
Abstract
The delayed death of CA1 neurons after global brain ischemia is associated with induction of apoptosis genes and is inhibited by protein synthesis inhibitors, suggesting that the degeneration of CA1 pyramidal neurons is an active process that requires new gene expression. The transient global ischemia model has been extensively used to identify enzymes and other proteins underlying delayed neuronal cell death. The expression of protein kinase C (PKC) subspecies after 20 minutes of global brain ischemia produced by a four-vessel occlusion model in the rat was studied. From the multiple PKC subspecies studied, only PKCδ mRNA was significantly up-regulated in CA1 pyramidal neurons at 24 hours and in activated microglia at 3 to at least 7 days after ischemia. The induction of PKCδ mRNA was also found in the cortex at 8 hours and 3 days after ischemia. This cortical but not hippocampal induction was regulated by an α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid/kainate receptor antagonist, 6-nitro-7-sulfamobenzo[f]quinoxaline-2,3-dione, and glucocorticoids. An N-methyl-
Transient global ischemia causes neuronal death in various brain areas, including the CA1 sector of the hippocampus, dorsolateral part of the striatum, thalamus, and cortex (Pulsinelli et al., 1982; Jin et al., 1999). The cell death in the CA1 region is selective for neurons, robust, and delayed when compared with the ischemic damage in other brain regions (Pulsinelli et al., 1982; Jin et al., 1999). Several studies have demonstrated that apoptosis, an important physiologic process requiring the activation of an intrinsic genetic program, is involved in ischemic cell death in the brain, particularly in the CA1 pyramidal cells (MacManus et al., 1993; Du et al., 1996; Jin et al., 1999). However, several hallmarks of apoptosis are not seen in CA1 neurons after global ischemia (MacManus et al., 1999), suggesting that the cell death involves atypical features of apoptosis or occurs mainly through necrosis. Altogether, the precise intrinsic mechanisms underlying delayed neuronal cell death have not yet been elucidated, despite extensive studies. Because numerous genes involved in either apoptosis or neuronal survival are induced by brain ischemia (Koistinaho and Hökfelt, 1997) and inhibitors of protein synthesis provide protection against brain ischemia (Goto et al., 1990), expression of novel proteins is likely to play a role in delayed neuronal death.
Protein kinase C (PKC) is a family of serine-threonine kinases that have differential subcellular localization and requirements for their activation (Tanaka and Nishizuka, 1994) and are likely to mediate unique functions in various cell types. Protein kinase C undergoes translocation from one intracellular compartment to another on activation (Mochly-Rosen and Gordon, 1998), and isozyme specificity is partly mediated by association of each PKC isozyme with specific anchoring proteins (Mochly-Rosen and Gordon, 1998). Ischemic insult induced by various global and focal brain ischemia models has been reported to result in a rapid loss of total PKC activity (Crumrine et al., 1990; Louis et al., 1991; Wieloch et al., 1991; Cardell and Wieloch, 1993; Busto et al., 1994). In addition, PKC activity is reduced in cultured neurons exposed to neurotoxic levels of excitatory amino acids, and this inactivation has been suggested to be part of an N-methyl-
The aim of the present study was to demonstrate the pattern of postischemic changes in the expression of PKC subspecies at the mRNA and protein level after transient global forebrain ischemia and to determine the effects of pretreatment with dexamethasone, a synthetic glucocorticoid, MK-801, an NMDA receptor antagonist, and 6-nitro-7-sulfamobenzo[f]quinoxaline-2,3-dione (NBQX), an α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid (AMPA)/kainate receptor antagonist, on PKC expression in the rat brain.
MATERIALS AND METHODS
Four-vessel occlusion
Transient forebrain ischemia was induced by a four-vessel occlusion model according to the method of Pulsinelli et al. (1982) with modifications. In brief, male Wistar rats (250 to 270 g) were anesthetized with 5% halothane and placed in a stereotaxic frame. The anesthesia was maintained on 1.0% halothane in 70% N2O/30% O2, and both vertebral arteries were occluded permanently by electrocauterization. On the following day, the rats were reanesthetized and the femoral artery was cannulated for recording of physiological parameters before and during ischemia. Rectal temperature was continuously monitored and kept at 37 to 37.5°C using a heating pad. The common carotid arteries were exposed bilaterally, and the anesthesia was discontinued. Forebrain ischemia was induced by occluding the common carotid arteries with microvascular clips for 20 minutes. At various time points, the animals were decapitated and tissues processed for in situ hybridization, immunoblotting, or Northern blotting or the brains were perfusion-fixed for immunohistochemistry. Sham-operated animals underwent vertebral artery occlusion but during the following day were anesthetized without occlusion of the common carotid arteries.
Drug treatments
MK-801 hydrogen maleate (Research Biochemicals, Natick, MA, U.S.A.; 3 mg/kg intraperitoneally) and dexamethasone sodium phosphate (Decadron; MSD Isotopes, Haarlem, the Netherlands; 3 mg/kg intraperitoneally) were given 30 minutes before ischemia (n = 3/group), and the dexamethasone-treated animals received additional doses (3 mg/kg) 1 and 3 hours after ischemia. 6-Nitro-7-sulfamobenzo[f]quinoxaline-2,3-dione (Tocris Cookson, Bristol, U.K.) was given intraperitoneally as two equal injections of a total dose of 60 mg/kg 15 minutes before and 1 hour after ischemia. Control animals received an intraperitoneal injection of saline 30 minutes before ischemia.
In situ hybridization
For in situ hybridization studies, rats were decapitated at 4 hours, 8 hours, 24 hours, 3 days, and 7 days after ischemia (n = 3/group). The brains were rapidly frozen and sectioned at 10-μm thickness. The synthetic oligonucleotides for PKC subspecies were the same as previously used (Miettinen et al., 1996). Oligonucleotides with length and GC ratio similar to the corresponding antisense oligonucleotides but without homology to any known gene sequences were used as controls. The probes were end-labeled with 35S-ATP using terminal deoxynucleotidyl transferase (MBI Fermentas, Vilnius Lithuania) and purified using a Probe Quant G50 column (Pharmacia Biotech, Sweden). The brain sections were hybridized overnight in hybridization solution containing 10 × 106 cpm/mL probe, 40 μL of 5 mol/L dithiothreitol, 50 μL of 10 mg/mL salmon sperm DNA, and 900 μL of hybridization cocktail [50 mL of formamide, 20 mL of 20 × saline-sodium citrate (SSC), 2 mL of 50 × Denhardt's reagent, 10 mL of 0.2 mol/L sodium phosphate buffer (pH 7.4), 10 g of dextran, 4 mL of 25% sarcosyl]. The sections were washed in 1 × SSC at 55°C for 2 hours, rinsed 2 × 5 minutes in deionized water at room temperature, dehydrated for 30 seconds in 60 and 90% ethanol, air dried, and covered with Kodak XAR-5 film for 21 days.
