
Abstract
Twenty-five years after the discovery of protein kinase C (PKC), the physiologic function of PKC, and especially its role in pathologic conditions, remains a subject of great interest with 30,000 studies published on these aspects. In the cerebral circulation, PKC plays a role in the regulation of myogenic tone by sensitization of myofilaments to calcium. Protein kinase C phosphorylates various ion channels including augmenting voltage-dependent Ca2+ channels and inhibiting K+ channels, which both lead to vessel contraction. These actions of PKC amplify vascular reactivity to different agonists and may be critical in the regulation of cerebral artery tone during vasospasm. Evidence accumulated during at least the last decade suggest that activation of PKC in cerebral vasospasm results in a delayed but prolonged contraction of major arteries after subarachnoid hemorrhage. Most of the experimental results in vitro or in animal models support the view that PKC is involved in cerebral vasospasm. Implication of PKC in cerebral vasospasm helps explain increased arterial narrowing at the signal transduction level and alters current perceptions that the pathophysiology is caused by a combination of multiple receptor activation, hemoglobin toxicity, and damaged neurogenic control. Activation of protein kinase C also interacts with other signaling pathways such as myosin light chain kinase, nitric oxide, intracellular Ca2+, protein tyrosine kinase, and its substrates such as mitogen-activated protein kinase. Even though identifying PKC revolutionized the understanding of cerebral vasospasm, clinical advances are hampered by the lack of clinical trials using selective PKC inhibitors.
The cerebral circulation gathers a number of vasoactive factors during subarachnoid hemorrhage (SAH)-induced vasospasm. Smooth muscle cells of the cerebral circulation have the ability to detect and respond to these extracellular signals during both normal and pathophysiologic conditions. These primary signals—for example, factors released by hemolysate or activated platelets—trigger a variety of secondary signals that act in a coordinated and orchestrated manner to ensure a physiological response—a change in blood vessel diameter in the case of cerebral vasospasm as is the condition being reviewed.
Interaction of blood-borne substances with the cell membrane activates a series of enzymatic activities that cause the donation of phosphate groups to initiate a series of intracellular substrate phosphorylations. This process is mediated by protein kinases, of which the cell possesses a number of varieties that are each unique in their temporal activation, substrate specificity, and intracellular location. Protein phosphorylations take place on one of three amino acid groups on target polypeptide chains—serine, threonine or tyrosine. The phosphate group is derived from the hydrolysis of adenosine triphosphate (ATP). Notably, protein phosphorylations can occur at more than one site of the polypeptide chain, necessitating the participation of more than one kinase, thus promoting the convergence of a number of intracellular signals.
Protein kinase C (PKC) was discovered in 1977 by Nishizuka and colleagues using rat brain extracts (Inoue et al., 1977) and is classed as a serine/threonine kinase. The dependence of kinase activity on ambient Ca2+ concentrations and its residence in the cytoplasmic fraction caused it to be named PKC (although since then Ca2+ -independent isoforms have been described). Binding of effector molecules to membrane receptors results in the activation of phospholipase C (PLC) through a G protein–dependent regulation. Activated PLC hydrolyzes phosphatidylinositol-4, 5-bisphosphate to diacylglycerol (DAG) and inositol-1, 4,5-trisphosphate (IP3). The water-soluble IP3 activates Ca2+ release channels on the sarcoplasmic reticulum. Release of endogenous Ca2+ binds to PKC and exposes the phospholipid binding site, whereby the cytosolically located PKC then translocates to the cell membrane to interact with DAG to fully activate the enzyme. According to recent findings, cytosolic PKC also can bind reversibly to the anionic plasma membrane phospholipids (through its C1A domain) or to phosphatidylserine (through its C1B domain), implying that PKC can be membrane bound without prior activation (Johnson et al., 2000). Isolated smooth muscle PKC has an absolute requirement for phosphatidylserine and DAG, and half-maximal activation occurs with physiologically relevant concentrations (∼0.5 μmol/L) of Ca2+ (Andrea and Walsh, 1992).
CLASSIFICATION OF PROTEIN KINASE C ISOFORMS
For a significant period of time after its discovery, PKC was considered to be a single protein causing a myriad of diverse cellular responses. With the advent of molecular cloning techniques, at least 12 distinct members of mammalian PKC isoforms have been discovered (see Table 1) (Coussens et al., 1987; Horowitz et al., 1996; Morgan and Leinweber, 1998; Meier and King, 2000). Each PKC isoform is a separate gene product, with the exception of PKC2β1 and PKC-β2, which are products of alternative splice variants of the same gene product. The structure of PKC reveals 4 conserved (C1–4) and 5 variable (V1-V5) regions. Isoenzyme differences are largely because of changes in the conserved region. The structure and function of the PKC isoforms is reviewed elsewhere (Mellor and Parker, 1998; Morgan and Leinweber, 1998; Ron and Kazanietz, 1999; Meier and King, 2000). Based on their amino acid profile, the PKC family of isoenzymes can be classified according to the regulatory domain structure and activator needs of individual members, as shown in Table 1 and Fig. 1. The C-terminal portion of the enzyme represents the catalytic domain. The ATP biding site (C3) is strictly conserved among the PKC isoforms and within the various protein kinases (Ruegg and Burgess, 1989). Thus, inhibitors of PKC targeted at the ATP binding site (for example, staurosporine, H-7, and so on) also will effectively inhibit other kinases such as myosin light chain kinase (MLCK) and tyrosine kinase (Table 2) (Hidaka and Hagiwara, 1987; Ruegg and Burgess, 1989; Singer, 1990). The C4 region, representing the catalytic core of the enzyme, also is preserved in the various PKC isoforms and other protein kinases. It is the C4 region that contains the protein kinase domain for the recognition of substrates to be phosphorylated by PKC. The other two conserved regions of PKC are the C1A and C1B areas for DAG binding (separate in conventional PKC [cPKC] and novel PKC [nPKC] but fused in atypical PKC [aPKC]) and the C2 region for Ca2+ and phospholipid binding. However, the novel isoform of PKC contains a C2 site that is unable to bind Ca2+. Pharmacologic agents that target the C1 regulatory domain (for example, calphostin C) are able to inhibit binding to the activator site by phorbol esters or DAG, thus conferring a relative degree of selectivity of such compounds as competitive inhibitors (Table 2). The ability of phorbol esters to activate PKC (Castagna et al., 1982) has been used extensively to investigate postreceptor signaling in the regulation of cell function. An additional utilitarian role of phorbol esters is their ability to cause down-regulation of PKC after prolonged exposure to either isolated cells or intact tissue (Morgan and Leinweber, 1998; Kanashiro and Khalil, 1998).
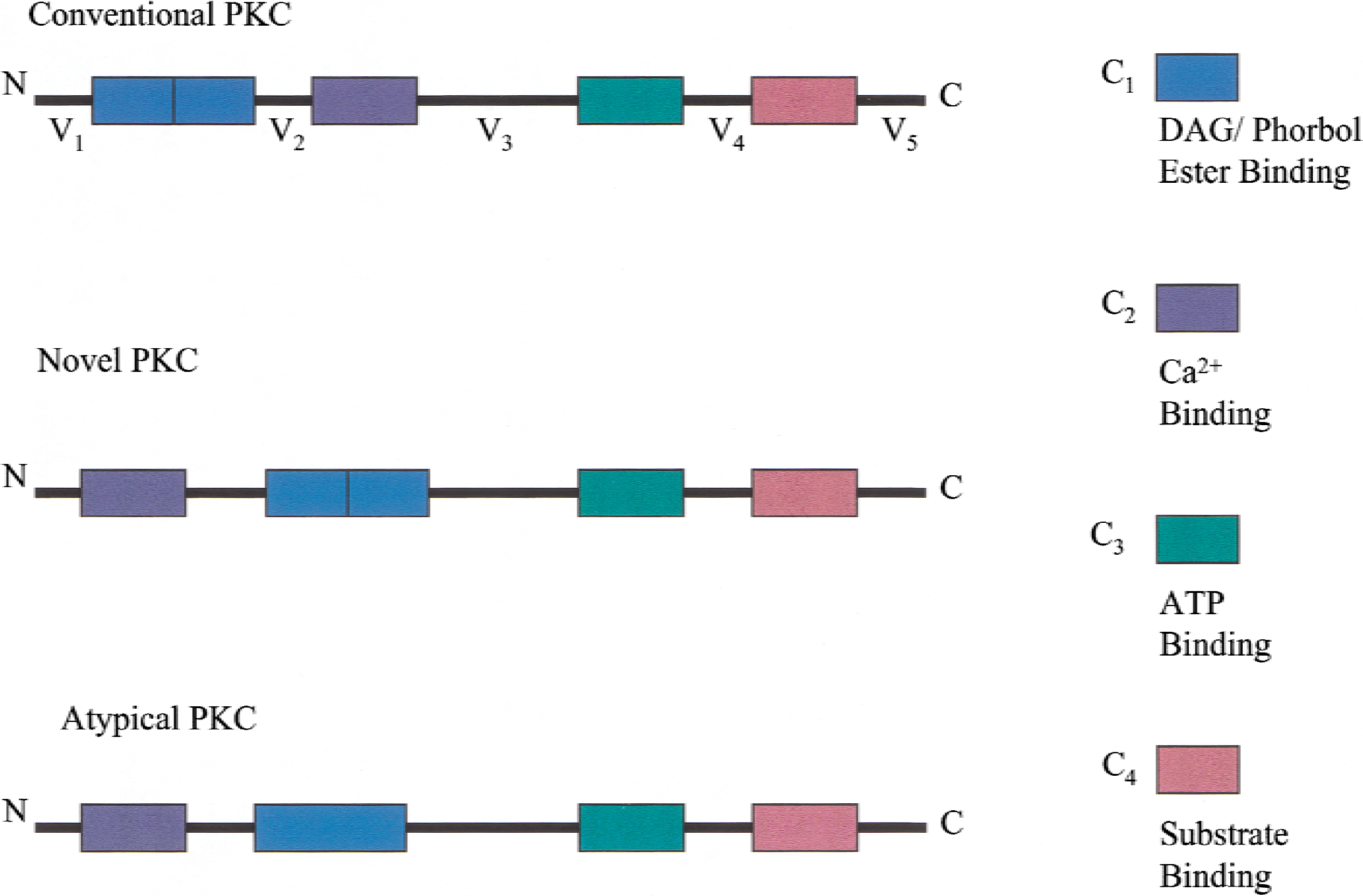
Primary structure of protein kinase C (PKC) isoenzymes showing classification into 3 major groups having 4 conserved (C1–C4 domains) and 5 variable regions (V1–V5 domains). The enzyme is divided into N- (regulatory) and C- (catalytic) terminals. The regulatory domain consists of the C1 and C2 regions; the C1 domain binds DAG and phorbol esters and is the pseudosubstrate region that can inhibit enzymatic activity by binding to the C4 catalytic domain. Conventional PKC and novel PKC contain two regions of phorbol ester binding, whereas atypical PKC contains one region. The C1 region is also the site for calphostin binding. The C2 region represents the Ca2+ binding site in conventional PKC but is absent in novel PKC and atypical PKC. The C1 and C2 regions exist as a single unit in atypical PKC and thus are unable to bind DAG. Between the C2 and C3 regions is the hinge region, a site of cleavage by calpain and trypsin. The C3 and C4 regions are the catalytic domains that bind adenosine triphosphate (ATP) to some inhibitors (H7, staurosporine).
