mol/L HCl, rinsed with phosphate-buffered saline solution and air dried. Succinate dehydrogenase activities for three different brain regions (cortex, striatum, hippocampus) were densitometrically quantified in serial brain sections (40 μm) at 1-mm intervals.
Photometric determination of SDH activity was performed in 50 mmol/L Tris/HC1 (pH = 7.4), 25 mmol/L sucrose, 50 μmol/L 2,6-dichloroindophenol, and 10 mmol/L succinate at 604 nm and 25°C after homogenization of brain slices. Protein content of the samples was measured according to Bradford (1976).
Chemiluminescence measurement of reactive oxygen species in acute brain slices subjected to 3-NPA
To evaluate whether 3-NPA induces the generation of reactive oxygen species (ROS), a lucigenin-enhanced chemiluminescence technique, which is especially sensitive to superoxide anion radicals, was used (Dirnagl et al., 1995). Acute vibratome brain slices (400 μm) were prepared, equilibrated at room temperature (30 minutes) and then maintained for 90 minutes at 34°C. Afterward slices where transferred into a recirculation system with a submerged chamber perfused with Krebs-Henseleit-buffer at 34°C and at a pO2 > 500 mm Hg. Lucigenin (100 μmol/L) was added to the perfusion buffer leading to the generation of photons on reaction with ROS. The perfusion chamber was positioned underneath a cooled (−20°C) photo-multiplier (Hamamatsu R943-02, dark count of 3 cps. at −20°C). Photon counts (sampled over 6-second intervals) were amplified and stored by a Hewlett Packard Universal counter connected to a Pc. Lucigenin was allowed to equilibrate in the intracellular space for 1.5 to 2 hours. 3-NPA (300 μmol/L) was added to the perfusion buffer in a concentration presumably reached in the brain when 20 mg/kg are injected intraperitoneally. The integral over a 4-hour counting period was calculated after correction for darkcount (n = 6).
Analysis of manganese superoxide dismutase expression by Northern blotting
Cortical tissue samples were obtained at the following time points after 3-NPA injection: 6, 13, 24 and 72 hours. Northern blot loaded with 20 μg total RNA each lane, isolated with TRIZOL reagent (Gibco BRL), was hybridized with polymerase chain reaction (PCR)-generated and fluorescein labeled manganese (Mn) superoxide dismutase (SOD) probe (positions 2862–7953 according to accession number X56600; primers: 5′ GAACAATCTGAACGTCACCGAGGAGAAGT A-3′ and 5′-CTGAAGATAGTAAGC-GTGCTCCCACACATC-3′). Detection by the Gene Images system (Amersham) was performed as recommended by the manufacturer.
Measurement of 3-NPA induced cerebral CUZn-and Mn-SOD activity
After 3-NPA treatment rats were rapidly decapitated in deep anesthesia after 1 hour and after 1, 3, 5, and 7 days. Cortical tissue samples were dissected out rapidly from the removed brain over ice (approximately 50μL each). Brain tissue was immediately frozen in liquid nitrogen and stored at −80°C until SOD measurement. The activities of Cu/Zn- and Mn-SOD, respectively, were determined by direct photometric measurement of the superoxide anion according to the method of Bolann et al. with minor modifications (Bolann and Ulvik, 1991; Bolann et al., 1996). The decrease of superoxide anion was directly monitored photometrically at 250 nm, 4°C, and pH = 9.5. Mn-SOD activity was distinguished from Cu/Zn-SOD by addition of 10 mmol/L KCN. Three animals were used in each group. Activity of Cu/Zn-SOD was calculated as units per milligram of protein according to Marklund (1976) and Bolann and Ulvik (1991) where one unit is defined as the activity which is sufficient to achieve a pseudo-first -order rate constant of 0.1 S–1 in 1 mL sample volume. One hundred percent activity of Cu/Zn-SOD from rat brain cortex lysate corresponded to 41.7 units/mg protein. Vmax activity of Mn-SOD was calculated as
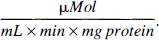
One hundred percent activity of Mn-SOD from rat brain cortex lysate corresponded to
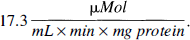
Statistical analysis
All data are reported as mean values ± SD. Multiple comparisons between groups (infarct volumes, physiologic variables, rCBF) were made using analysis of variance with Bonferroni's post hoc comparison (SPSS Inc. Chicago IL). P values of .05 were considered statistically significant.
Drugs
All drugs, unless otherwise noted, were purchased from Sigma Chemicals (Deisenhofen, Germany).
RESULTS
3-NPA induces tolerance to focal cerebral ischemia without producing tissue damage
Single dose intraperitoneal treatment with 3-NPA induced strong protection (73% reduction compared to the control group, P < .05) against subsequent permanent focal cerebral ischemia only when given 3 days before induction of cerebral ischemia. The 3-day pretreatment group was also significantly different to the 12-hour, 2-day, and 7-day pretreatment groups (Fig. 1). For all further experiments we chose the 3-day preconditioning interval. In the transient focal cerebral ischemia experiments, 3-NPA pretreatment reduced infarct sizes from 145 ± 26 mm 3 (n=9) to 93 ± 41 mm 3 (n= 10, 35% reduction, P < .01).