Image analysis
The mRNA expression levels of PKC subspecies were quantified by measuring the optical density of the specified brain regions using a digital image analysis system (MCID 4; Imaging Research, St. Catherine's, Ontario, Canada). The gray levels corresponding to the 14C plastic standards (Amersham, Buckinghamshire, U.K.) lying within the exposure range of the film were detected and used as a fourth-degree polynomial approximation to construct a gray level to activity transfer. Densitometric measurements were done from four sections per each animal, and the data were assessed with the two-tailed Mann-Whitney U/Wilcoxon rank sum W test.
Northern blotting
Total RNA was isolated from frozen cortical and hippocampal tissues using Trizol reagent according to the manufacturer's instructions. Samples of 20 μg/lane were electrophoresed through a formaldehyde/1.2% agarose gel and transferred to a nylon membrane (Hybond-N; Amersham) by capillary blotting. The membrane was hybridized with a 32P random prime-labeled (Ready To Go DNA labeling kit; Pharmacia Biotech, Sweden) PKCδ cDNA (specific activity 1 × 109 cpm/μg) in 5 × SSC/5 × Denhardt's/50% deionized formamide/1% sodium dodecyl sulfate (SDS) overnight at 42°C. It was washed in 2 × SSC/0.1% SDS twice for 5 minutes at room temperature, in 0.2 × SSC/0.1% SDS twice for 5 minutes at room temperature, and for 10 minutes at 42°C and autoradiographed at −80°C. After stripping, the membrane was rehybridized with a 32P-labeled β-actin cDNA (specific activity 8 × 108 cpm/μg), as described above.
Immunohistochemistry
For immunohistochemistry, rats were perfused with 4% paraformaldehyde at 12 hours, 72 hours, and 7 days following the ischemia (n = 3/group), and the brains were postfixed for 4 hours in 4% paraformaldehyde. The vibratome sections (50 μm) were incubated for 48 hours in the primary PKCδ antibody (rabbit, 1:1,000; GibcoBRL, Life Technologies, or Santa Cruz Biotechnology, Santa Cruz, CA, U.S.A.), and the bound antibody was visualized with the avidin—biotin—peroxidase method (Vectastain kit; Vector Labs, Burlingame, CA, U.S.A.) using 3,3'-diaminobenzidine as the peroxidase substrate. For double-staining studies, the brains were fixed 8 and 72 hours after ischemia and immersed in 20% sucrose buffer for 72 hours, frozen in liquid nitrogen-cooled isopentane, and cut at 14-μm thickness in a cryostat. The sections were incubated for 48 hours at room temperature in a mixture of PKCδ (1:250; Santa Cruz Biotechnology) and mouse monoclonal OX-42 (1:250; Serotec, Oxford, U.K.) antibodies or PKCδ (1:250) and mouse monoclonal glial fibrillary acidic protein (1:250; Boehringer-Mannheim, Indianapolis, IN, U.S.A.) or PKCδ (1:250) and mouse monoclonal microtubule-associated protein-2 (1:250; Boehringer-Mannheim) or PKCδ (1:250) and SMI-31, the antibody for phosphorylated heavy and medium molecular weight neurofilaments (1:5,000; Sternberger Monoclonals, Baltimore, MD, U.S.A.) diluted in 0.1 mol/L sodium phosphate buffer (pH 7.4), containing 0.3% Triton X-100 and 1% bovine serum albumin. Fluorescein isothiocyanate-conjugated anti-rabbit and tetramethylrhodamine isothiocyanate-conjugated anti-mouse IgGs (Jackson ImmunoResearch, West Grove, PA, U.S.A.) were used as secondary antibodies. The sections were coverslipped in glycerol/phosphate-buffered saline (3:1) and examined in a Leica DMRB microscope equipped with rhodamine and fluorescein isothiocyanate filter sets.
Immunoblotting
Three days after ischemia, sham-operated (n = 2) and ischemic (n = 4) rats were killed, and the hippocampi were quickly dissected out. The tissue was homogenized in 1 mmol/L Tris-HCl buffer (pH 7.4), containing 320 mmol/L sucrose, 1 mmol/L ethyleneglycol bis(aminoethylether)tetraacetate, 5 mmol/L dithiothreitol, 1 mmol/L sodium vanadate, 50 mmol/L NaF, 0.2 mmol/L phenylmethylsulfonyl fluoride, 10 μg/mL leupeptin, and 10 μg/mL aprotinin. Fifty microliters of the buffer was added per 10 mg of wet tissue. Cell debris was removed by 10-minute centrifugation at 1,000 g. The cytosolic and membrane fractions were separated by ultracentrifugation for 60 minutes at 100,000 g. The pellet, containing membranes, was solubilized in homogenization buffer plus 1% Triton X-100, and insoluble fraction was removed by centrifugation for 10 minutes at 10,000 g. Sodium dodecyl sulfate polyacrylamide gel electrophoresis Laemmli sample buffer was added to the cytosol and membrane fractions. The samples were incubated at 100°C for 5 min. Electrophoresis was carried out in a 10% SDS-polyacrylamide gel in MiniProtean (Bio-Rad) apparatus. A sample containing 20 μg of total protein was loaded per lane. Separated proteins were transferred onto Hybond-P membrane (Amersham-Pharmacia Biotech) in a MiniTransBlot (Bio-Rad) wet blotting apparatus according to the manufacturer's instructions. The membranes were processed according to the manufacturer's instructions. Protein kinase Cδ was detected using monoclonal (Transduction Laboratories; 1:5,000) or rabbit polyclonal (Gibco; 1:2,000) and secondary horseradish peroxidase-labeled antibody (Amersham-Pharmacia Biotech; 1:10,000 dilution), using an ECL Plus kit (Amersham-Pharmacia Biotech). The membranes were directly scanned on a Storm (Molecular Dynamics, Sunnyvale, CA, U.S.A.) fluoroimager, and the detected bands quantified using Image Quant software.
RESULTS
Expression of protein kinase C mRNA after transient forebrain ischemia
In situ hybridization results revealed a basal expression of all PKC subspecies in control rats, as has been reported earlier (Miettinen et al., 1996). Protein kinase Ca showed the highest expression in cortical layers 2 and 3, whereas PKCβ, -ϵ, and -ζ were more homogeneously distributed in all cortical layers. The expression of PKCγ was seen in cortical layers 1, 3, and 5. A low expression of PKCδ was detected in cortical layers 2, 3, and 5. The PKC isoforms α, β, γ, ϵ, and ζ showed mRNA expression in CA1 to CA3 areas of the hippocampus and in the dentate gyrus, whereas PKCδ appeared to be the only PKC isoform studied that was not expressed at a detectable level in the hippocampus (Fig. 1A). A strong basal hybridization signal of PKCδ mRNA was seen in the thalamus (Fig. 1).