Classification of the 12 different PKC isoenzymes located in mammalian tissue

Three major subclasses have been described, and members of each subclass share similar profiles with regard to regulation of enzymatic activity. PKC-λ is the mouse analogue of PKC-ι. The recently identified PKC-μ is activated by phorbol esters but bears minimal sequence resemblance to other PKC isoenzymes. PKC, protein kinase C; DAG, diacylglycerol.
List of commonly used cell permeable activators and inhibitors of PKC

PKC, protein kinase C; MLCK, myosin light chain kinase; MAPK, mitogen-activated protein kinase; PTK, protein tyrosine kinase.
The PKC protein sequence also contains five variable domains. The V1 region is a pseudosubstrate site representing an autoinhibitory region that binds to the catalytic C4 site in the tertiary protein conformational state of the enzyme. Protein kinase C is normally held inactive because of the interaction with the pseudosubstrate. Upon activation, conformational changes decrease the affinity of the V1 region for the catalytic C4 segment, therefore removing autoinhibition of the enzyme. The functions of the V2, V3, and V4 regions are not established and the V5 region may regulate interactions with other polypeptides (Morgan and Leinweber, 1998).
Separating the catalytic and regulatory domains is the V3 hinge region between C2 and C3 sequences (Fig. 1). The hinge region is a site for endogenous Ca2+ -dependent protease activity, such as that by chymotrypsin and calpain. Cleavage of PKC at the V3 region produces a catalytic form of the enzyme known as PKM (because it is recovered in the membranous fraction). Introduction of the PKM fragment to permeabilized vascular smooth muscle induces a sustained contraction (Collins et al., 1992) without a need for increases in Ca2+ or myosin phosphorylation (Menice et al., 1997). In the later phases of SAH, there is a decrease in PKC accompanied by a corresponding augmented PKM activity in cerebral arteries (Sato et al., 1997).
These isoforms have been grouped into three related families (Table 1), based on their differences in structure, substrate requirements, expression, and localization (Dempsey et al., 2000; Jaken and Parker, 2000; Moscat and Diaz-Meco, 2000). Protein kinase C isoforms are classified according to their regulatory domains as follows:
Conventional PKC: calcium dependent with C1 binding sites for DAG (or phorbol esters) and a C2 binding site for calcium and phospholipid.
Novel PKC: DAG (or phorbol ester)-sensitive but C2 site unable to bind calcium.
Atypical PKC: insensitive to DAG but activated by phosphatidylserine, ceramides, and phorbol ester.
A recent addition is PKD (or PKC-μ) that has unique features relating it to the PKC family of enzymes, but the activator profile and functional significance of this isoform is unknown (Johannes et al., 1994; Hayashi et al., 1999; Matthews et al., 2000). Although PKD enjoys minimal sequence homology with other PKC isoenzymes, it does share some homology in the catalytic domain. PKD binds phorbol esters with high affinity in a phospholipid-dependent manner, but has no Ca2+ requirement (Walsh et al., 1996; Morgan and Leinweber, 1998).
The amount of mature conventional PKC isoforms is largely determined by phosphorylation of the enzymes by PDK-1, a phosphoinositide-dependent kinase 1 (Dutil et al., 1998). Individual PKC isoenzymes translocate in unique patterns upon activation and phosphorylate specific intracellular proteins—making phosphorylation and localization the key determinants of PKC regulation of cell function (Dempsey et al., 2000). Clearly, the amount and activity of PKC and its isoenzymes is cell specific and is regulated by a series of endogenous processes (PDK-1, translocation, and so on), and will largely determine the susceptibility to treatment modalities, as temporal changes in these processes during vascular injury are likely (Dutil et al., 1998).
SUBSTRATES
Activated PKC is a highly promiscuous enzyme, having a number of target proteins for phosphorylations either in vitro or in vivo (Bhalla and Lyengar, 1999). In relation to its role in smooth muscle contraction, activated PKC phosphorylates a variety of proteins involved in excitation–contraction coupling. PKCε binds to actin (possibly both actin isoforms: contractile [α, γ contractile] and cytoskeletal [β, γ cytoskeletal]?) in a persistent manner in neural (Prekeris et al., 1996) and cardiac (Huang et al., 1997) tissue. However, in smooth muscle, this may not be as important because with agonist activation, PKCε preferentially redistributes to the cell membrane rather than to actin-rich areas (either contractile or cytoskeletal) (Morgan and Leinweber, 1998).
Caldesmon is thought to play a critical role in regulating actin–myosin interaction in smooth muscle (Andrea and Walsh, 1992; Horowitz et al., 1996). Caldesmon is an actin and calmodulin binding protein that inhibits actin-activated MgATPase of the excitation-contraction cycle; this thin filament associated protein is found in smooth muscle and nonmuscle tissue (Fig. 2). Caldesmon inhibition of actomyosin ATPase is relieved by phosphorylation—for example, after activation by PKC or protein tyrosine kinase (Fig. 2) (Horowitz et al., 1996). Emerging evidence exists that caldesmon phosphorylation itself may be regulated by mitogen-activated protein kinase (MAPK) (Adam and Hathaway, 1993), a signaling pathway activated by PKC. It is possibly through such a mechanism that smooth muscle can maintain steady-state contraction in the absence of significant myosin phosphorylation or elevation in intracellular Ca2+ (Horowitz et al., 1996; Laporte et al., 1994). However, other reports note significant increases in myosin phosphorylation during PKC-stimulated contraction (Chatterjee and Tejada, 1986; Barany et al., 1992; Rembold and Murphy, 1988a).
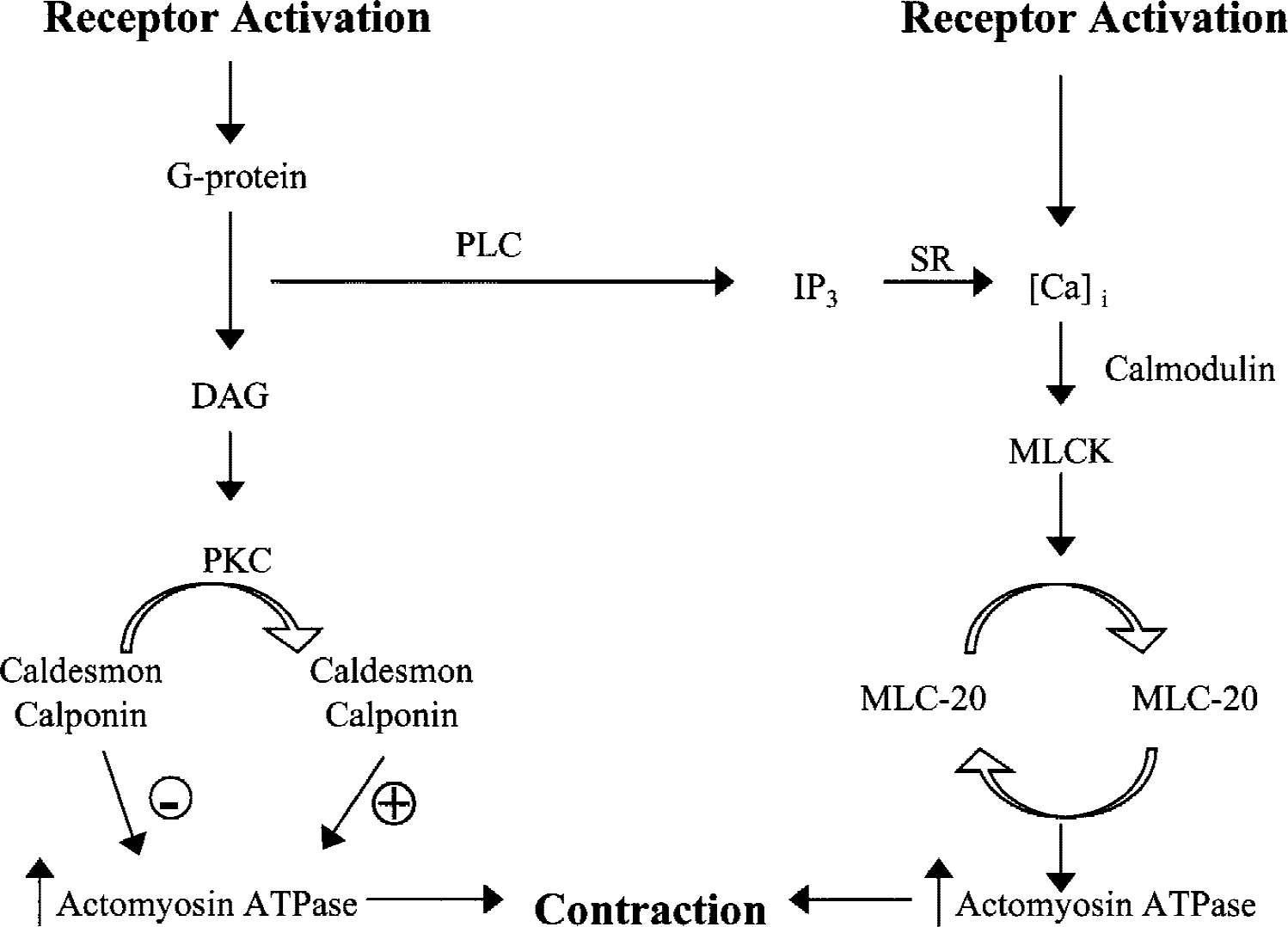
Proposed signaling pathways for vascular contraction. Receptor agonists bind to membrane receptors and activate phospholipase C (PLC) activity through their interaction with G proteins. PLC activity leads to the production of two intracellular messengers—inositol-1, 4,5-trisphosphate (IP3) and diacylglycerol (DAG). After receptor activation, intracellular Ca2+ [Ca2+]I rapidly increase as a result of Ca2+ release from intracellular stores (sarcoplasmic reticulum [SR]) and entry from the extracellular space. Raised intracellular Ca2+ binds to calmodulin, which leads to activation of myosin light chain kinase (MLCK) and subsequent phosphorylation of myosin light chain (MLC-20) and increased actomyosin ATPase activity and contraction. The activation of protein kinase C (PKC) by DAG leads to phosphorylation of thin filament associated proteins (caldesmon/calponin) and disinhibition of actomyosin ATPase, resulting in greater force production in the absence of added Ca2+ entry or light chain phosphorylation. Dephosphorylated caldesmon and calponin have inhibitory actions on actomyosin ATPase.
Although there is strong evidence suggesting caldesmon as an important target protein for PKC phosphorylation during tonic force generation by smooth muscle, a number of other proteins are also implicated (Leinweber et al., 1999). For example, calponin, which also inhibits myosin MgATPase activity, is another contractile protein phosphorylated by PKC in smooth muscle (Winder and Walsh, 1990) (Fig. 2). Calponin is also an actin and calmodulin binding protein but is found exclusively in smooth muscle. The extent of PKC-induced calponin phosphorylation is closely related to the level of vascular tone, suggesting a role for this actin-associated protein in excitation–contraction coupling (Mino et al., 1995).