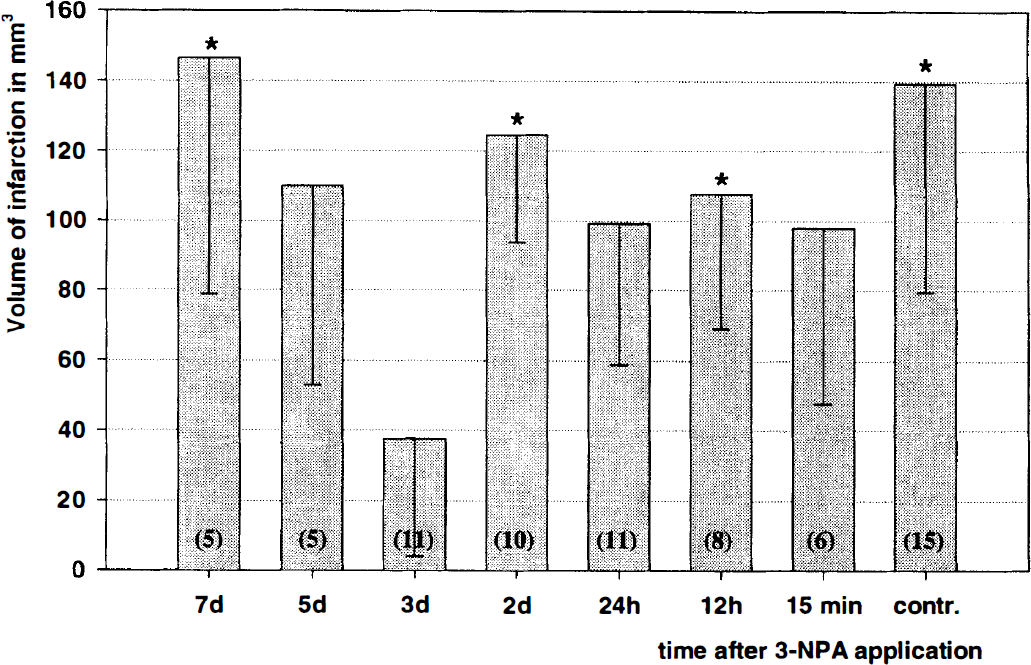
Temporal profile of 3-Nitroproprionic acid (3-NPA) induced tolerance to ischemia. Permanent focal cerebral ischemia was induced at different time points after 3-NPA preconditioning (single systemic injection). X-axis indicates time interval between 3-NPA injection and induction of focal cerebral ischemia. Infarct volumes were evaluated 4 days after common carotid artery and middle cerebral artery occlusion. Number of animals given in brackets. Data given as mean ± SD. *Denotes significant difference compared to 3d NPA group; P < 0.05.
Three days after intraperitoneal injection of a single dose of 3-NPA (20 mg/kg), brains did not show any evidence for cell damage on light microscopical examination in any brain region. No evidence for DNA strand breaks indicative of apoptosis were found on DNA gel electrophoresis. Within the first 3 hours after 3-NPA administration (period of mild acidosis) animals slightly hyperventilated and showed depressed motor activity. No signs of delayed movement impairment (e.g., hyperactivity or hypoactivity, ventral or lateral recumbency, as noted after chronic 3-NPA treatment) (Borlongan et al., 1995) was detected during the 7 -day observation period.
Physiologic data. Treatment with 20 mg/kg 3-NPA led to a short and mild decrease in arterial blood pH 45 minutes after injection (blood pH minimum at about 7.30) in rats anesthetized with halothane for monitoring purposes, which normalized within 20 minutes. Rectal temperature measurements and blood glucose measurements directly after 3-NPA injection and during the entire 7-day observation period revealed no differences in preconditioned compared to control rats.
During surgery physiologic parameters of control and preconditioned animals showed no statistically significant differences (Table 1). Animals treated with cycloheximide (CHX) or dimethylthiourea (DMTU) (data not shown) showed no significant differences in physiologic variables. Common carotid artery/middle cerebral artery occlusion decreased temperature in the ipsilateral M. temporalis by about 1° C in all animals, without differences between experimental groups. Because of a stress response during surgery all animals showed increased blood glucose levels, which normalized when controlled 24 hours later (no differences in between groups). The increased hematocrit found in all animals 24 hours after CCA/MCAO is most likely due to reduced water uptake after surgery (Table 1).