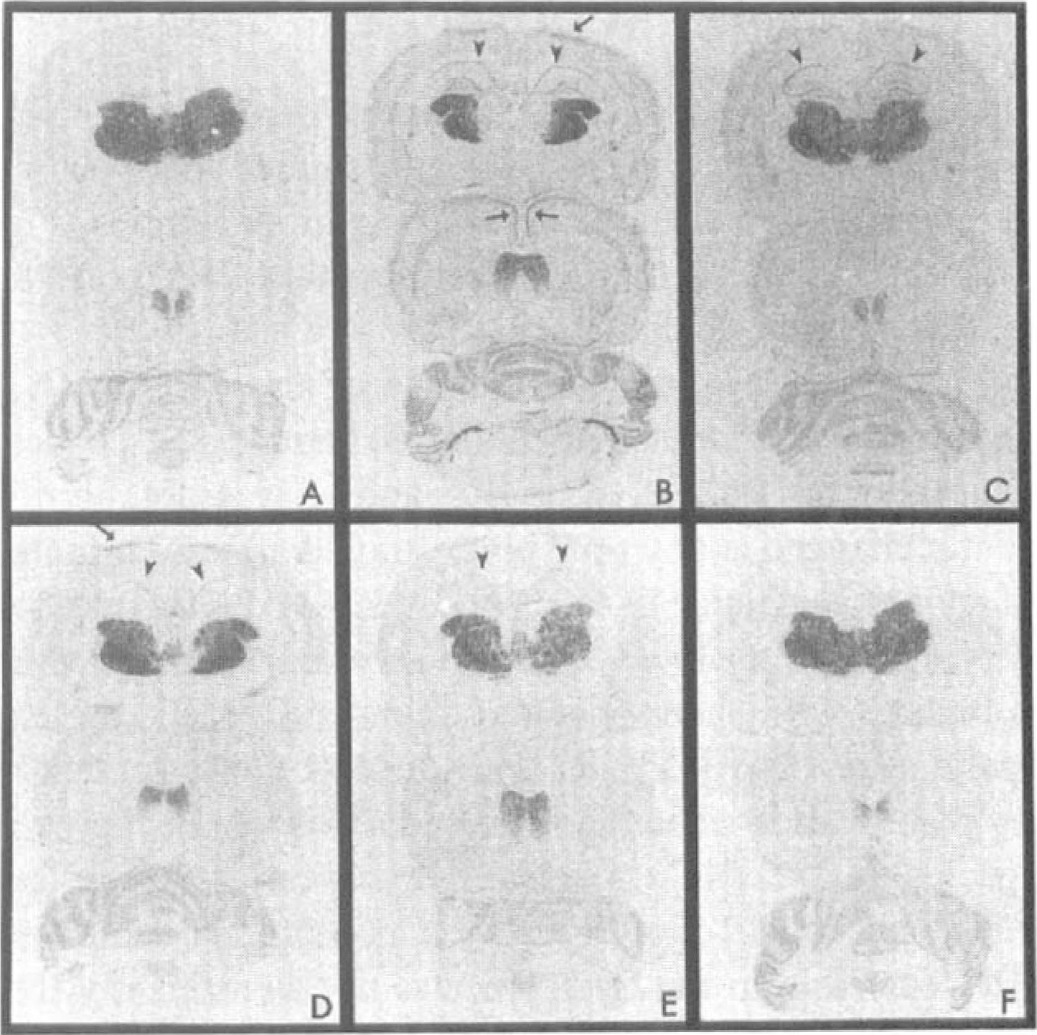
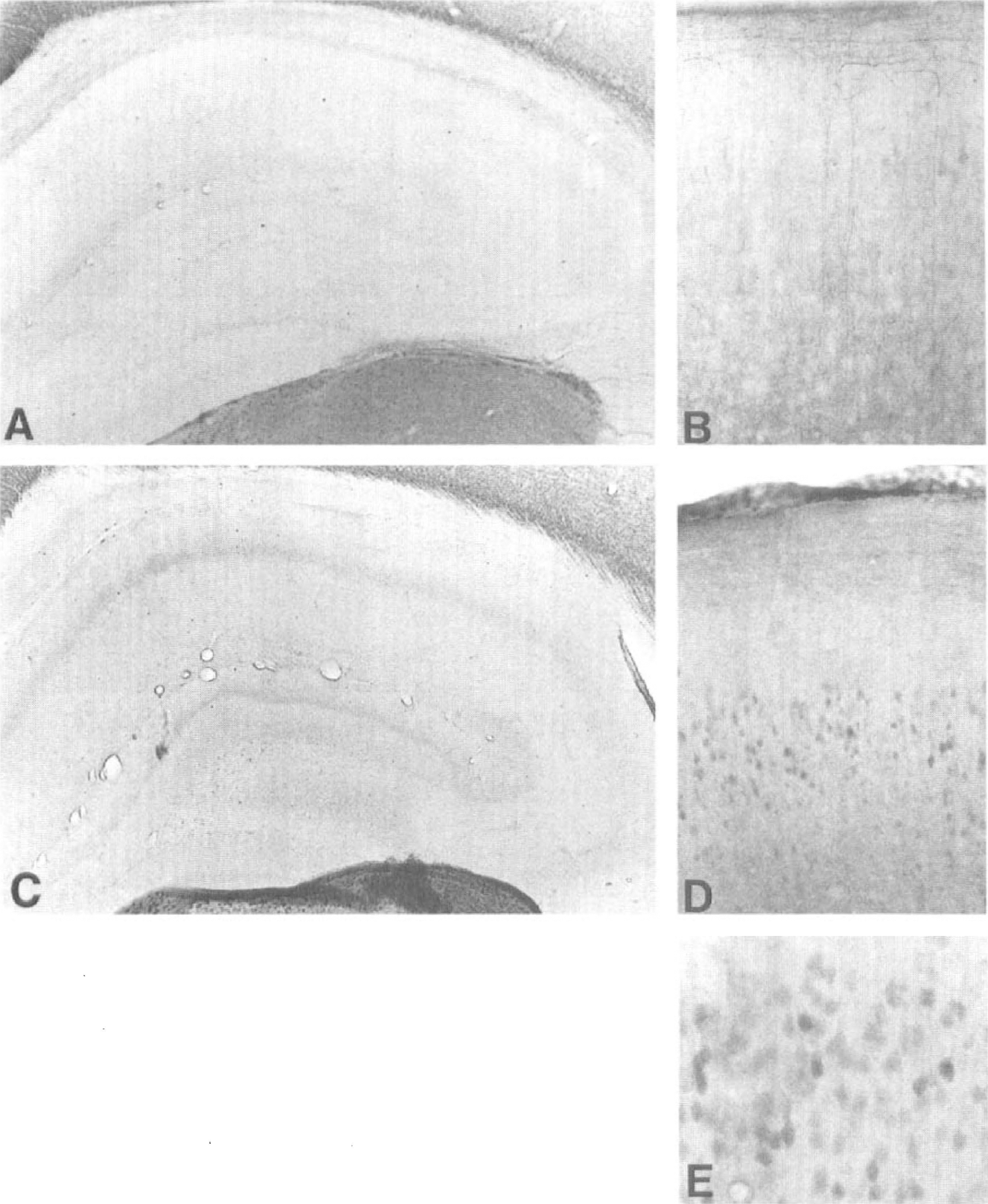
Transient global ischemia induces early and prolonged expression of protein kinase δ (PKCδ) mRNA in the cortex (arrows) and hippocampal CA1 area (arrowheads). In situ hybridization autoradiographs show expression of PKCδ mRNA in the rat brain at hippocampal (
Figure 2 shows the time course of the PKC mRNA expression after transient global ischemia. The cortical expression of PKCα, -β, -γ, -ϵ, and -ζ was not significantly changed at any time point studied after 20 minutes of global ischemia. In CA3, the level of PKCα mRNA was significantly lower at 24 hours after ischemia when compared with sham-operated animals. In CA1, the expression of PKCβ, -γ, -ϵ, and -ζ mRNA was gradually decreased after ischemia. The reduction of all the subspecies except PKCα reached a significant level by day 7. The mRNA level of PKCα was also slightly decreased after ischemia in the CA1 but was not significantly different from values in sham-operated animals.