Myosin light chain phosphatase (MLC20 phosphatase) is crucially important in dephosphorylating myosin light chain, thus leading to smooth muscle relaxation (Somlyo and Somlyo, 1994). Activated PKC deceases MLC20 phosphatase activity (Msuo et al., 1994), thus allowing force production without additional changes in intracellular Ca2+ or myosin phosphorylation patterns. A consensus has recently developed that small G proteins, such as rho and its associated rho-kinase serve to inhibit MLC20 phosphatase activity during normal and pathophysiologic conditions (Fig. 3) (Noda et al., 1995; Uehata et al., 1997).
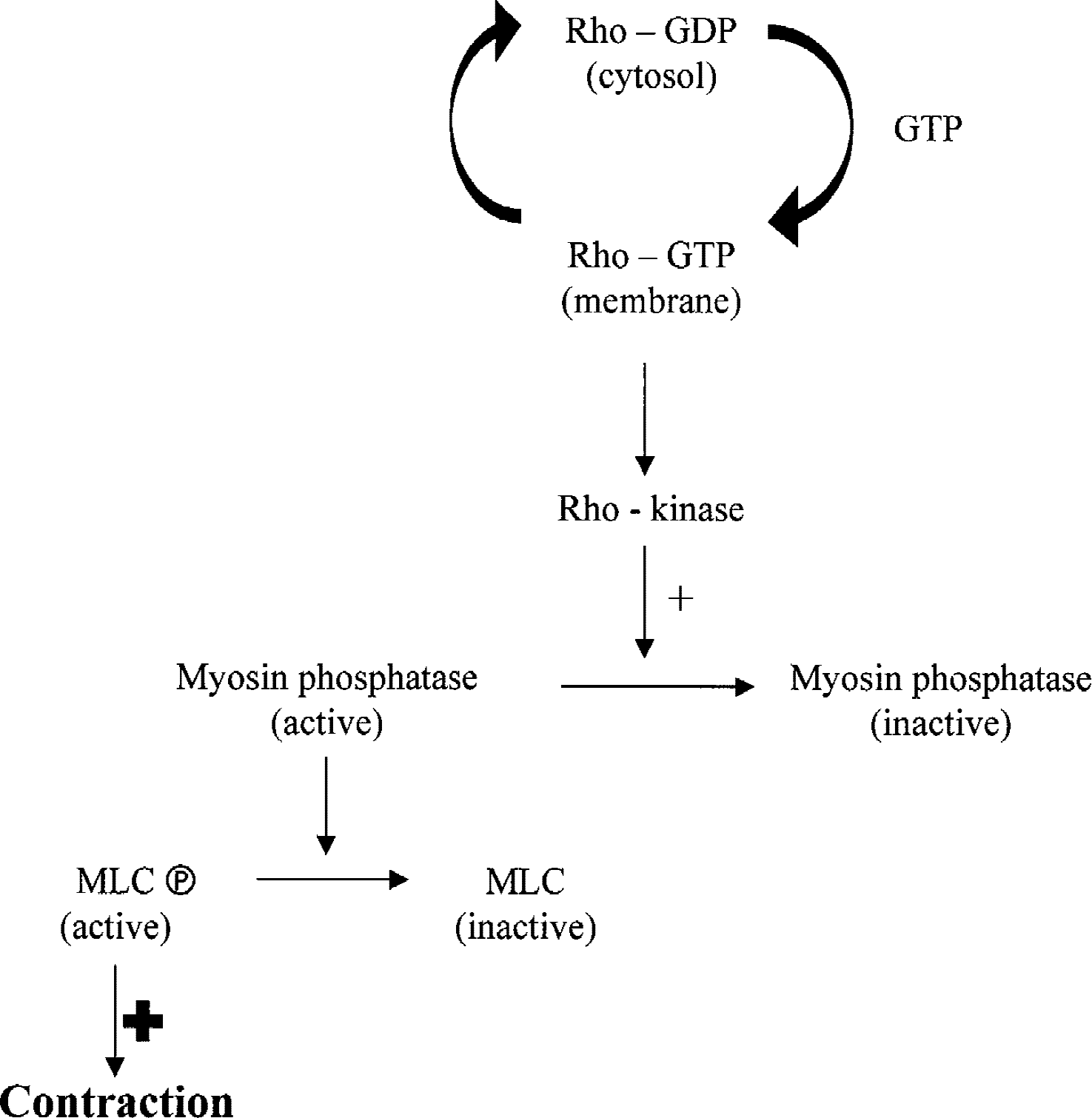
Activated myosin light chain phosphatase causes dephosphorylation of myosin light chain (MLC), leading to relaxation. Receptor-mediated signaling causes protein kinase C (PKC) stimulation, leading to activation of low molecular weight small GTPases such as rho; the activity of rho is regulated by exchange of guanine nucleotides with the rho-GTP form being active at the membrane. Rho-GTP in turn activates rho-kinase that causes sustained phosphorylation of MLC through inhibition of MLC phosphatase activity. This increased phosphorylation of MLC occurs independently of Ca2+ -dependent activation of enzymes such as myosin light chain kinase, leading to the phenomenon of Ca2+ sensitization. GTP, guanosine triphosphate.
Although the evidence implicating PKC in vascular smooth muscle contraction is impressive, the physiologic relevance of the proposed mechanisms remains to be established, especially for the specific roles of the various PKC isoforms. With increasing use of peptide mapping and the known preferences for PKC substrates (basic residues closely situated to Ser/Thr), it is likely that database searches will reveal several additional proteins available for PKC phosphorylation. It is apparent that colocalization of PKC and substrate in the cell is crucial for physiologic relevance.
DRUG ACTION
After its discovery nearly a quarter of a century ago, a number of pharmacologic tools have been developed with the intention of either activating or inhibiting PKC. More recent efforts focus on isoform-specific ligands but have had limited success, because peptide fragments that are relatively specific inhibitors of the various isoforms lack utility during physiologic conditions because of restricted membrane permeability and limited cytoplasmic availability (Csukai and Mochley-Rosen, 1999; Meier and King, 2000). Table 2 summarizes the properties of more commonly used ligands for PKC. The specificity and chemical nature of many commonly used cell permeable protein kinase inhibitors has been documented in considerable detail by Davies et al. (2000). It is estimated that there are possibly 2,000 kinases encoded by the human genome, and that each kinase is likely to phosphorylate 15 proteins (Cohen, 1999). Because most protein kinases belong to the same superfamily, this would add to the burden of targeting small cell-permeable inhibitors to specific kinase (Cohen, 1999; Davies et al., 2000).
Calphostin C has an IC50 of 50 nmol/L and is at least 100 times more selective for PKC than other kinases. It has limited clinical use because it is not isoenzyme specific, it requires light for activation, and it covalently modifies the enzyme (Mochley-Rosen and Kauvar, 1998). Agents such as H7 and staurosporine target the ATP binding site, and thus lack specificity within the kinase family; they can also inhibit voltage-gated Ca2+ channels in vascular smooth muscle. Staurosporine inhibits both serine/threonine and tyrosine protein kinases, and the rank order for inhibition is phosphorylase kinase > PKC > cyclic adenosine monophosphate (cAMP)-dependent kinase, MLCK (Table 2).
The most promising inhibitors of PKC isoforms act at the catalytic domain for ATP binding (Way et al., 2000). Although the selectivity for PKC is poor, the structure of staurosporine has lent itself for the development of two classes of “isoform selective” inhibitors: indolocarbazole (for example, CGP 41251 and UCN 01) and bisindolocarbazole (for example, Ro317208, Ro 320432, LY 33531) inhibitors. These inhibitors reduce activity of both cPKC and nPKC isoforms equipotently with minimal evidence for isoform specificity but with the advantage of selectivity for PKC inhibition that than other kinases (Way et al., 2000). The most promising lead compound appears to be LY 33531, which shows impressive selectivity (60-to 70-fold) for PKCβ isoforms compared with PKCα (Way et al., 2000) (Table 2). Of the proposed selective inhibitors reported to date, LY 33351 has been used most extensively in the study of the role of PKC isoforms in diseases such as diabetes, cardiac hypertrophy, cardiac failure, and retinal angiogenesis (Way et al., 2000).
Another useful approach is inhibition of PKC isoform translocation, which derives from the fact that each activated isoform anchors to specific proteins within the cell; receptors for activated proteins (RACKs) is one such anchoring protein (Csukai and Mochly-Rosen, 1999; Way et al., 2000). The interaction of PKC and RACK is isoform selective and is largely mediated by the C2 region of cPKC, with the result being that peptide fragments of this domain have been developed as selective inhibitory compounds (Csukai and Mochly-Rosen, 1999; Mochly-Rosen and Kavar, 1998 Mochly-Rosen and Kavar, 2000). Peptide translocation inhibitors of PKCε (εV1 and εV1–2), targeting the V1 domain, have been used to demonstrate an important role for PKCε activation in ischemic preconditioning in rat and rabbit cardiomyocytes (Way et al., 2000), leading to the possibility of using activators of PKCε (for example, Pseudo ε RACK) in the therapy of myocardial ischemia. The role of PKC isoforms in cerebral ischemia and SAH-induced vasospasm currently is being investigated intensely by many laboratories.
SMOOTH MUSCLE CONTRACTION
It has been known for some time that smooth muscle is able to generate considerably greater force than striated muscle, given that the smooth muscle content of myosin is 80% less (Murphy et al., 1974). Thus, smooth muscle myosin is capable of generating five times more force than skeletal muscle, given the myosin content of the two muscle types (vanBuren et al., 1994). The duration of interaction of myosin with actin is similar in the two muscle types (Harris and Warshaw, 1993).
Phosphorylation of the 20-kDa myosin light chain (MLC20) by Ca2+ -dependent activation of MLCK is universally accepted as the key pathway for vascular contraction. In many cases though, MLC20 phosphorylation and cytoplasmic Ca2+ levels decline to near resting values, whereas peak force production is maintained. Receptor activation leading to PIP2 hydrolysis and PLC activation leads to vascular contraction having a lower Ca2+ contraction compared with receptor-independent contraction (Mulvany, 1996). These and similar findings led many to suggest that another important pathway is involved in receptor-induced vascular contraction (Fig. 2).
Upon receptor activation, DAG causes the translocation of PKC to the cell membrane (Haller et al., 1990, 1998), where it presumably phosphorylates a variety of target proteins. The early increases in MLC20 phosphorylation and Ca2+ are transient, followed by more sustained phophorylations of several proteins including caldesmon, desmin, and others (Takuwa et al., 1988; Rembold and Murphy, 1998b). Levels of phosphatases (1 and 2A) are decreased in a rabbit model of SAH (Fukami et al., 1995) and thus may result in extended phosphorylation of MLCK, caldesmon, and calponin.