Physiologic variables of the permanent and transient group
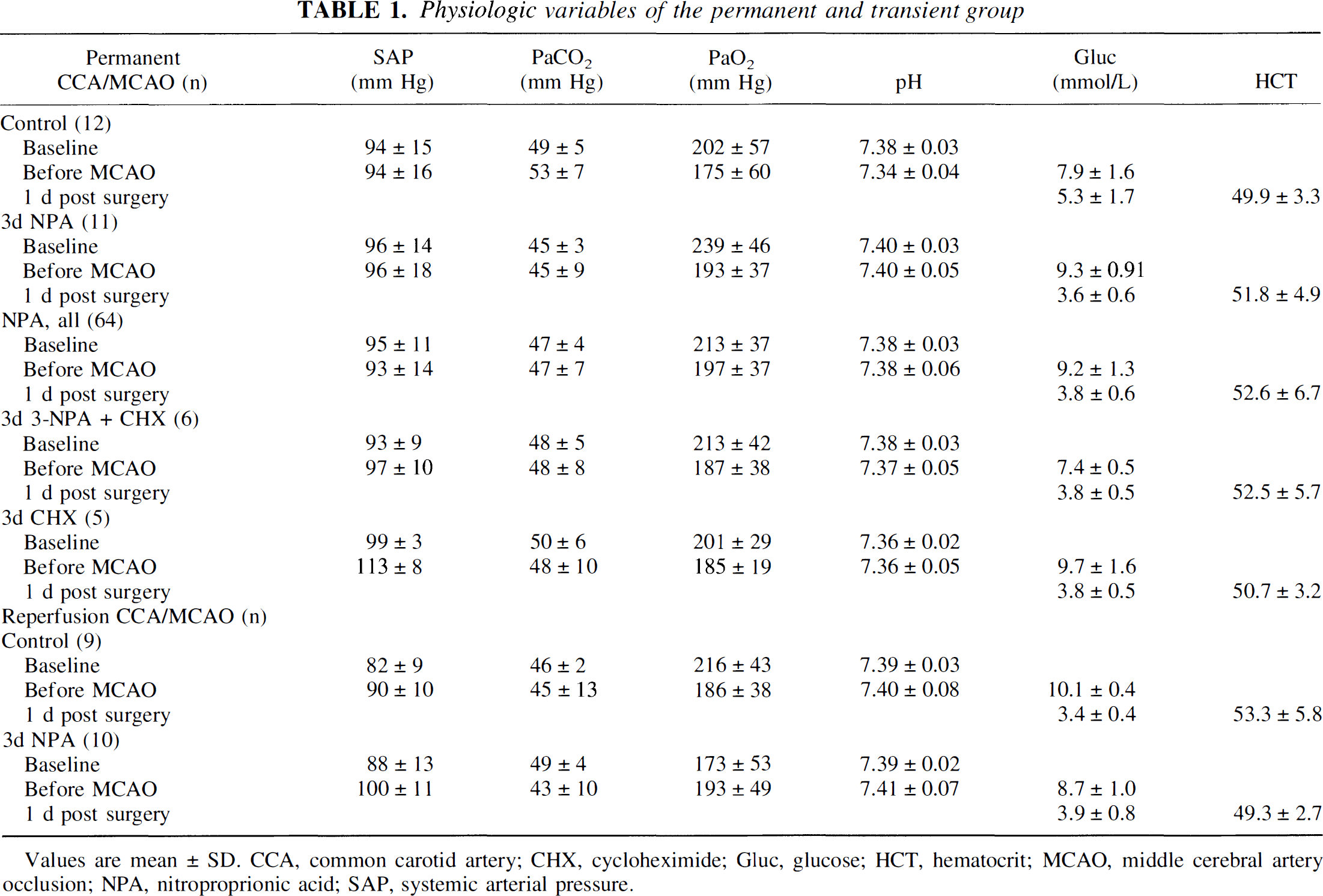
Values are mean ± SD. CCA. common carotid artery; CHX. cycloheximide; Glue, glucose; HCT, hematocrit; MCAO, middle cerebral artery occlusion; NPA, nitroproprionic acid; SAP, systemic arterial pressure.
LDF measurements of rCBF were performed to test whether 3-NPA pretreatment may have affected the severity of ischemia or extent of reperfusion. No differences between control rats and preconditioned animals (3-day 3-NPA) were found, neither in the permanent, nor in the transient ischemia group. In the permanent occlusion group during CCA/MCAO the rCBF of control and preconditioned animals decreased to 13 ± 6% and 13 ± 7%, respectively. After reopening of the temporary contralateral CCA occlusion rCBF slightly increased to 33 ± 10% and 19 ± 3% (control versus preconditioned; not significant). In the transient ischemia group, rCBF decreased to 11 ± 1% (control) and 12 ± 5% (preconditioned). During the first 15 minutes of reperfusion, rCBF increased to 88 ± 24% (control) and 87 ± 22% (preconditioned).
Spatial distribution and time profile of cerebral succinate dehydrogenase inhibition by 3-NPA
Succinate dehydrogenase activity decreased to about 70% 1 hour after 3-NPA application. Thereafter SDH activity slowly recovered within 7 days. No significant difference between the various gray matter regions was detected. Quantitative, photometric measurements of brain section homogenates highly corresponded to the densitometrically determined SDH activities (Fig. 2).
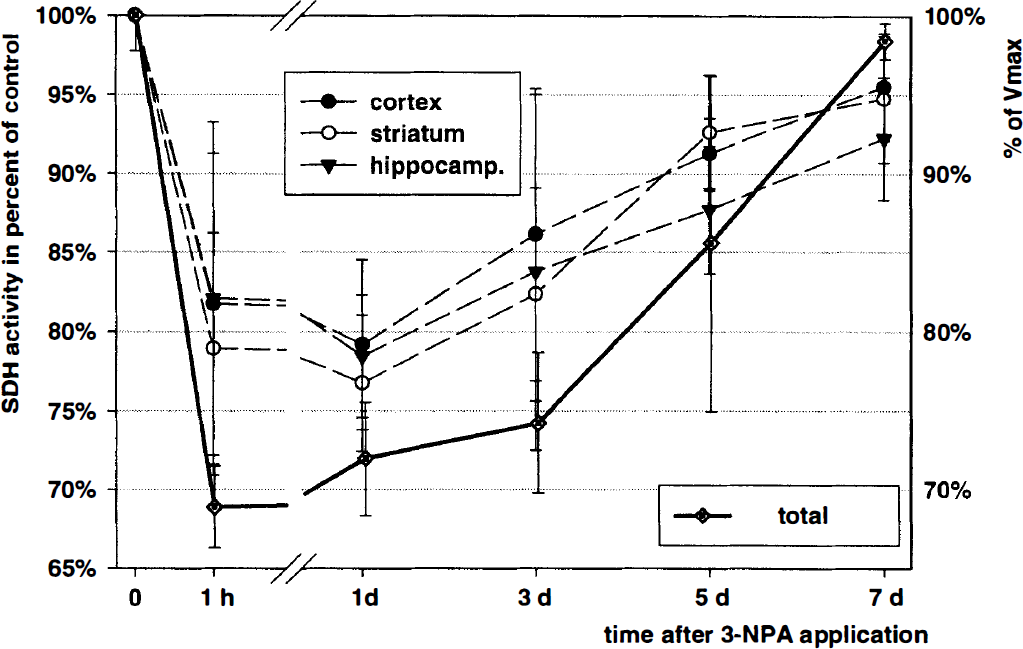
Temporal and spatial profile of the inhibition of the succinate dehyrogenase (SOH) activity after a single dose of 3-Nitroproprionic acid (3-NPA) (20 mg/kg). Left y-axis: SOH activity as percent of control in different brain regions (cortex, hippocampus, and striatum). Three animals were used at each time point. Approximately eight brain sections per animal were analyzed using a histoblot technique. Right y-axis: absolute SOH activity was photometrically determined as percent of Vmax in homogenates of three different coronal brain sections per animal. Three animals were used at each time point. Data given as mean ± SD.