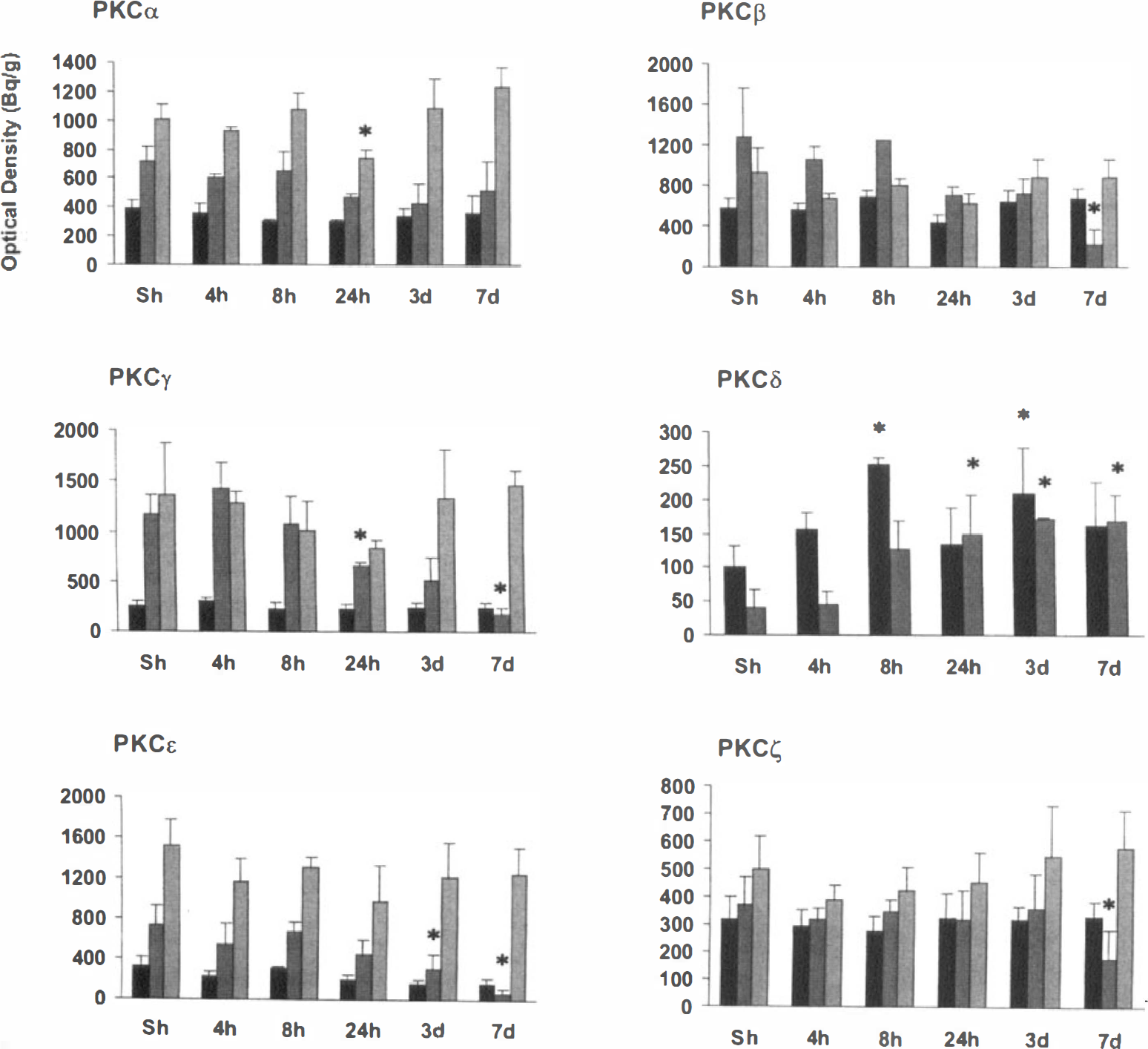
The mRNA expression of protein kinase C (PKC) subspecies in the frontoparietal and cingulate cortex and CA1 and CA3 areas (columns from left to right) at the dorsal hippocampal level measured on x-ray film in situ hybridization autoradiographs. Values represent the means ± SD from three to four animals. *P < 0.05, statistically significant difference in the expression level between ischemic and sham-treated animals (two-tailed Mann-Whitney U/Wilcoxon rank sum W test). The expression level of the PKCδ mRNA in the CA3 area was detected occasionally at 8- and 24-hour time points but was never seen 3 days after ischemia and was not measured due to the variation between the animals. (■) Cortex; (■) CA1; (□)CA3.
The only PKC subspecies that showed induction in response to ischemia was PKCδ. Its mRNA expression showed a two-phase increase in the cortex, being significantly higher at 8 hours and 3 days after ischemia when compared with control values (Fig. 2). The expression of PKCδ was increased mainly in the most superficial layers of the frontoparietal and cingulate cortex (Figs. 1B and 1D) and remained up-regulated until 7 days (Fig. 2). In the CA1 area, PKCδ mRNA was also increased 8 hours after ischemia and remained significantly increased until 7 days (Figs. 1B to 1E and 2). The expression of PKCδ was occasionally detected in the CA3 area at 8- and 24-hour time points (Figs. 1B and 1C) but not at 3 days after ischemia (Fig. 1D).
We next studied whether NMDA- and AMPA-type glutamate receptors and glucocorticoids regulate the ischemia-induced expression of PKCδ, the only subspecies found to be up-regulated in the present study. Values for physiologic parameters determined before the ischemia were all within the normal range and did not significantly differ between the treatment groups (Table 1). An NMDA receptor antagonist, MK-801 (3 mg/kg), had no effect on ischemia-induced PKCδ expression at the 8-hour time point but reduced cortical PKCβ mRNA expression by 20% (Fig. 3). An AMPA receptor antagonist, NBQX (60 mg/kg), reduced the ischemia-induced expression of PKCδ in the cortex by 70% but did not reduce significantly the expression in CA1. In addition, NBQX reduced hippocampal expression of PKCα mRNA by ~35% but did not affect other PKC subspecies. Treatment with dexamethasone, a synthetic glucocorticoid (3 × 3 mg/kg), inhibited significantly the induction of PKCδ mRNA in the cortex but not in the hippocampus and did not affect other PKC subspecies.