Phosphorylation of contractile proteins during receptor-induced vascular smooth muscle contraction causes intracellular sensitization to Ca2+, thus allowing maintained force production with otherwise ineffective concentrations of Ca2+ (Nishimura and van Breemen, 1989). Similar findings also have been made with phorbol esters, potent activators of PKC (Laher et al., 1989; Masuo et al., 1994). These effects are currently proposed to be mediated by rho-kinase inhibition of MLC phosphatase activity (Kitazawa et al., 1991; Hirata et al., 1992) (see Fig. 3). Agonist stimulation of vascular smooth muscle activates heterometric G proteins (for example, linked to PLC) and monomeric low molecular weight small GTPases such as the rho family of proteins. Inactive rho is located in the cytosol and is bound to guanosine diphosphate. Activation causes guanosine diphosphate to be exchanged for GTP, leading to stimulation of rho-kinase and translocation to the plasma membrane. Rho-kinase maintains MLC phosphorylation by inhibiting the activity of MLC phosphatase (Nagumo et al., 2000) (Fig. 3). Inhibitors of rho-kinase such as Y-27632 have been used to elucidate the extent of Ca2+ sensitization in hypertension (Uehata et al., 1997) and coronary artery spasm (Kandabashi et al., 2000). An important recent finding is that rho-kinase modulates the expression of preproendothelin, thereby contributing to the genesis of a number of vascular diseases (Hernandez-Perera et al., 2000). The role of rho/rho-kinase in Ca2+ sensitization and maintained force production is described in detail by Fukata et al. (2001).
CEREBRAL ARTERY TONE
The cerebral circulation is able to maintain a relatively constant blood flow over a range of arterial pressures (Busija and Heistad, 1984). This autoregulatory capacity of the circulation to the brain is largely because of the intrinsic tone of large and small arteries. It is against this intrinsic tone that other vasoactive factors—for example, metabolic, neural, endothelial, and so on—act to regulate arterial diameter and hence local blood flow. Thus, an understanding of the mechanisms regulating intrinsic tone is important, as intrinsic tone is likely exacerbated in disease states such as in cerebral vasospasm. The intracellular signaling pathways that regulate cerebral arterial diameter are summarized by Harder and collaborators (Harder et al., 1998).
It was Bayliss (1902) who first observed that a distending mechanical stimulus to the arterial wall is able to elicit vascular contraction, a response known as the myogenic response because it occurs independently of neural, metabolic, or endothelial factors (Bevan and Laher, 1991). Raised intravascular pressure causes a membrane depolarization (Harder, 1984; Knot and Nelson, 1998), allowing the entry of Ca2+ through voltage-gated, L-type, Ca2+ channels that are sensitive to Ca2+ antagonists such as nifedipine and nimodipine (Brayden and Nelson, 1992; McCarron et al., 1997). The myogenic response in cerebral arteries has a lower Ca2+ requirement than other forms of tone (Gokina and Osol, 1998; Gokina et al., 1999), consistent with findings previously made in other blood vessels (Laher and Bevan, 1989; D'Angelo et al., 1997).
The central role of PKC in myogenic regulation of blood vessel tone was first reported by Laher and Bevan in 1986. Subsequent reports extended this to include rat cerebral (Osol et al., 1991), mesenteric (Karibe et al., 1997), skeletal (Hill et al., 1990), and human coronary (Miller et al., 1997) arteries. Thus, it is proposed that raised intravascular pressure acts on a mechanoreceptor to stimulate PLC activity leading to DAG activation of PKC, resulting in increased cerebral artery tone (Osol et al., 1993) that is related to intracellular Ca2+ sensitization of myofilaments (Gokina and Osol, 1998). The role of PKC in the physiologic regulation of cerebral artery tone appears to be firmly established (Bevan and Laher, 1991; Schubert and Mulvany, 1999), and it is becoming increasingly clear that PKC may play a dominant role in pathophysiologic regulation of cerebral artery tone during SAH-induced vasospasm (Matsui et al., 1991a, 1991b; Nishizawa et al., 1992a, 1992b). Thus, intrinsic myogenic tone of cerebral arteries in a canine model of double SAH is elevated 3.7 times compared with control. In similar conditions, the contribution of passive elements—matrix proteins, collagen deposition, wall fibrosis, myofibroblast infiltrates, and so on—is elevated by 1.7 times in SAH (Butler et al., 1996).
Activation of PKC either by phorbol esters or by physiologic activators such as DAG produces a sustained contraction in normal dog cerebral arteries (Nishizawa et al., 1990). In basilar arteries of beagles, phorbol 12, 13-diacetate (PDA, a synthetic PKC activator) produces a slowly developing but sustained contraction that is unaffected by a variety of neurotransmitter antagonists such as atropine, methysergide, diphenhydramine, phentolamine, and indomethacin. However, 4β-phorbol, a phorbol without PKC-activating properties, induced only weak contractions. Protein kinase C inhibitors such as staurosporine and H-7 abolished the contractile effect of PDA (Sugawa et al., 1991). Similar effects also occurred in cerebral arteries from other species—for example, bovine cerebral arteries using PDA and 1-oleoyl-2-acetyl-sn-glycerol (OAG, an activator of DAG) (Watanabe et al., 1998)-–and in canine basilar arteries using phorbol 12, 13-dibutyrate (PDBu) (Nishizawa et al., 1996).
In addition to the prolonged contractile effect of PKC in cerebral arteries, there are two other distinguishing features of PKC-induced contraction. First, PKC activity is more pronounced in cerebral arteries because PDBu produces larger contractile effects in rabbit basilar arteries than in coronary, femoral, and mesenteric arteries (Minato et al., 1997). Second, sizeable contractile effects occur to PKC in cerebral arteries bathed in low concentrations of extracellular Ca2+ (Sugawa et al., 1991).
The effects of PKC in regulating Ca2+ sensitivity through phosphorylation of myofilament proteins are widely recognized; recent efforts have focused on other aspects of PKC in the modulation of vascular tone. Through phosphorylation of various ion channels, PKC is able to ultimately increase the extent of contraction (Shearman et al., 1989). Formation of active PKC augments dihydropyridine-sensitive, Ca2+ conductance in several vascular smooth muscle preparations (Fish et al., 1988; Galizzi et al., 1987). Augmentation of Ca2+ influx by PKC is inextricably linked to inhibition of a variety of K channels, leading to membrane depolarization of vascular smooth muscle (Bonev and Nelson, 1996; Aiello et al., 1996; Minami et al., 1993). K+ channel activity is highly regulated by local Ca2+ gradients beneath the plasma membrane (Bullbring and Tomita, 1987). These micodomains of Ca2+ can result from the regulated Ca2+ uptake by the superficially located sarcoplasmic reticulum as proposed by van Bremen et al. (1995) and are shown to occur in cerebral arteries (Asano et al., 1998).
The advent of recent imaging technology allowed the exciting discovery of another means of generation of local Ca2+ gradients—through the spontaneous release of “Ca2+ sparks” from the sarcoplasmic reticulum, first described in cerebral arteries (see Jaggar et al., 2000 for a comprehensive review on Ca2+ sparks). These Ca2+ sparks serve as feedback regulators of outward K+ currents (through Ca2+ -activated K+ channels), making feasible the intriguing possibility of elevations in local Ca2+ concentrations leading to vasodilation (Jaggar et al., 2000). Activation of PKC leads to inhibition of Ca2+ -activated K+ channels (Minami et al., 1993) and Ca2+ spark frequency (Bonev et al., 1997) in coronary and cerebral arteries, respectively. It is unclear if the depolarization recorded in vasospastic cerebral arteries (Harder et al., 1987) is related to decreased Ca2+ spark activity, for example, subsequent to PKC activation by hemolytic products.
Vasospasm is characterized by a delayed onset and a prolonged vasoconstriction lasting several weeks, suggesting that the contractile profile of spasmogens in normal cerebral arteries may offer limited representation of the arterial environment in SAH, which is inundated by hemolytic products for days. Testing the effect of PKC in spastic arteries from an animal model is a necessary and important second step. Matsui et al. (1991b) found that the spontaneous tonus in spastic basilar artery (harvested from a beagle double hemorrhage model of experimental SAH on day 7) showed a greater sensitivity to H-7, a PKC inhibitor, than to W-7, a Ca2+ /calmodulin inhibitor, which indicated augmented PKC activity in chronic vasospasm (Matsui et al., 1991b). Nishizawa et al. (1996) reported greater PKC regulation of resting tension of spastic canine basilar artery than that in control animals.
The hypersensitivity of spastic arteries to different agonists is related to a general, nonspecific “amplifying” role of PKC in vascular sensitivity. In rabbit basilar artery, the constrictor effect of thrombin was augmented by collagen-activated human platelets and this augmentation was abolished by pretreatment of arteries with staurosporine, a PKC inhibitor (Germann et al., 1991). The enhanced nonspecific sensitivity of cerebral arteries in SAH to a variety of constricting agents thus may be modulated by PKC (Laher et al., 1989, 1990) and more depolarized vascular smooth muscle cells in the media of spastic arteries (Waters and Harder, 1987). Cerebral arteries in a rodent model of SAH have an enhanced responsiveness to ET (Alafaci et al., 1990). The endothelial peptide ET augments the responses to a number of vasoconstrictors by increasing the Ca2+ sensitivity of contractile proteins, for example, in coronary and cerebral arteries (Tanaka et al., 1995; Obara et al., 1999). Interestingly, enhanced vascular tone occurs in the absence of greater MLC phosphorylation, and the resultant enhanced vascular response is only partially inhibited by Ca2+ channel inhibitors but is completely sensitive to calphostin, H-7, and staurosporine inhibitors of PKC (Obara et al., 1999; Nakayama et al., 1991). Oxyhemoglobin is a potent releaser of ET in cerebral arteries; both the release of ET and induction of mRNA for ET-1 are mediated by PKC (Kasuya et al., 1993). Whether myosin phosphatase inhibition by rho-kinase modulates the enhanced contractile activity observed in SAH (for example, after endothelin release) is currently unknown.
PROTEIN KINASE C IN CEREBRAL VASOSPASM
Even though cerebral vasospasm was first described 50 years ago, knowledge of its pathogenesis and clinical ischemic neurologic deficits was not accelerated until the 1970s. Appreciation of the etiology of SAH, the application of early intervention, the use of computer-assisted tomography and positron emission tomography have all contributed to the authors' understanding of cerebral vasospasm. The constituents of a subarachnoid blood clot, especially oxyhemoglobin, are the principal pathogenetic agents.
Oxyhemoglobin releases free radicals, initiates and propagates lipid peroxidation, releases vasoactive agents, produces perivascular nerve damage, and induces structural damage in the arterial wall (Zhang et al., 1998). Other vasoactive compounds from blood clots, from cerebrospinal fluid, and from the arterial wall also may be involved. Early studies focused on eicosanoids, free radicals, 5-HT, catecholamines, histamine, leucotrienes, and so on, and resulted in variable outcomes in different experimental models. A transformation from these inconsistent, multiple receptor activation, cytotoxicity, neurogenic-vascular control mechanisms to a simple but unifying theory that describes the ability of smooth muscle to maintain prolonged contraction is needed. A major advance occurred in the 1980s with the frenzied study of the role of PKC in arterial contraction.