Measurement of energy-rich phosphates
ATP values decreased about 35% within the first 30 minutes after intraperitoneal 3-NPA injection (Fig. 3). Control values in nmol/mg protein: ATP 29.9 ± 5.5, ADP 10.8 ± 1.7, AMP 3.1 ± 0.1. This ATP decrease was accompanied by a two-fold increase of ADP and a 40% increase of AMP and coincided with the point when mild blood acidosis was measured (see above). ATP levels recovered fast and reached control values 2 hours after 3-NPA injection. Guanosinphosphates (control values in nmol/mg protein: GTP 4.7 ± 0.8, GDP 2.6 ± 0.2, GMP O.5 ± 0.08) showed similar kinetics, with a 33% decrease in GTP accompanied by an increase in GDP and GMP (Fig. 3).
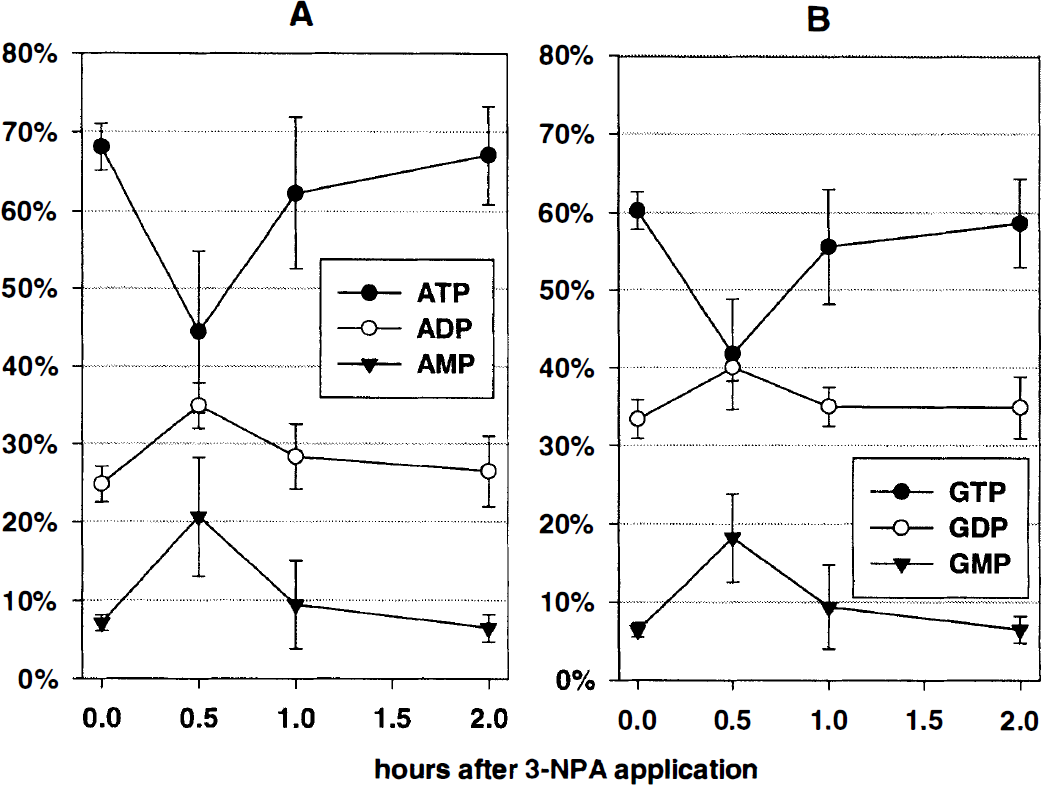
Temporal profile of energy rich adenosine
3-NPA induces reactive oxygen species generation
Superfusion of 3-NPA (300μmol/L) onto acute brain slices led to a significant rise in photon counts in lucigenin-enhanced chemiluminescence, indicating a burst of OFR. Increased photon counts were detected already 1 hour after the 3-NPA challenge and reached a maximum 2 hours later. Thereafter, photon counts decreased and control values were reached within the next 2 hours (Fig. 4). During the 4-hour observation period 3-NPA treated slices (n = 6) showed an integral of 61,546 ± 27,808 photon counts in 4 hours, whereas untreated slices showed only an integral of 14,872 ± 8,093 counts in 4 hours (n = 11, P < .001).