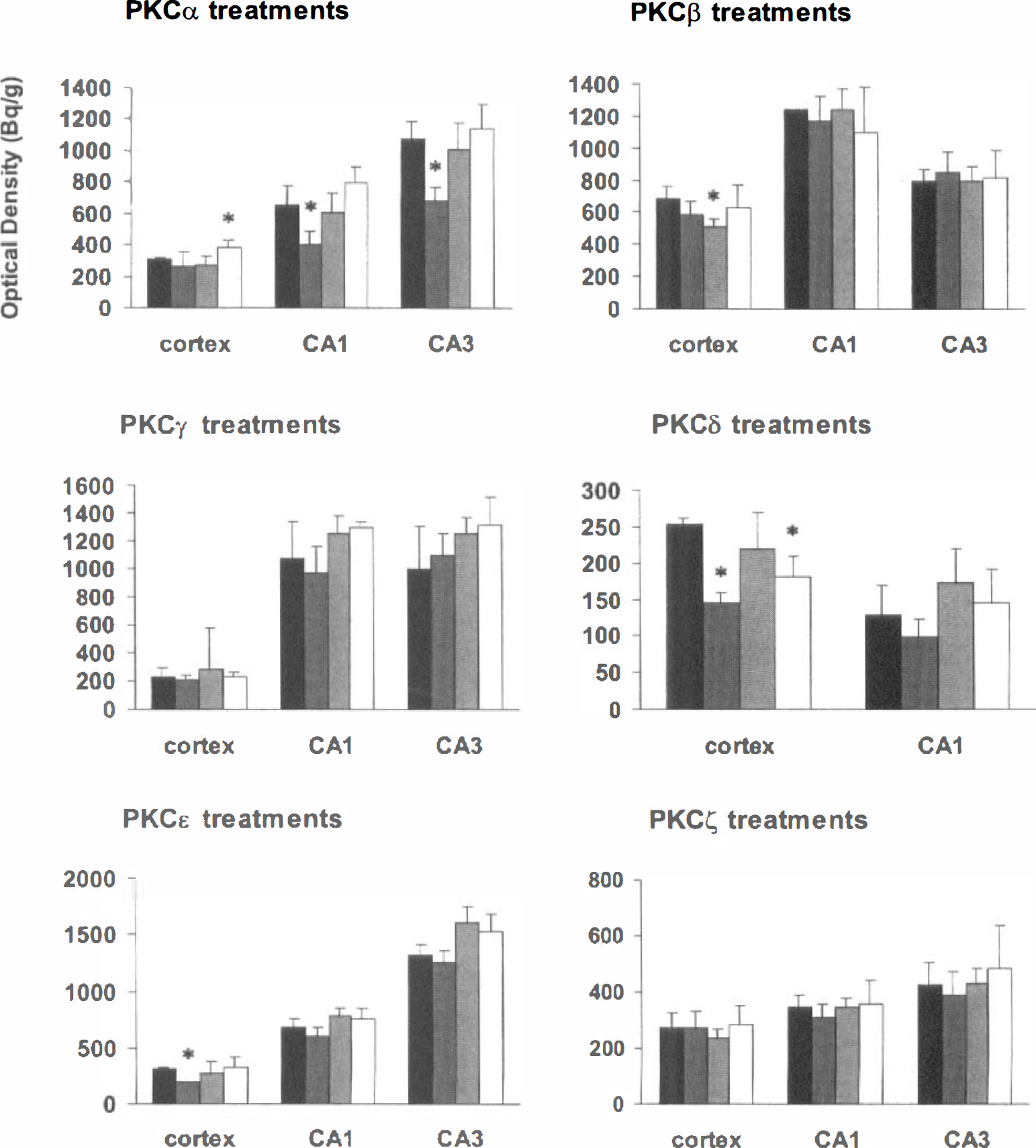
The effects of pretreatments with saline, 6-nitro-7-sulfobenzo[f-]quinoxaline-2,3-dione (NBQX), MK-801, and dexamethasone (columns from left to right) on the mRNA expression of the protein kinase C (PKC) subspecies in the cortex and CA1 and CA3 areas at the dorsal hippocampal level 8 hours after ischemia measured on x-ray film in situ hybridization autoradiographs. Values represent the means ± SD from three to four animals. *P < 0.05, statistically significant difference in expression level between saline- and drug-treated ischemic animals (two-tailed Mann-Whitney U/Wilcoxon rank sum W test). (■) Saline; (■) NBQX; (□) MK-801; (□) Dexamethasone.
Physiologic parameters
All values are mean ± SD. There are no statistically significant differences between the groups.
The induction of the PKCδ mRNA was confirmed by Northern blotting, which demonstrated a specific band of 3.1-kb size for PKCδ (Fig. 4). Supporting the results of in situ hybridization, Northern blotting showed that PKCδ mRNA was induced in the samples from the cortex and hippocampus 24 hours after forebrain ischemia when compared with control samples. Control hybridization of the same blots with β-actin cDNA probe demonstrated equal amounts of loaded RNAs (Fig. 4).
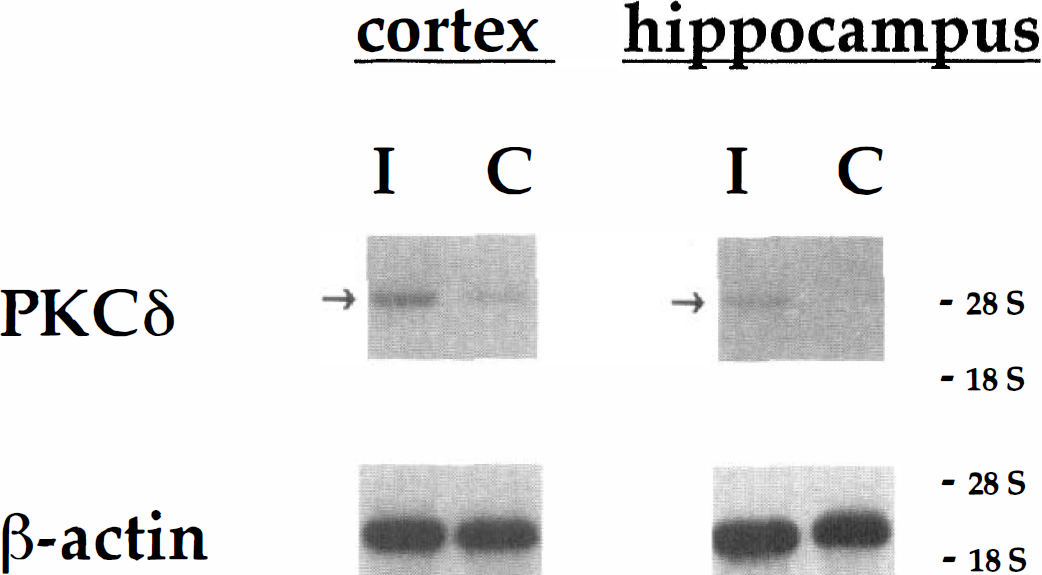
Northern blotting analysis of total RNA from the rat brain 24 hours after transient global brain ischemia. The blot demonstrated a specific band of 3.1-kb size for a 32P-labeled protein kinase Cδ (PKCδ) cDNA probe. The PKCδ mRNA was induced in the samples from the cortex and hippocampus (arrows) after forebrain ischemia when compared with control samples. A 32P-labeled β-actin cDNA probe was used to demonstrate equal amounts of loaded RNAs.