The possible role of PKC in cerebral vasospasm was initially proposed in the late 1980s to early 1990s when it was observed that activation of PKC induced a potent and long-lasting contraction of canine basilar arteries (Nishizawa et al., 1990; Asano et al., 1990; Sugawa et al., 1991). The evidence supporting a central role for PKC in cerebral vasospasm is gaining momentum, as summarized in Table 3. An important finding is that the activity of PLC (beta, gamma, delta) is greater in the CSF from patients after SAH; moreover, there is a correlation between PLC activity and the clinical presentation of such patients (Nakashima et al., 1993).
Summary of studies implicating a role for PKC in cerebral vasospasm

Experimental vasospasm was produced in dogs and other animals. Most studies report that protein kinase C (PKC) activities were elevated in the spastic arteries (↑), even though some studies showed that the activity of PKC was reduced (↓) or not changed (↔). The PKC inhibitors staurosporine and H-7 reduced cerebral vasospasm and diacylglycerol (DAG) or PKC levels. VAS is a free radical scavenger. DHM, double hemorrhage model; MLC, myosin light chain.
Protein kinase C inhibitors reversed the vasoconstriction by various agonists in normal or spastic arteries (Matsui et al., 1991b), and attenuated experimental cerebral vasospasm in a beagle double hemorrhage model of SAH (Matsui et al., 1991a). For example, Matsui et al. (1991a) demonstrate that the tonus of vasospastic arteries from a canine double hemorrhage model of SAH was not relieved by inhibitors of calmodulin (W-7 or R25571) or voltage-gated Ca2+ channels; application of PKC inhibitors (H-7 or staurosporine) caused significant vasodilation of these arteries. These initial studies suggested that chronic cerebral vasospasm represented a sustained and pathologic smooth muscle contraction, and paved the way for further examination signaling pathways for PKC in cerebral vasospasm. It is speculated that PKC may play a role as a key factor linking several other signaling pathways such as calmodulin, MLCK, nitric oxide (NO), intracellular Ca2+, protein tyrosine kinase (PTK), or MAPK (Sugawa et al., 1991; Takuwa et al., 1993; Nishizawa et al., 1995; Marton et al., 1996; Kim et al., 2000; Vollrath et al., 1998; Fujikawa et al., 1999; Zubkov et al., 1999), and the summed outcome is a prolonged vasoconstriction.
Three key elements are required to establish the role of PKC in cerebral vasospasm. First, the levels of PKC or its endogenous activator DAG should be elevated during vasospasm. Second, PKC inhibitors should reverse vasospasm. Third, PKC activators should produce angiographic vasospasm and histologic changes in cerebral arteries.
Levels of DAG and protein kinase C
Varying levels of DAG, PKC, or intact PKC in spastic arteries of animal models of SAH are reported in the literature. The time course of PKC activation does not always correlate with the arterial narrowing, suggesting that calpain may be needed in addition to PKC activation.
The level of DAG significantly increases in canine basilar arteries on days 2, 4, and 7, whereas it is unchanged on days 1 and 14 in a canine double hemorrhage model of SAH (Matsui et al., 1991a). A linear correlation between DAG content and angiographic diameter of the basilar artery occurs in this model of SAH. Translocation of PKC has been observed spastic canine basilar arteries harvested 7 days after experimental SAH (Nishizawa et al., 1992a). Protein kinase C precipitation in the membrane fraction in spastic arteries was enhanced compared with that observed in control tissue, where the PKC precipitation in the cytosolic fraction PKC activity was significantly decreased on days 4 and 7, but the membrane fraction was unaltered during this period (Matsui et al., 1993). Cytosolic PKC precipitation in arteries harvested at day 14 returned toward control levels with the remission of vasospasm (Takuwa et al., 1993). Furthermore, even though the PKC content was increased, its elevation did not correlate with the time course of vasospasm (Sako et al., 1993). Membrane-bound PKC increased most 3 times more than control levels on day 4 and returned to baseline values by day 10. Cytosolic PKC decreased significantly from 4 hours to day 14 after SAH. Total content of PKC decreased on day 2 and from days 7 to 14. Thus, when compared with angiographic vasospasm, PKC activation preceded maximum vasospasm (occurring on day 7) by several days, and PKC content normalized whereas vasospasm persisted through days 14 to 21 (Sako et al., 1993).
Severe vasospasm occurs on day 4 of a canine two-hemorrhage model and persisted at least until day 7; the extent of which correlated with angiographic narrowing of cerebral arteries (Nishizawa et al., 2000a). The early phases of vasospasm correlated with changes in the translocation of PKC δ from the cytosol to the membrane, whereas the later phases of vasospasm coincided with the translocation of PKC α (Nishizawa et al., 2000b). Thus, these data make clear that it is necessary to study the temporal and spatial changes in the activities of various PKC during SAH-induced vasospasm, with the goal of identifying the targets of phosphorylation during the genesis of maintained arterial narrowing.
The predominant isoforms of PKC in canine basilar artery, both cytosolic and membrane fractions are PKCα, PKCδ, PKCη, PKCε, and PKCζ (Nishizawa et al., 2000b; Takuwa et al., 1993). PKCγ and PKCδ were not found, and only trace amounts of PKCβ were detected (Takuwa et al., 1993). PKCα, PKCβ, and PKCζ were localized predominantly in the cytosol, whereas a relatively larger amount of PKCε was located in the membrane. In spastic arteries, the cytosolic content of PKCα decreases on day 7 and returns to normal values on day 14. However, membrane PKCα was not altered throughout the experimental period. Levels of PKCε in both cytosolic and membrane fractions are markedly reduced on day 7 and return to control levels by day 14. There were no significant changes of PKCζ in either cytosol or membrane fractions (Takuwa et al., 1993). A possible reason for the discrepancy between these two reports (Sako et al., 1993; Takuwa et al., 1993) may be related to the down-regulation of PKC in spastic arteries by a sustained elevation of DAG (Matsui et al., 1991a). However, the clinical relevance of an early elevation of PKC such as that occurring on day 4 (Sako et al., 1993) with a delayed vasoconstriction on day 7 or beyond needs to be established. The method of monitoring PKC and defining intact PKC (translocation of PKC) also requires attention (Peterson, 1996).
In an attempt to resolve the discordant data reported above, Sato et al. (1997) examined the dissociation of the catalytic domain of PKC (PKM, 45 kDa) in spastic arteries in a canine double hemorrhage model of SAH. Membrane-bound PKC is cleaved at a specific site by calpain to form PKM and then is released into the cytosol to phosphorylate substrates. The regulation of PKM by μ-calpain (a Ca2+ -dependent neutral protease activated continuously during vasospasm) was investigated. In spastic arteries, immunoreactivity of 80 kDa PKCα was decreased in the cytosolic fraction and was increased in the membrane fraction (contrary to the findings of Takuwa et al., 1993), suggesting a translocation of PKCα from the cytosol to the membrane. PKM activity was absent in control basilar arteries but was enhanced in spastic arteries, indicating formation of PKM caused by cleavage of PKCα by μ-calpain. The increase of PKM with time is consistent with the time course of angiographic vasospasm. Calpain is classified into two homologous isoenzymes with different Ca2+ requirements: μ- andmgr;-and m-calpains active at micro-and millimolar concentrations of Ca2+, respectively. Because μ-calpain but not m-calpain was implicated in this study, it is likely that an elevation of [Ca2+]i in the micromolar range occurs in spastic arteries. These findings could explain some early claims that H-7, a potent PKC inhibitor whose binding site is in the catalytic domain, inhibits PKCα and PKM (Minami et al., 1992; Sato et al., 1997). However, it remains to be demonstrated whether a prolonged elevation of [Ca2+]i in spastic arteries may activate μ-calpain and therefore cleave PKCα.
Transient elevation of [Ca2+]i has been documented in isolated cerebral smooth muscle cells (Zhang et al., 1995). The situation is less clear when intact cerebral arteries are studied, with concentrations reported to be at higher (Butler et al., 1996), unchanged (Yamada et al., 1994) or lower (Sakaki et al., 1989) levels than control.
Effect of protein kinase C inhibitors
The use of PKC inhibitors in reversing cerebral vasospasm has yielded inconsistent data. In a canine double hemorrhage model of SAH, Matsui et al. (1991a) demonstrated that the PKC inhibitors staurosporine and H-7 reversed day 7 vasospasm by topical application. However, the preexisting spasm was not ameliorated by local application of inhibitors of neurotransmitter receptors (atropine, methysergide, phentolamine, and diphenhy-dramine), calmodulin, or calcium channels (Matsui et al., 1991a; Minami et al., 1992). H-7 effectively reduces tone in both spastic and normal basilar arteries, whereas calphostin C is less effective in dilating spastic than in normal basilar arteries (Matsui et al., 1991a). However, the relaxant effect of calpeptin (a selective inhibitor of calpain) is more pronounced in spastic arteries. Calpain dissociates PKC into catalytic and regulatory domains by limited proteolysis, and activates PKC irreversibly even in the absence of Ca2+ and phospholipids. This is in keeping with the findings of Sato et al. (1997) confirming that calpain cleavage of PKC into PKM modulates vasospasm in SAH. Calpain also activates MLCK and prolongs the activation of cAMP-dependent protein kinase.
However, PKC inhibitors fail to reverse cerebral vasospasm in a rabbit model of SAH. On day 3 after experimental SAH, topical application of staurosporine produced minimal relaxation in rabbit spastic basilar arteries, whereas cromakalim (a potassium channel activator) completely reversed vasospasm (Zuccarello et al., 1996). Contrary to this are findings by Quan and Sobey (2000) that report a diminished response to inhibitors of potassium channels in a rodent model of SAH. It is likely that experimental conditions (species, and so on) contributed to these differences in the efficacy of PKC inhibitors in reversing SAH.
Sako et al. (1993) mimicked cerebral vasospasm in dogs by intracisternal injection of a phorbol ester (12–0-tetradecanoylphorbol-13-acetate (TP)). TP induced a dose-dependent, slowly developing severe constriction of the basilar artery. A single injection of TP (5 nmol/L in cerebrospinal fluid [CSF]) causes sustained contraction lasting more than 3 days that returns to control conditions by day 7. Preinjection of staurosporine abolished the vasoconstriction caused by TP. A greater concentration of TP (10 nmol/L in CSF) produced more pronounced angiographic narrowing of the basilar artery that led to sudden cardiac arrest and death within 2 hours. Multiple injections of TP (5 nmol/L) produced sustained contractions and elevations of PKC of the basilar artery that persisted for more than 10 days (comparable with vasospasm occurring in a double hemorrhage model of SAH). However, a dissociation of PKC activity and vasospasm is evident when comparing vasospasm induced by TP and double hemorrhage in dogs. In the SAH model, membrane-bound PKC activity increases on day 4 and abates by day 14, whereas angiographic vasospasm peaked on day 7 and persists at a moderate level until day 14. The authors concluded that cerebral vasospasm is unlikely caused by activation of the PKC pathway only.