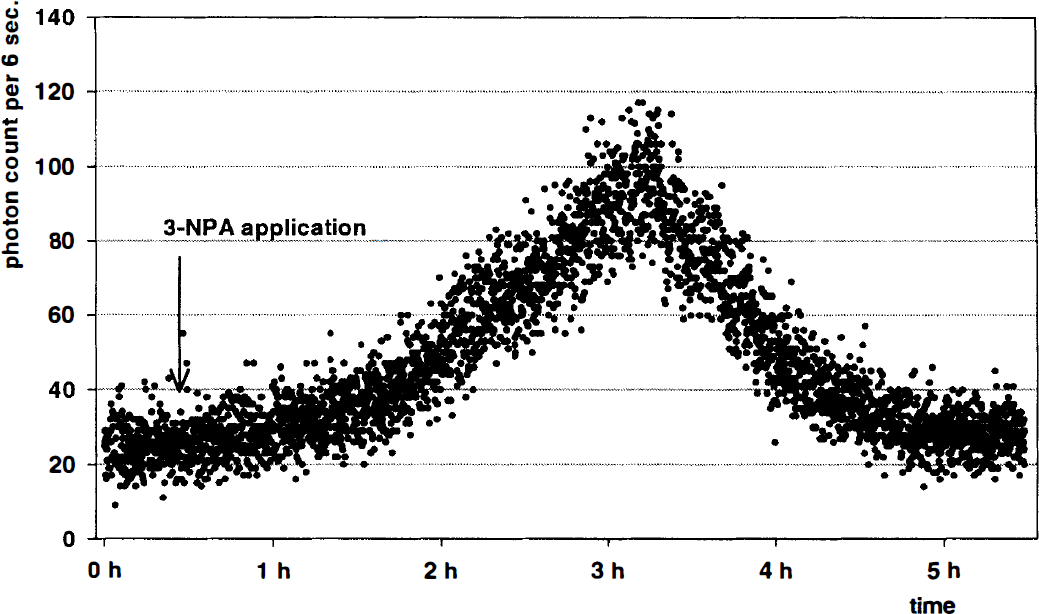
Typical chemiluminescence recording of an acute brain slice, superfused with a perfusion medium containing the chemiluminescence enhancer lucigenin. After an equilibrium period 3-nitroproprionic acid (3-NPA) was added to the perfusion medium (arrow) in a dose equivalent to the in vivo experiments (300 μmol/L). Emitted photons were counted as marker for the generation of reactive oxygen species, revealing a burst in reactive oxygen species production. Photons were counted in 6-second intervals.
Inhibition of oxygen free radicals production blocks tolerance induction
The ROS scavenger DMTU (especially effective in scavenging hydroxyl radicals), administered 30 minutes 3 before 3-NPA, led to infarct (176 ± 42 mm3) which were not statistically significantly different from control animals which had not received 3-NPA preconditioning. DMTU alone, given 3 days in advance to CCA/MCAO had no effect on infarct volumes (142 ± 51 mm3) (Fig. 5).
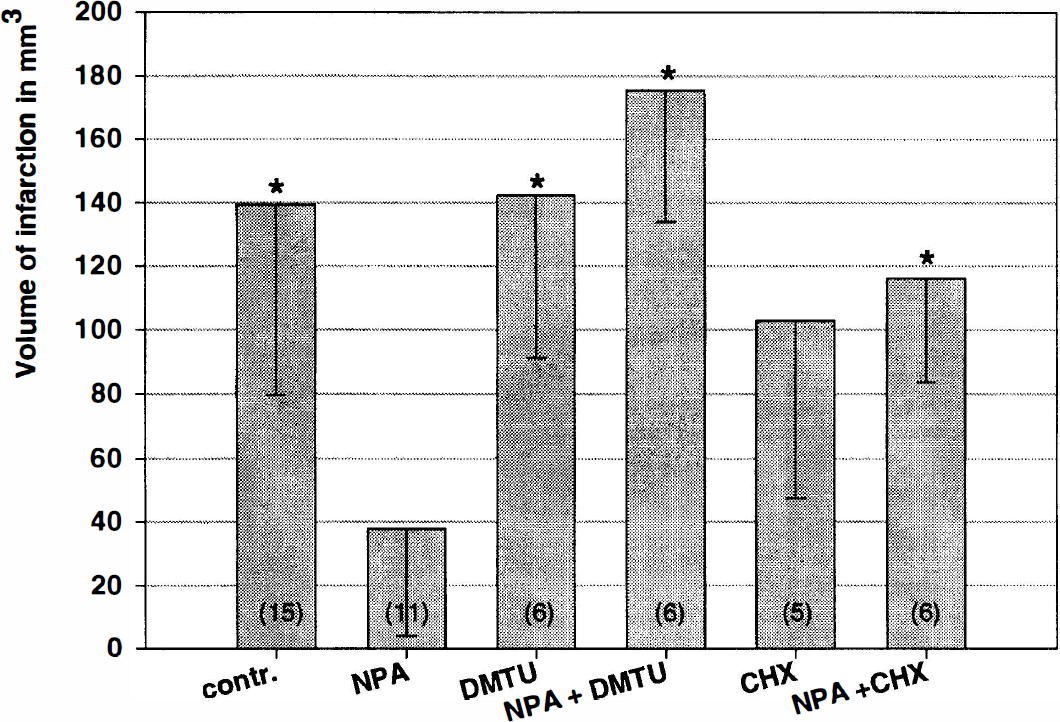
3-Nitroproprionic acid (3-NPA) induced tolerance to focal cerebral ischemia can be blocked by co-application of cycloheximide (CHX) or the free radical scavenger dimethylthiourea (DMTU), when administered 30 minutes before 3-NPA preconditioning 3 days prior to the focal cerebral ischemia. CHX or DMTU alone administered at the same time point showed no effect on infarct volume. Number of animals given in brackets. Data given as mean ± SD. *Denotes significant difference to 3-NPA, P < 0.05.