Immunohistochemistry
The time course of PKCδ immunoreactivity in the cortex and hippocampus was studied in animals that survived for 12 hours, 72 hours, or 7 days after ischemia. In control animals, few PKCδ-positive neurons were detected in the frontoparietal and cingulate cortex (Fig. 5B), whereas no immunoreactive neurons were seen in the hippocampus (Fig. 5A). In addition, a large number of immunostained neurons were seen in the thalamus (not shown). Twelve hours after ischemia, the number of PKCδ-positive neurons increased both in cortical layers 2 to 3 (Figs. 5D and 5E) and in layer 5, and some immunostained neurons were observed in the CA1 region of the hippocampus (Fig. 5C). At 3 days, a strong band of immunoreactive microglia-like cells was seen in the CA1 pyramidal cell layer (Figs. 6A, 6B, and 6D), which was still present 7 days after ischemia (Fig. 6C).
Protein kinase Cδ (PKCδ) immunoreactivity in the control rat hippocampus
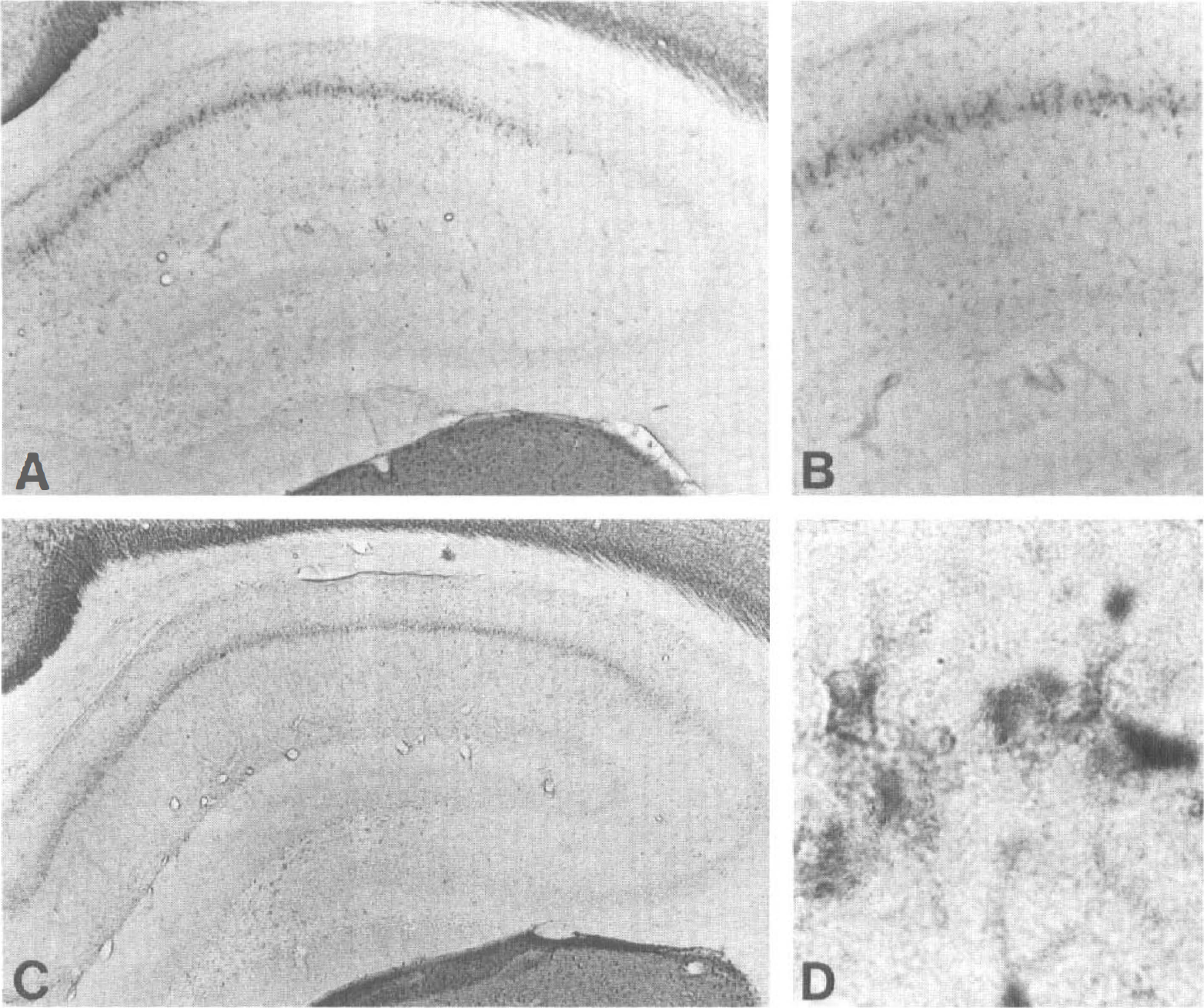
A strong protein kinase Cδ (PKCδ) immunoreactivity was seen in the hippocampal CA1 area both 3
The double-staining experiments showed a colocalization of PKCδ and microtubule-associated protein-2 antibodies in neurons of the CA1 pyramidal cell layer 8 hours after ischemia (Figs. 7A and 7B). The PKCδ-positive cells did not stain with glial fibrillary acidic protein (Figs. 7C and 7D) or OX-42 antibody (not shown) at 8 hours. At 24 hours after ischemia, the PKCδ immunoreactivity colocalized with SMI-31, the antibody detecting phosphorylated neurofilaments (Figs. 7E and 7F). By 3 days, the microtubule-associated protein-2 immunostaining had disappeared from the CA1 area (not shown), and the PKCδ-positive cells double-stained with OX-42 (Figs. 7G and 7H) but not with glial fibrillary protein (Figs. 7I and 7J) or SMI-31 (not shown), indicating that they represented activated microglia.