Histology
Another striking difference between cerebral vasospasm induced by PKC activators and SAH are variances in histologic changes. In a canine model of SAH, the basilar artery (day 10) undergoes marked degenerative changes characterized by myonecrosis (Sako et al., 1993). The vascular endothelial cells have visibly deformed nuclei, cytoplasmic vacuoles, and malformed tight junctions. Smooth muscle cells are characterized by a loss of nuclei and organelles, the appearance of vacuoles and dense bodies, and fragmentation of myofilaments. The stroma is filled with collagen fibers, dense particles, and thick basement membranelike substances. However, in dogs subjected to multiple TP injections and studied on day 10, marked intimal corrugation and deformity of the smooth muscle cells of the basilar artery were shown, but without evidence of myonecrosis. The morphology of endothelial cells after TP injection was not described (Sako et al., 1993).
RELATION OF PROTEIN KINASE C WITH OTHER SIGNALING PATHWAYS
Myosin light chain kinase
There are at least two components in agonist-induced contraction in smooth muscle: a Ca2+ /calmodulin pathway to initiate the response, and a PKC pathway, along with the Ca2+ /calmodulin pathway, to sustain vascular tone (Rasmussen et al., 1987). In cerebral vasospasm, spasmogens are thought to elevate intracellular Ca2+ to form a Ca2+ /calmodulin complex leading to activation of MLCK and the phosphorylation of MLC. However, several early studies failed to demonstrate the participation of Ca2+ /calmodulin pathway in cerebral vasospasm. The role of MLCK in cerebral vasospasm remains uncertain and its relation to PKC activation remains unsettled.
The concentration of calmodulin significantly decreases in spastic arteries in a double hemorrhage model of SAH (Sakaki et al., 1989). In beagle basilar arteries, Sugawa et al. (1991) demonstrated that activation of PKC produced a slowly developing, sustained contraction that was independent of MLC phosphorylation. The relaxant effect of W-7, a calmodulin inhibitor, was markedly diminished in basilar artery segments with severe spasm (harvested from a beagle double hemorrhage model of SAH on day 7), indicating a minimal role for the Ca2+ /calmodulin system in this model of SAH (Matsui et al., 1991b). In a similar canine double hemorrhage model of SAH, the turnover of phospholipids such as phosphatidylinositol (PI), phosphatidylcholine (PC), and phosphatidylethanolamine (PE), the time course of PKC activity, and the phosphorylation of 20 kDa MLC were followed. The phosphorylation of 20 kDa MLC was not augmented in spastic (day 4 or 7) basilar arteries (Matsui et al., 1993; Takuwa et al., 1993). These results suggest that a different mechanism (such as PKC phosphorylation independent of changes of the 20-kDa MLC activation) may modulate arterial constriction during cerebral vasospasm.
The interrelations of the PKC and calmodulin systems also were investigated in vitro using isometric tension recordings (Nishizawa et al., 1992b). It was demonstrated that the tonic contraction of canine basilar artery was PKC dependent, but the initiation of contraction by calmodulin was necessary for the subsequent PKC-dependent tonic contraction. However, calmodulin inhibitors such as trifluoperazine also inhibited PKC-induced contraction, prompting the authors to suggest the use of trifluoperazine in the early stages of cerebral vasospasm as a means of suppressing both the calmodulin and PKC systems (Nishizawa et al., 1992b). The selectivity of trifluoperazine was not validated and its effect was not compared with PKC inhibitors in that study. An earlier study reported that trifluoperazine provided mild prophylactic protection against cerebral vasospasm but with minimal therapeutic value (Peterson et al., 1989).
However, recent studies suggested that MLCK does play a role in the active contraction of spastic arteries. Cerebrospinal fluid from vasospastic SAH patients, but not those without vasospasm, contains an extractable substance that modulates MLC phosphorylation in vitro (Pyne et al., 2000). Butler et al. (1996) reported an elevation of phosphorylation of MLCK accompanied by elevation of [Ca2+]i in their study of canine anterior spinal artery in a double hemorrhage model of SAH. The data reported by Butler et al. (1996) differ from the findings of Takuwa et al. (1993) but are consistent with those reported by Harada et al. (1993). The report of Butler et al. (1996) also supports a previous study that ML-9 (a relatively selective antagonist of MLCK) partially reversed vasospasm in dogs (Kokubu et al., 1989). However, there is evidence against a possible role for MLCK in vasospasm (Sato et al., 1997). For example, PKC-catalyzed phosphorylation of MLCK decreases MLCK activity and may result in relaxation other than contraction (Sato et al., 1997).
The status of thin (actin) and thick (myosin) filament regulation of smooth muscle contraction was studied in a double SAH canine model (Kim et al., 2000). Even though there was a trend for an increase in phosphorylation of the regulatory light chain, the changes were not statistically significant (contrary to a previous report from the same group—Butler et al., 1996). However, significant degradation of calponin was observed in spastic arteries, and a nonspecific kinase inhibitor, HA1077, inhibited vasospasm and calponin degradation.
Protein tyrosine kinase and mitogen-activated protein kinase
Some investigators have suggested that thin filament-associated regulatory proteins (such as caldesmon and calponin) are involved in smooth muscle contraction independent of MLC phosphorylation (see above). Caldesmon and calponin interact with F-actin, tropomyosin, and/or myosin to inhibit actin-activated myosin ATPase (Childs et al., 1992). Activation of caldesmon or calponin by PKC removes the inhibition of actomyosin ATPase and results in contraction. One of the pathways for PKC-induced activation of caldesmon is by activation of MAPK, a dual substrate of serine/threonine and tyrosine kinases (Figs. 3 and 4).
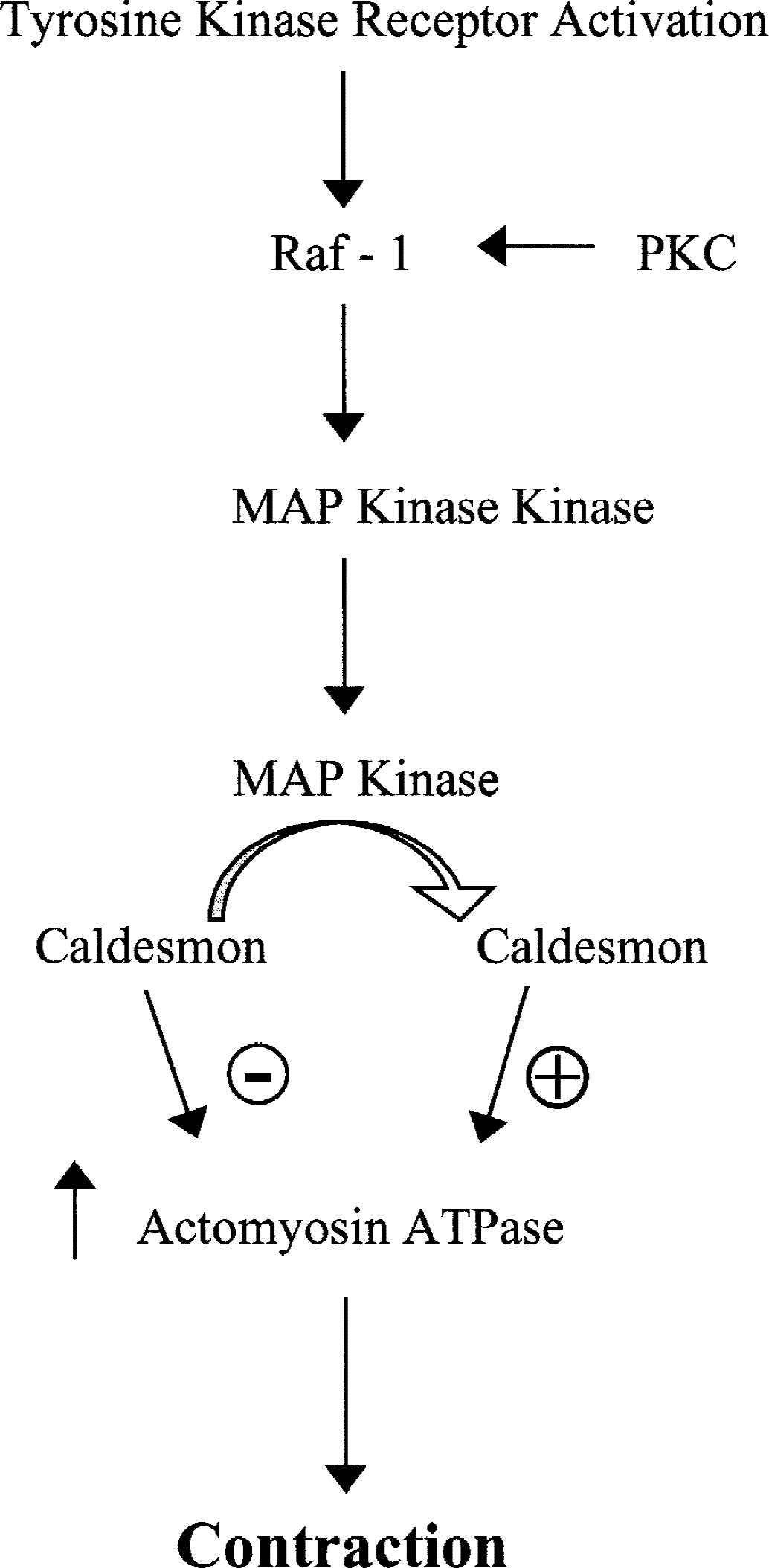
Activation of small GTPases such as Raf-1 occurs by tyrosine kinase receptor activation and protein kinase C (PKC) stimulation. A downstream target for Raf-1 is mitogen-activated protein (MAP) kinase kinase, which activates MAP kinase causing caldesmon phosphorylation, thus relieving its inhibition of actomyosin ATPase and causing vascular contraction. GTP, guanosine triphosphate; ATP, adenosine triphosphate.
Tyrosine kinase activity is important for cell growth and oncogenesis (Hollenberg, 1994) and for the in vivo constriction of cerebral arteries to serotonin (Fig. 4) (Kitazono et al., 1998). The role of PTK and MAPK in cerebral vasospasm is currently being investigated with some vigor (Table 4). The effect of hemolysate and oxyhemoglobin (causative agents for cerebral vasospasm) on tyrosine phosphorylation was studied in cultured endothelial, smooth muscle, and fibroblast cells. Marton et al. (1996) demonstrated that hemolysate induced a transient tyrosine phosphorylation of 66 and 120 kDa proteins in cultured endothelial cells. Removal of external Ca2+ prolonged this effect of hemolysate, indicating tyrosine phosphorylation was involved in hemolysate-induced Ca2+ signaling (especially for Ca2+ entry). Indeed, depletion of intracellular Ca2+ stores by thapsigargin, a Ca2+ pump inhibitor, produced a long-lasting tyrosine phosphorylation of up to 30 minutes. Preincubation of cells with genistein and tyrphostin A23 (two structurally different PTK inhibitors) abolished Ca2+ entry without marked changes of Ca2+ released from internal stores (Marton et al., 1996). Similar findings were reported in cerebral smooth muscle and fibroblast cells by Iwabuchi et al. (1999). Hemolysate activated tyrosine phosphorylation of 70 and 110 kDa proteins in rat basilar smooth muscle cells (Iwabuchi et al., 1999). The PTK inhibitors genistein and tyrphostin A51 reduced both Ca2+ release (from internal stores) and entry (from the extracellular space), thereby suggesting differences in regulation of Ca2+ mobilization by PTK in rat basilar smooth muscle cells as compared with that of bovine endothelial cells (Marton et al., 1996). In cultured canine cerebral smooth muscle cells, oxyhemoglobin induced tyrosine phosphorylation, including c-Src (60 kDa), with a time course that paralleled the contraction to oxyhemoglobin. Genistein abolished tyrosine phosphorylation induced by oxyhemoglobin (Vollrath et al., 1998). In cultured human dermal or canine basilar fibroblasts, hemolysate activates tyrosine phosphorylation of 64 and 120 kDa proteins in a concentration-and time-dependent manner (Patlolla et al., 1999). Tyrosine phosphorylation and lattice compaction induced by hemolysate are attenuated by the PTK inhibitors genistein and tyrphostin A51 (Patlolla et al., 1999).