Protein synthesis inhibition blocks tolerance induction
The unspecific protein synthesis inhibitor CHX (1 mg/kg intraperitoneally) administered 30 minutes before 3-NPA treatment resulted in infarct volumes (116 ± 32 mm3) identical to control animals without preconditioning. Cycloheximide alone (i.e., without 3-NPA), given 3 days in advance to CCA/MCAO, had no effect on infarct volumes (102 ± 55 mm 3) (Fig. 5).
3-NPA did not induce Mn-SOD mRNA, nor brain Cu/Zn-SOD or Mn-SOD activity
Northern blotting SOD did not show any significant regulation of cortical Mn-SOD mRNA expression 6, 13, 24 or 72 hours after intraperitoneal injection of 20 mg/kg 3-NPA (Fig. 6). To determine SOD activity, cortical tissue samples from different animals were collected at 1 hour, 24 hours, 3 days, 5 days, and 7 days after the 3-NPA challenge. Significant changes in mitochondrial Mn-SOD, or cytosolic Cu/Zn-SOD activity were not detected (Table 2).
Relative Cu/Zn-SOD and Mn-SOD activity in cortical brain samples collected at different time points after 3-NPA administration (1 d, 3 d, 5 d, and 7 d)
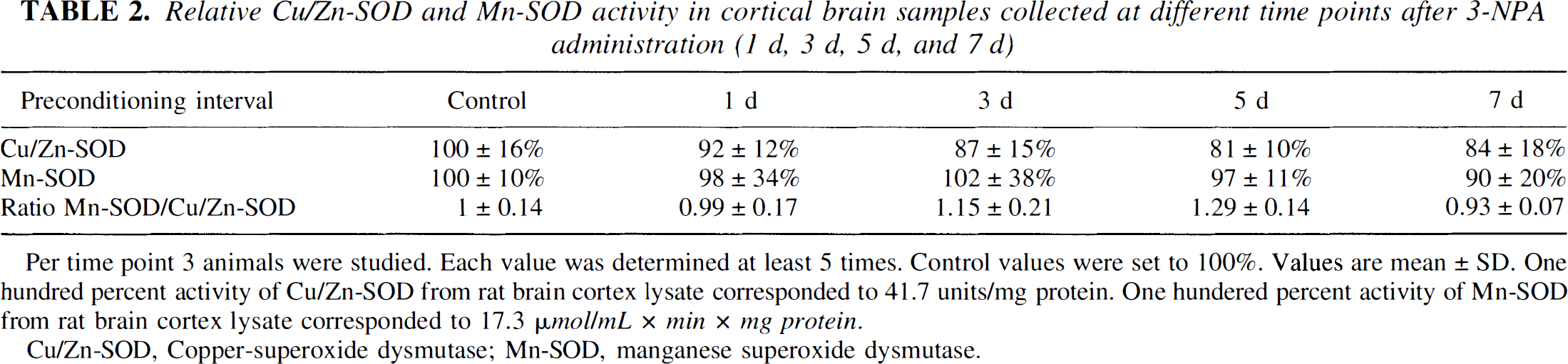
Per time point 3 animals were studied. Each value was determined at least 5 times. Control values were set to 100%. Values are mean ± SD. One hundred percent activity of Cu/Zn-SOD from rat brain cortex lysate corresponded to 41.7 units/mg protein. One hundered percent activity of Mn-SOD from rat brain cortex lysate corresponded to 17.3 μmol/mL × min × mg protein.
Cu/Zn-SOD, Copper-superoxide dysmutase; Mn-SOD, manganese superoxide dysmutase.
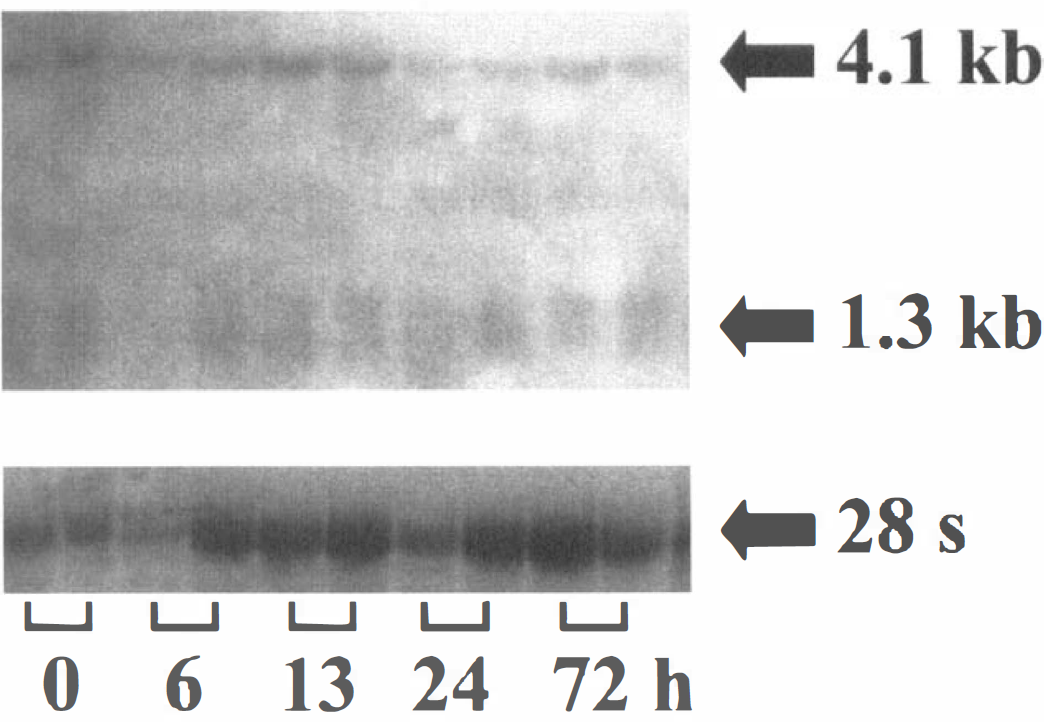
Northern blot: total RNA (20 μg per lane) was isolated from rat cortical brain tissue at different time points (two rats each) after intraperitoneal 3-NPA injection. Northern blot (upper part) was detected with Fluorescein-labelled Mn-SOD specific probe. In accordance with Ho et al., (1991) two prominent splicing variants were detected at 4.1 kb and 1.3 to 1.6 kb. Below: total RNA stained with ethidium bromide before blotting.