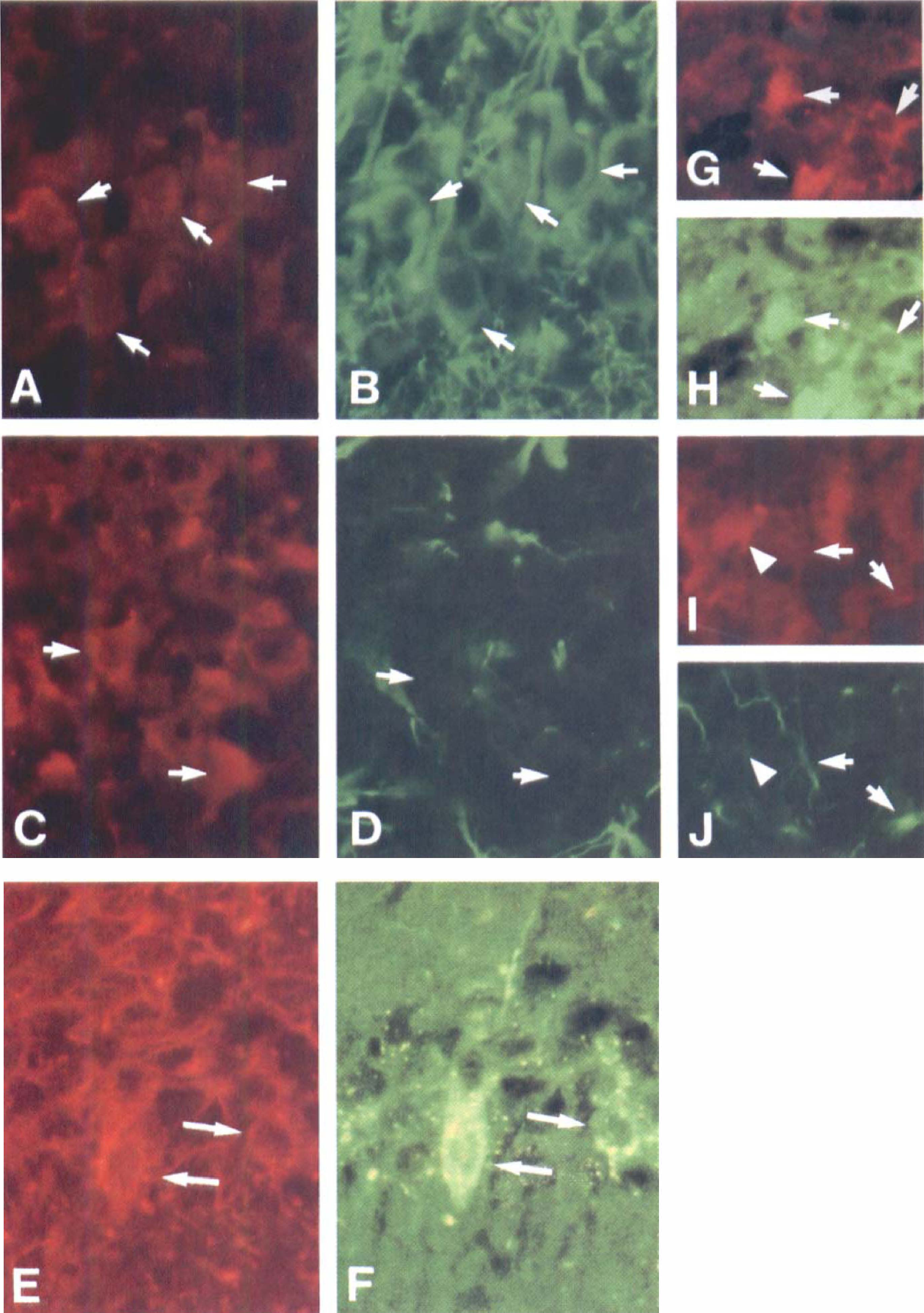
Protein kinase Cδ (PKC8)-immunoreactive cells (
Immunoblotting
The immunoblots demonstrated the presence of PKCδ in both cytosolic and membrane fractions in each hippocampus (Fig. 8A). In three of four animals, a 70% increase in the membrane fraction was seen (Fig. 8B), thus possibly reflecting increased activity of the PKCδ present in the hippocampus 3 days after ischemia.
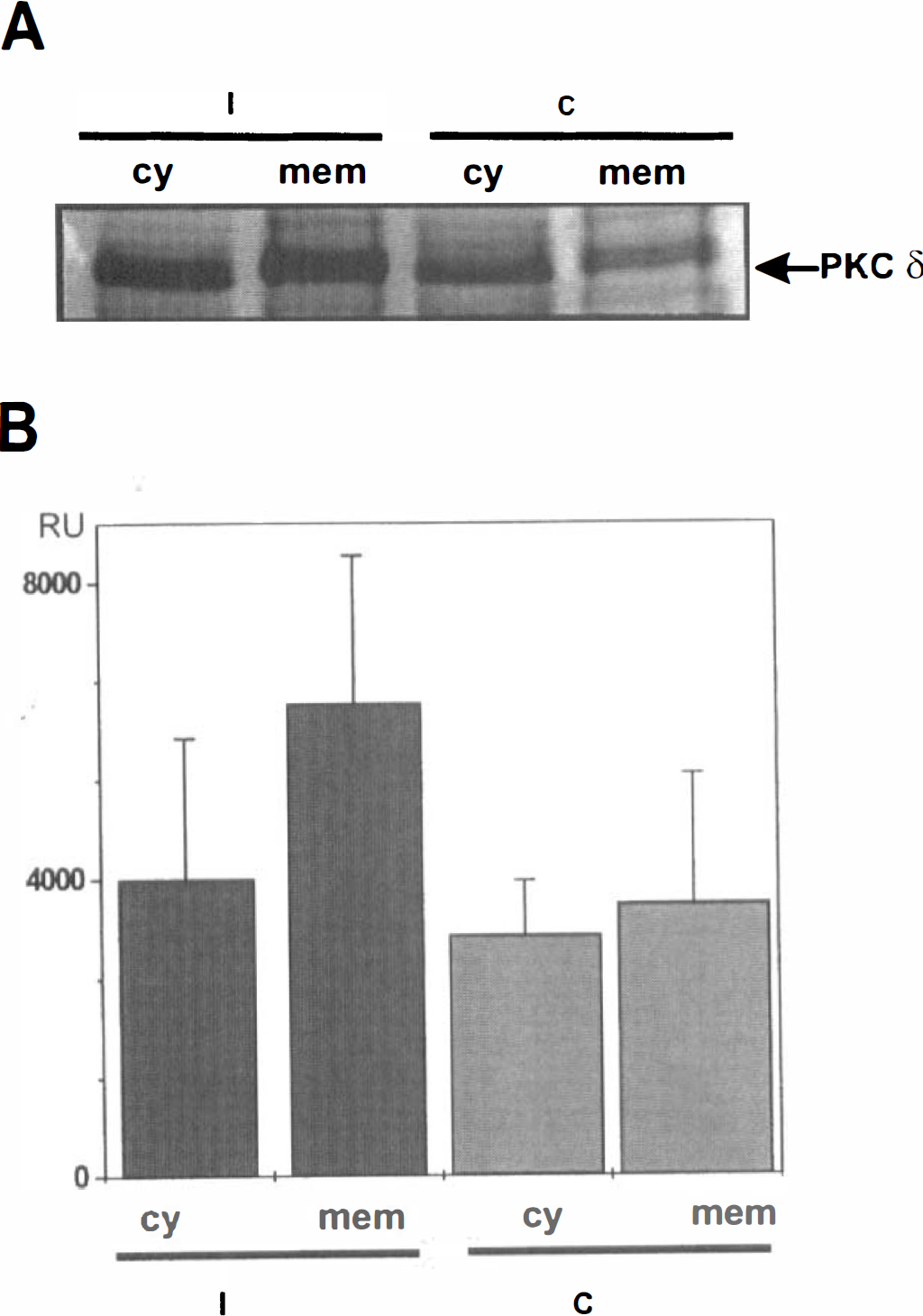
Up-regulation of protein kinase Cδ (PKCδ) in rat brain on ischemia.