Role of PTK-MAPK in cerebral vasoconstriction and vasospasm
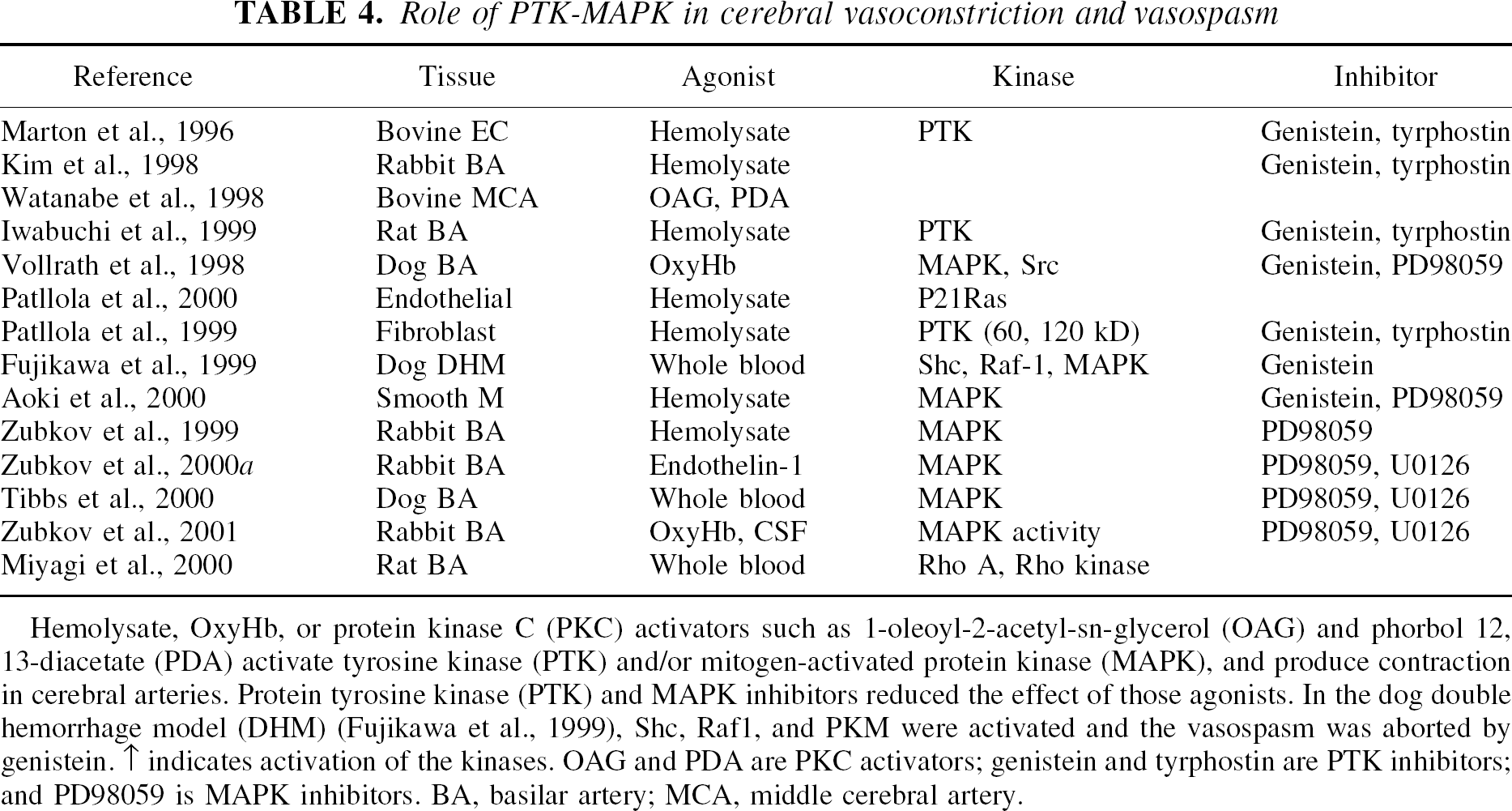
Hemolysate, OxyHb, or protein kinase C (PKC) activators such as 1-oleoyl-2-acetyl-sn-glycerol (OAG) and phorbol 12, 13-diacetate (PDA) activate tyrosine kinase (PTK) and/or mitogen-activated protein kinase (MAPK), and produce contraction in cerebral arteries. Protein tyrosine kinase (PTK) and MAPK inhibitors reduced the effect of those agonists. In the dog double hemorrhage model (DHM) (Fujikawa et al., 1999), Shc, Raf1, and PKM were activated and the vasospasm was aborted by genistein. ↑ indicates activation of the kinases. OAG and PDA are PKC activators; genistein and tyrphostin are PTK inhibitors; and PD98059 is MAPK inhibitors. BA, basilar artery; MCA, middle cerebral artery.
The relaxant effect of PTK inhibitors was studied in bovine arteries using isometric force recordings, where genistein and tyrphostin depressed contractions caused by 5-HT, prostaglandin F2α, endothelin-1, and thromboxane in a concentration-dependent manner (Watanabe et al., 1998). In contrast, pervanadate, an inhibitor of phosphoprotein tyrosine phosphatase, augmented contractions to those agents. PTK inhibitors also significantly depressed contractions induced by activation of PKC, indicating that PTK resides distal to PLC and PKC and is involved in sustained contraction of cerebral arteries (Watanabe et al., 1998). Hemolysate also causes concentration-dependent contractions of rabbit basilar arteries that are sensitive to the PTK inhibitors genistein and tyrphostin A23, but not diadzein (an inactive analogue for genistein), as demonstrated by Kim et al. (1998). Similarly, genistein reversed oxyhemoglobin-induced contraction of canine cerebral arteries (Vollrath et al., 1998).
In a canine double hemorrhage model of SAH, Fujikawa et al. (1999) demonstrated that Shc (an adaptor protein that links Grb2 with Src), Raf1 (a serine kinase that links Ras to MEK), and MAPK were activated in basilar arteries after SAH. Topical application of the PTK inhibitor genistein reversed, in a concentration-dependent manner, both tyrosine phosphorylation and cerebral vasospasm. Genistein also reduced the generation of PKM (a catalytic fragment of PKCα), which was increased in vasospasm. This study also confirmed a previous report from the same group (Sato et al., 1997) for an important role of PKM in vasospasm, and furthermore established the link between PKC, PTK, and MAPK (Fujikawa et al., 1999). Mitogen-activated protein kinase inhibitors were used in a double hemorrhage model in dogs and partially reversed the angiographic vasospasm in major arteries (Tibbs et al., 2000) and histologic vasospasm of penetrating arteries (Zubkov et al., 2000b).
The studies discussed above make clear the role of PTK in the effects of hemolysate or oxyhemoglobin in [Ca2+]i and tension regulation in models of SAH, and more importantly links contraction and proliferation—key features of cerebral vasospasm (Zhang et al., 1998). Mitogen-activated protein kinase, as a dual substrate of serine/threonine and tyrosine kinases, may serve as a “common pathway” to relay signals from the membrane to the nucleus (Figs. 4 and 5). Activation of MAPK induces contraction that is mediated by caldesmon, an actin-binding inhibitory protein. It has been established that MAPK phosphorylates caldesmon in vascular smooth muscle cells (Childs et al., 1992), and the immunoreactivity of caldesmon in cerebral arteries after SAH is markedly decreased (Takenaka et al., 1993); reduced caldesmon levels after SAH may result in prolonged contraction. There is evidence supporting a role for MAPK in signal transduction during cerebral vasospasm. In cultured canine cerebral smooth muscle cells, MAPK (especially Erk2 (42 kDa)) is activated by oxyhemoglobin (Vollrath et al., 1998). The MAPK kinase inhibitor PD98059 abolished this effect of oxyhemoglobin and the contractile action of oxyhemoglobin (Zubkov et al., 1999). Similarly, hemolysate activates MAPK phosphorylation (Erk1 and Erk2) in a concentration-and time-dependent manner in rabbit basilar artery (Zubkov et al., 1999). The contractile effects of hemolysate, oxyhemoglobin, and bloody CSF in rabbit basilar artery are attenuated by a recently described MAPK inhibitor (U0126) (Zubkov et al., 2001). Mitogen-activated protein kinase phosphorylation was confirmed in spastic arteries in a canine double hemorrhage model of SAH (Fujikawa et al., 1999).