DISCUSSION
Pretreatment with 3-NPA induces profound tolerance to focal cerebral ischemia
For the first time we were able to show that it is feasible to induce protection against focal cerebral ischemia using an inhibitor of the oxidative phosphorylation. 3-NPA pretreatment 3 days before induction of focal cerebral ischemia reduced infarct volumes by 73% (permanent ischemia) or 35%, respectively (transient ischemia). Compared with other models of induced ischemic tolerance in the brain, 3-NPA induces the highest degree of protection shown so far. We can only speculate why protection in permanent focal cerebral ischemia was more pronounced.
It is widely accepted that reperfusion, despite its potentially beneficial effects, may also challenge the ischemic tissue. Oxygen free radicals, invasion of activated leukocytes, or plasma extravasation have been implicated in this so-called reperfusion paradox. Preconditioned neurons may thus be overwhelmed by mechanisms of damage activated by reperfusion.
The preconditioning dose of 3-NPA did not generate any detectable parenchymal damage, as shown by standard light microscopy, nor did it produce apoptosis or neurological abnormalities. This is in accordance with a study by Alexi et al. (1998), in which rats receiving a single dose of 20 mg/kg intraperitoneally did not show any signs of brain damage. In contrast, repeated low doses of 3-NPA induce selective striatal cell loss in animals (Beal et al., 1993), and high concentrations of 3-NP A have been shown to induce apoptotic cell death of neurons in vitro (Behrens et al., 1995).
We found that development of tolerance was a time-consuming process. Statistically significant protection was only seen 3 days after 3-NPA. A preconditioning interval of 2 days was not sufficient to generate tolerance, nor was a 5-day interval (Fig. 1).
In the brain this temporal pattern of tolerance induction is a consistent finding regardless of the preconditioning stimulus (Prass et al., 1998). Usually maximum protection can be found 2 to 5 days after tissue challenge, whereas tolerance disappears 7 days after the stimulus. A narrow time window of tolerance induction was found in a recent study in which maximum protection was induced by 3-NPA intraperitoneally 3 days before induction of global cerebral ischemia in the gerbil (Sugino et al., 1999), which is in good agreement with our findings.
Histologic infarct evaluation was performed 4 days after induction of focal cerebral ischemia, a time point when in this model the infarct has fully matured. However, it cannot be ruled out that 3-NPA may only have delayed infarction. Although unlikely, future studies must investigate even longer survival times (>2 weeks).
From our data it can be safely concluded that the preconditioning effect of 3-NPA is not caused by effects on body or brain temperature, blood glucose, or other physiologic variables. The fact that CBF changes during ischemia and reperfusion where not different in preconditioned versus untreated animals argues against an involvement of rCBF in preconditioning. Since LDF does not measure absolute rCBF, it might be argued that an increase in baseline rCBF may have escaped our investigation. However, delayed effects on rCBF have not been reported. In addition, we have preliminary evidence that 3-NPA also preconditions purified cortical neurons in culture against oxygen glucose deprivation (data not shown), strengthening the notion that development of tolerance is independent of hemodynamic effects.
3-NPA effects on energy metabolism
3-NPA is a suicide inhibitor of the SDH. In eucaryontic cells, SDH is present as complex II in oxidative phosphorylation and in the tricarboxylic acid cycle. Systemic administration of 20 mg/kg led to SDH inhibition by about 30% within 1 hour, which is in agreement with the literature (Alexi et al., 1998). Succinate dehydrogenase activity regeneration was a rather slow process, full SDH activity was not regained until the day 7 (Fig. 2).
Succinate dehydrogenase inhibition by 3-NPA decreased ATP and GTP to a minimum of about 35% of the normal levels in the first 30 minutes, which is in accordance to in vitro findings of Erecinska and Nelson (1994), and recovered within 2 hours. This degree of metabolic inhibition was not associated with tissue damage.
Inhibition of oxidative phosphorylation induces nonaerobic glycolysis and, therefore, production of lactate. Other groups showed an increase in lactate production within the first 3 hours after 3-NPA application in cell culture (Zeevalk et al., 1995) and in vivo (Jenkins et al., 1996), which is consistent with the time pattern found in our study.