DISCUSSION
The main findings of this study were that of the multiple subspecies of the PKC family, only the PKCδ isoform is up-regulated in the vulnerable regions of the rat brain after transient global brain ischemia and that AMPA receptors and glucocorticoids regulate this specific induction in the cortex but not in the hippocampus. The PKCδ induction in neurons precedes neuronal death in the CA1 area, and it is also found in the activated microglia present in the CA1 region at the time the delayed neuronal death takes place (Pulsinelli et al., 1982).
Although many studies have suggested that activation of PKC is involved in neuronal cell death (Hara et al., 1990; Mattson, 1991; Kharlamov et al., 1993), loss of PKC activity is also an essential step in excitotoxic cell death (Durkin et al., 1997). The controversy of the previous studies is most likely due to different activation pathways and roles of each PKC subspecies in neuronal survival. The fact that neither dexamethasone, which is thought to enhance ischemic death of CA1 neurons (Koide et al., 1986), nor NBQX, which provides protection against ischemic death of the CA1 neurons (Small and Buchan, 1997), altered the early expression of PKCδ mRNA in the present study indicates that PKCδ induction in these neurons does not correlate with neuronal survival. Instead, the late peak in PKCδ expression with an association with membrane translocation was localized in activated microglia in the areas of delayed neuronal death. The role of microglial activation in delayed neuronal death is not clear, but activated microglia are involved in excitotoxicity and inflammation, both of which are components of ischemia pathogenesis (Rogove and Tsirka, 1998; Dirnagl et al., 1999).
To our knowledge, there is no previous study available on expression of the various PKC subspecies after transient global ischemia in the rat brain. The induction of PKCδ and -ζ mRNAs takes place in the gerbil forebrain 6 and 24 hours after 10 minutes of global brain ischemia (Savithiry and Kumar, 1994). In the same ischemic model, PKCγ mRNA is transiently up-regulated at 3 hours after 5 minutes of forebrain ischemia, while no significant changes in the amounts of PKCα and -βI mRNA occur (Zablocka et al., 1998). In the present study, most of the PKC mRNAs were eventually decreased in the CA1 area, most likely as a consequence of local neuronal degeneration. Because most of the PKC subspecies are expressed in the neurons (Huang et al., 1988; Hosoda et al., 1989; Saito et al., 1989; Ito et al., 1990), the decline in the expression of PKC isoforms after transient forebrain ischemia may be secondary to neuronal cell death. The remaining expression level of PKCα may therefore occur in either nonneuronal cells (Sieber et al., 1998) or nonvulnerable CA1 neurons.
The expression of Ca-dependent PKC isoforms was not changed in the cortex at any postischemic time points, whereas the expression of the PKCα mRNA was lower at 24 hours after ischemia in the CA3 area and the mRNA levels of PKCβ and -γ and the Ca-independent isoforms PKCϵ and -ζ were gradually decreased in the CA1 after ischemia. Wieloch et al. (1991) reported that PKCβII and -γ but not PKCα isoforms are redistributed from the cytosol to cell membranes and down-regulated in the striatal samples from rats subjected to the two-vessel occlusion plus hypotension model for 15 minutes, whereas 10 minutes of ischemia in gerbils was shown to cause a transient increase in PKCα and -γ protein expression at 4 hours of reperfusion in CA1 (Yokota et al., 1992). In addition, considering that rapid loss of total PKC activity after ischemic insult has been reported (Crumrine et al., 1990; Louis et al., 1991; Cardell and Wieloch, 1993; Busto et al., 1994), variation of the models and animals used in the expression studies of PKC subspecies may be responsible for the differences between the reported data. Nevertheless, the results suggest that if the Ca-dependent subspecies do play a role in ischemic cell death, it is more likely due to reduced than enhanced enzyme activity of these isoforms. Because the changes in the expression of classic PKCs took place only at the time when cell death was already proceeding, it is conceivable that Ca-dependent subspecies do not have an active role in ischemic neuronal death.
The compounds used to study NMDA- and AMPA-type glutamate receptors and glucocorticoids in this study did not influence the physiological parameters, thus not affecting the differences in the expression level of PKC isoforms. The expression of PKCδ was decreased significantly by the AMPA receptor antagonist NBQX in the cortical but not in the hippocampal CA1 region. In addition, NBQX reduced hippocampal expression of PKCα mRNA. Even though we cannot exclude the possibility that the effects of NBQX are at least partially due to decreased brain temperature (Nurse and Corbett, 1996), this is unlikely, because the influence of NBQX on the expression of other PKC subspecies was not consistent. Treatment with dexamethasone inhibited the induction of PKCδ mRNA in the cortex without affecting other PKC subspecies, whereas the NMDA receptor antagonist MK-801 had no effect on PKCδ expression but reduced the expression of PKCβ mRNA in the cortex. The effect of MK-801 on PKCδ after global brain ischemia is different from that in our previous report in which it inhibited the PKCδ induction after focal brain ischemia in the rat (Miettinen et al., 1996). In the focal brain ischemia model, several gene inductions may be caused by cortical spreading depression-like depolarizations and not by the ischemia-reperfusion phenomenon itself (Koistinaho and Hökfelt, 1997). The difference in the ability of MK-801 to inhibit PKCδ induction after focal ischemia may therefore be due to the blocked cortical spreading depression (Iijima et al., 1992).
In normal physiological conditions, PKCδ expression is found exclusively in neurons in the rat brain. In primary cultures, rat microglia have been reported to express PKCα, -δ, and ϵ isoforms (Nakai et al., 1999), whereas in the normal brain in vivo, no PKC immunoreactivity has been detected in these cells. Protein kinase C activation increases intracellular Ca2+ levels (Yoo et al., 1996) and mediates cytokine-induced superoxide production (Chao et al., 1995) in cultured human microglia, increases synthesis of nitric oxide (Yoon et al., 1994), and triggers microglia-induced neurotoxicity (McMillian et al., 1997) in rodent microglia. Whether PKCδ induction in microglia promotes neuronal cell death in brain ischemia requires further investigation.
Our results show that a damaging insult to the brain tissue is able to induce the PKCδ gene in cells in which it is not present under physiological conditions. Thus, totally new signal transduction pathways may become activated after appropriate challenge and contribute, for example, to inflammatory or neurotoxic responses in microglia.
Footnotes
Acknowledgment
The rat PKCδ cDNA was kindly provided by Profs. Y. Ono and Y. Nishizuka.