Metabolic impairment may be responsible for neuronal damage by high doses of 3-NPA (Brouillet et al., 1995). Recent data shows that the type of neuronal death induced by 3-NPA is dose-dependent (Pang and Geddes, 1997). In rat hippocampal cell cultures and in vivo intermediate doses of 3-NPA clearly induced apoptosis (Sato et al., 1997), whereas high doses led to NMDA-receptor dependent excitotoxic cell death. In the low concentration used here ATP depletion does not produce neuronal necrosis, nor apoptosis, as shown. We therefore speculate that 3-NPA, dependent on its concentration, can induce three different cellular responses: tolerance, apoptosis, or excitotoxic cell necrosis.
It is well established that reductions in energy metabolism (e.g., by hypothermia) are neuroprotective. One might therefore speculate that low doses of 3-NPA generate their neuroprotective effect simply by inhibiting cellular energy metabolism. However, for the following reasons we believe that this is unlikely: (1) Maximum SDH and ATP inhibition are already present 1 hour after 3-NPA application, a time point at which no significant protection was found. (2) Development of tolerance was a time-consuming process, with maximum protection when SDH inhibition and the decrease in energy rich phosphates was already recovering. (3) Inhibition of protein synthesis abolished preconditioning (see below). We conclude that impairment of the mitochondrial electron transport chain and the consecutive short impairment of energy production are not the primary cause, but rather the trigger for the induction of ischemic tolerance.
Cycloheximide, a protein synthesis inhibitor, prevents tolerance induction by 3-NPA
Independently of the stimulus used to induce tolerance against ischemia in the brain, the interval to maximum protection is between 24 and 72 hours (Prass et al., 1998). It is generally assumed that the neosynthesis of protective proteins is responsible for the delay in tolerance development. This is in line with our data because protein synthesis inhibition with CHX completely abolished 3-NPA preconditioning.
Cycloheximide itself protects against focal cerebral ischemia when given immediately before induction of focal cerebral ischemia (Kharlamov et al., 1997). Cycloheximide also has the capacity to protect cultured striatal and cortical neurons against the toxicity of 3-NPA (Behrens et al., 1995). These effects where mainly attributed to a CHX-induced blockade of apoptosis (Du et al., 1996) because apoptotic cell degeneration depends on intact protein synthesis. In rats CHX doses higher than 0.6 mg/kg are known to reduce brain protein synthesis for at least 24 hours (Pavlik and Teisinger, 1980). However, recent in vitro findings indicate that CHX, when given in low doses, besides inhibiting protein synthesis also acts as a potent inductor of neuroprotective proteins, such as bcl-2 and antioxidants (Furukawa et al., 1997). One might speculate that an early induction of free radical scavengers would have decreased the potential of 3-NPA to induce tolerance, not by inhibition of protein synthesis, but rather by diminishing the 3-NPA induced free radical burst which is essential for an efficient tolerance induction. The fact that the time interval between CHX and 3-NPA application was rather short (30 minutes) argues against this speculation, the induction of protective proteins by CHX takes several hours (Furukawa et al., 1997). However, we cannot rule out that other mechanisms than protein synthesis inhibition may have contributed to the CHX induced blockade of tolerance induction.
Involvement of free radicals in tolerance induction
Inhibition of mitochondrial respiration generates ROS formation (Garcia-Ruiz et al., 1995). We therefore speculated that ROS formation caused by 3-NPA might critically contribute to tolerance induction. With lucigenin-enhanced chemiluminescence, an assay particularly sensitive to superoxide anion, in acute brain slices we detected a burst of ROS induced by 3-NPA. Other studies have produced additional evidence for 3-NPA-induced oxidative stress (Beal et al., 1995; Fu et al., 1995). Further proof for the critical involvement of free radicals in the cascade of tolerance development is given by the fact that pretreatment with the free radical scavenger DMTU completely abolished tolerance induction (Fig. 5).
Biological tissues react to oxidative stress with the induction of ROS scavenging enzymes. It is well known that ROS scavengers protect the brain against various types of ischemia. Thus, it was tempting to speculate that it is the induction of these protective proteins which protect the brain after 3-NPA preconditioning. Superoxide dismutase isoforms were shown to be induced by preconditioning in several models of global cerebral (Kato et al., 1995) and myocardial ischemia (Steeves et al., 1994). 3-NPA neurotoxicity is attenuated in Cu/Zn-SOD transgenic mice (Beal et al., 1995). However, we found no evidence for changes in Cu/Zn-SOD or Mn-SOD activity, nor did we detect changes in Mn-SOD mRNA at any time point studied. This is in accordance with other studies which showed no changes in brain SOD content after up to 40 mg/kg 3-NPA (Fu et al., 1995; Prapurna et al., 1996). Our data therefore argues against the involvement of SOD in tolerance induction in our model. However, it does not exclude that other inducible OFR scavenging systems may have been involved, such as catalase, glutathion, ascorbic acid, tocopherols, uric acid, melatonin, or histidine related compounds.
Besides OFR scavengers, other potential protein candidates to explain 3-NPA induced tolerance have to be considered: antiapoptotic proteins such as bcl-2, bcl-x-long, neuronal apoptosis inhibitory protein (NAIP), heat shock proteins, and neurotrophic factors such as BDNF, among others.
CONCLUSION
The neosynthesis of thus far unknown protective proteins is responsible for profound ischemic tolerance induced by 3-NPA. It is likely that the formation of OFR is involved in this process, but SOD is not responsible for protection. Further research is needed to pinpoint the protective proteins. In contrast to previously described preconditioning protocols, strategies like the one described here may lead to clinically applicable, pharmacologic strategies to protect the brain against focal ischemia. In any event, 3-NPA preconditioning presents an unique model to study cellular responses to metabolic stress and the induction of endogenous, protective programs.