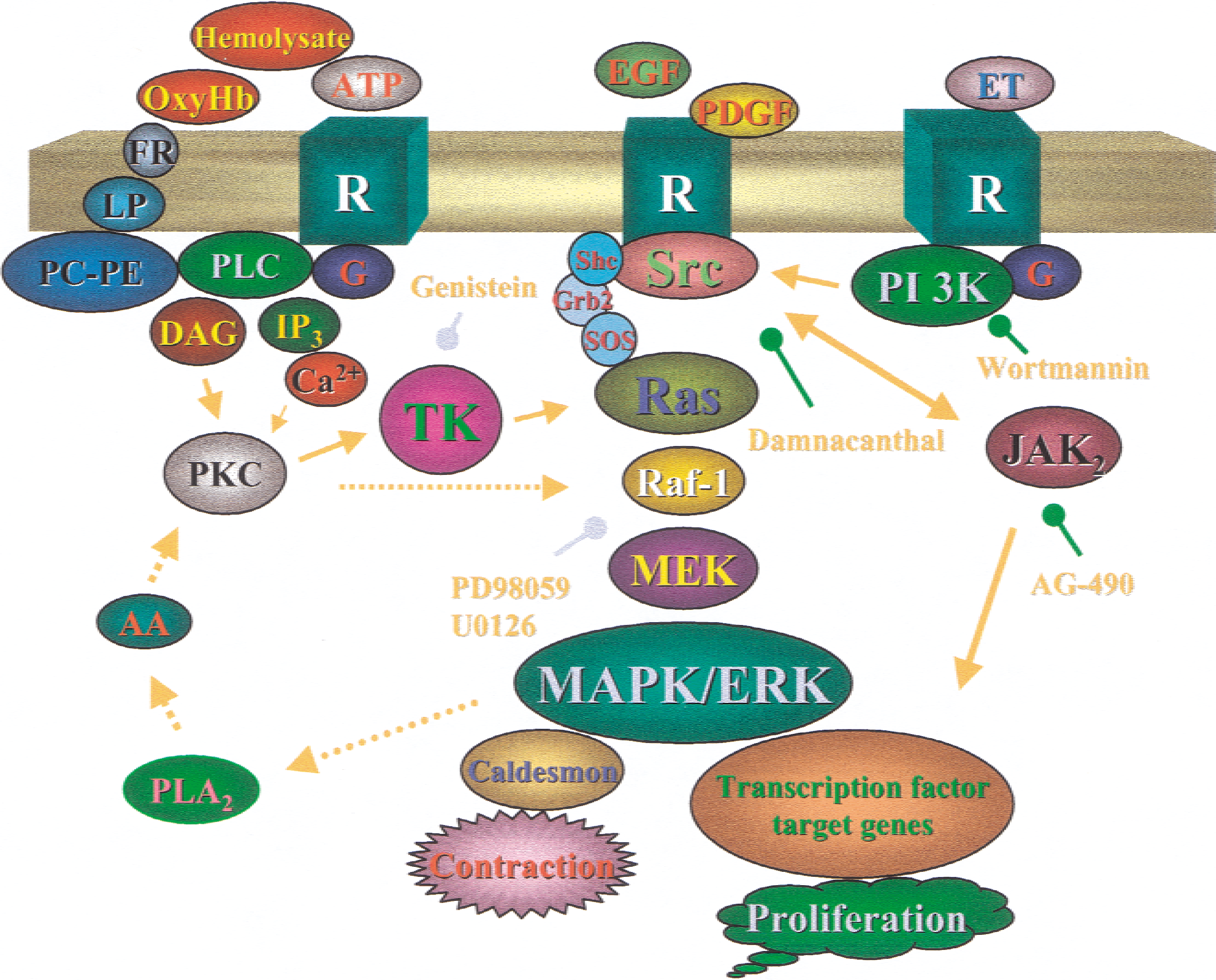
Schematic diagram showing a possible protein kinase C (PKC) network in mediating the signals during cerebral vasospasm. Activation of G protein–coupled receptors (G) by endogenous factors such as endothelin-1, 5HT, or adenosine triphosphate (ATP) leads to activation of phospholipase Cβ (PLCβ), generating inositol-1, 4,5-trisphosphate (IP3) and diacylglycerol (DAG), which are involved in intracellular Ca2+ mobilization and PKC activation, respectively. Ca2+ and PKC activates protein tyrosine kinase (TK), which in turn activates Src or Ras proteins, eventually leading to the activation of mitogen-activated protein kinase (MAPK). Growth factors such as platelet-derived growth factor (PDGF) or epidermal growth factor (EGF) released during vasospasm activate growth factor receptors (receptor tyrosine kinase [RTK]) and lead to Src and Ras activation, assisted with adapter proteins Grb2 or Shc and exchange factor son of sevenless (SOS). Ras stimulates Raf-1, leading to the activation of MAPK. PKC can activate Raf-1 directly. MAPK may activate internal PLA2 that generates arachidonic acid (AA), which activates PKC to enhance the feedback loop. Oxyhemoglobin generates free radicals (FR) and lipid peroxide (LP) and may activate the phosphatidylcholine (PC) and phosphatidylethanolamine (PE) pool to sustain a prolonged elevation of DAG. PKC and elevation of intracellular Ca2+ produce contraction of cerebral arteries. MAPK is involved in tissue proliferation and contraction. Yellow lines indicate signaling pathways. Green lines indicate sites of pharmacologic inhibitor action. Dotted lines represent pathways established in peripheral arteries that need to be confirmed in cerebral arterial smooth muscle.
Intracellular Ca2+
Contraction caused by PKC activation is largely independent of extracellular Ca2+. Activation of PKC by phorbol 12, 13-diacetate (PDA) induces similar maximum contraction in the presence or absence of extracellular Ca2+ in canine basilar arteries, even though the EC50 was approximately 10 times greater in the absence of Ca2+ (Sugawa et al., 1991). The contractile response of spastic arteries to extracellular Ca2+ was decreased, whereas the tone produced by 5-HT, prostaglandin F2α, and PDA was unchanged (Matsui et al., 1991b). The slow-developing sustained contraction induced by PDBu is of a similar magnitude in the presence or absence of external Ca2+ in bovine carotid artery, and is reduced by staurosporine but not by the MLCK inhibitor ML-9 (Whitney et al., 1995). The contraction induced by PDBu in rabbit basilar artery (but not in peripheral arteries) is highly dependent on extracellular Ca2+. Protein kinase C inhibitors such as H-7 and AJ-3941, which also act as Ca2+ channel blockers, reduced PDBu-induced contraction in rabbit basilar artery only in the presence of extracellular Ca2+. Other Ca2+ channel blockers such as diltiazem and nicardipine failed to inhibit the contraction under either condition (Minato et al., 1997).
A component of PKC activation in cerebral arteries is Ca2+ dependent. Under physiologic conditions, DAG is generated from the hydrolysis of phosphatidylinositol (PI) by PLC, a process that does not require an elevation of intracellular Ca2+. However, a sustained elevation of Ca2+ may increase the hydrolysis of PE and PC (phosphatidylcholine) and prolong DAG accumulation. Even though most studies using spasmogens (such as hemolysate) report an elevation of intracellular Ca2+, one in vivo study documents a decreased intracellular Ca2+ level 4 days after SAH compared with resting smooth muscle (Kohno et al., 1991). A likely source for DAG activation is lipid peroxides, because a free radical scavenger (AVS) suppressed DAG content as well as malondialdehyde concentration in CSF (Ohta et al., 1995).
Another aspect of calcium and contraction is the role of calcium sensitization, leading to a calcium-independent contractile component. It is currently thought that Rho A, a small G protein, plays a key role in agonist-induced myofilament calcium sensitization and cytoskeletal organization (Somlyo and Somlyo, 1994). Activation of RhoA reduces calcium requirement during prolonged contraction, by inactivating myosin phosphatase. Using reverse transcription-polymerase chain reaction, investigators demonstrated that RhoA and its kinases are increased in spastic arteries obtained from a rat double hemorrhage model with a time course that parallels development of cerebral vasospasm (Miyagi et al., 2000). In a double hemorrhage model of dogs, Rho-kinase, myosin light chain phosphorylation, and myosin-binding subunits all increased (Sato et al., 2000). The Rho-kinase inhibitor Y-27632 dilated spastic arteries and decreased the phosphorylation of myosin-binding subunits as well as of myosin light chain. Besides Rho, the activity of another small G-protein, p21Ras, increases for an extended time in rabbit basilar arteries incubated with hemolysate (Patlolla et al., 2000). A partially selective inhibitor of Rho-kinase, fasudil, prevented the development of endothelial injury and neutrophil infiltration in a canine double hemorrhage model (Satoh et al., 1999) and improved clinical outcome of patients with cerebral vasospasm (Tachibana et al., 1999; Nakashima et al., 1998).
Nitric oxide
Cerebral vascular tone is regulated by relaxant and contractile factors. Relaxant factors released from the endothelium include prostacyclin, endothelium-derived hyperpolarizing factor, and endothelium-derived relaxing factor, which has been identified as NO. Nitric oxide activates soluble guanylate cyclase in vascular smooth muscle cells and increases cyclic guanosine monophosphate (cGMP) that leads to relaxation (Nelson and Quayle, 1995). These relaxants modulate vascular tone by countering the effect of contractile agents such as endothelin. Because both NO-cGMP and PKC systems may participate in cerebral vasospasm, it was proposed by Nishizawa et al. (1995) that the NO-cGMP pathway serves as a negative feedback control on the PKC system. Thus, it is suggested that SAH disrupts the NO-cGMP feedback control mechanism, resulting in a prolonged PKC activation that leads to a pathologic contraction (Nishizawa et al., 1998). In a canine double hemorrhage model of SAH, severe vasospasm on days 5 and 7 is accompanied by corresponding reciprocal changes in cGMP (decreased) and PKC (increases) (Nishizawa et al., 1995). In a single hemorrhagic model where less severe vasospasm is produced, changes of cGMP and PKC are proportionately attenuated; causing Nishizawa et al. (1995) to suggest that these changes are not independent phenomena but are interrelated. Clearly, the causal role of NO in regulating PKC and thus modulating vasospasm needs to be established.
In a later study by the same group, attempts were made to establish the relation of NO with PKC in an in vitro setting (Nishizawa et al., 1996). Isometric tension was compared in normal and spastic canine cerebral arteries. Nitric oxide inhibitors generated PKC-dependent contraction in normal but not in spastic arteries. Both myogenic and nonmyogenic constriction was enhanced, in a PKC-dependent fashion, in spastic but not in normal arteries. From these results, Nishizawa et al. (1996) concluded that once cerebral vasospasm has developed, NO donors fail to dilate arteries because the NO negative feedback control mechanism was damaged. However, contrary to this proposal is the finding that NO donors reverse cerebral vasospasm in monkeys (Pluta et al., 1996). Nevertheless, the proposal by Nishizawa et al. (1995, 1996) concurs with the finding that that activation of PKC by phorbol 12, 13-diacetate (PDA) was regulated by cyclic GMP and phospholipase A2 (PLA2) but not by cAMP (Sugawa et al., 1991). An analogue of cyclic GMP, 8-romocyclic GMP, and quinacrine, a PLA2 inhibitor, attenuated the PDA-induced contraction in canine basilar arteries (Sugawa et al., 1991).
Vasospasm is also thought to be caused by an imbalance of contractile and relaxant eicosanoids such as prostacyclin and prostaglandins (White, 1990). It also was suggested that vasospasm may result from an imbalance between endothelial factors such as NO and endothelin (Sasaki and Kassell, 1990). Indeed, PKC is thought to regulate the endothelin production in cultured endothelial cells (Kasuya et al., 1993; Yakubu and Leffler, 1999). Endothelin-1 initiates vasospasm through PKC activation (Nishizawa et al., 2000a). Understanding the interaction of NO and PKC will add to an already complicated signal transduction pathway for cerebral vasospasm.
PERSPECTIVE
It is not unreasonable to conclude that during the latter phases of cerebral vasospasm there is a predominant role for a Ca2+ /calmodulin-independent regulation of cerebral vascular resistance (Fig. 6). There is persuasive data to support a key role for PKC in augmenting Ca2+ sensitivity of thin filaments (caldesmon/calponin), allowing vascular tone to be supported in the absence of increases in intracellular Ca2+. Levels of catalytic fragments of PKC are augmented in cerebral vasospasm and have a temporal relation to arterial narrowing. Protein kinase C also orchestrates a number of other cellular processes that ultimately lead to depolarization (for example, by directly inhibiting K+ channels and reducing Ca2+ sparks) and reduced myosin phosphatase activity (for example, by activating Rho-kinase). The positive identification of various PKC isoenzymes will create opportunities to study the intracellular translocation of the various isoforms in relation to vasospasm; this would require identification of the Ca2+ and cofactor requirements of the isoenzymes implicated in addition to the cellular targets that are phosphorylated—for example, other kinases, ion channels, pumps and exchangers, cytoskeletal elements, nuclear proteins. By so doing, cell permeable inhibitors specific for the PKC isoenzymes activated in cerebral vasospasm will find use in the management of this condition.
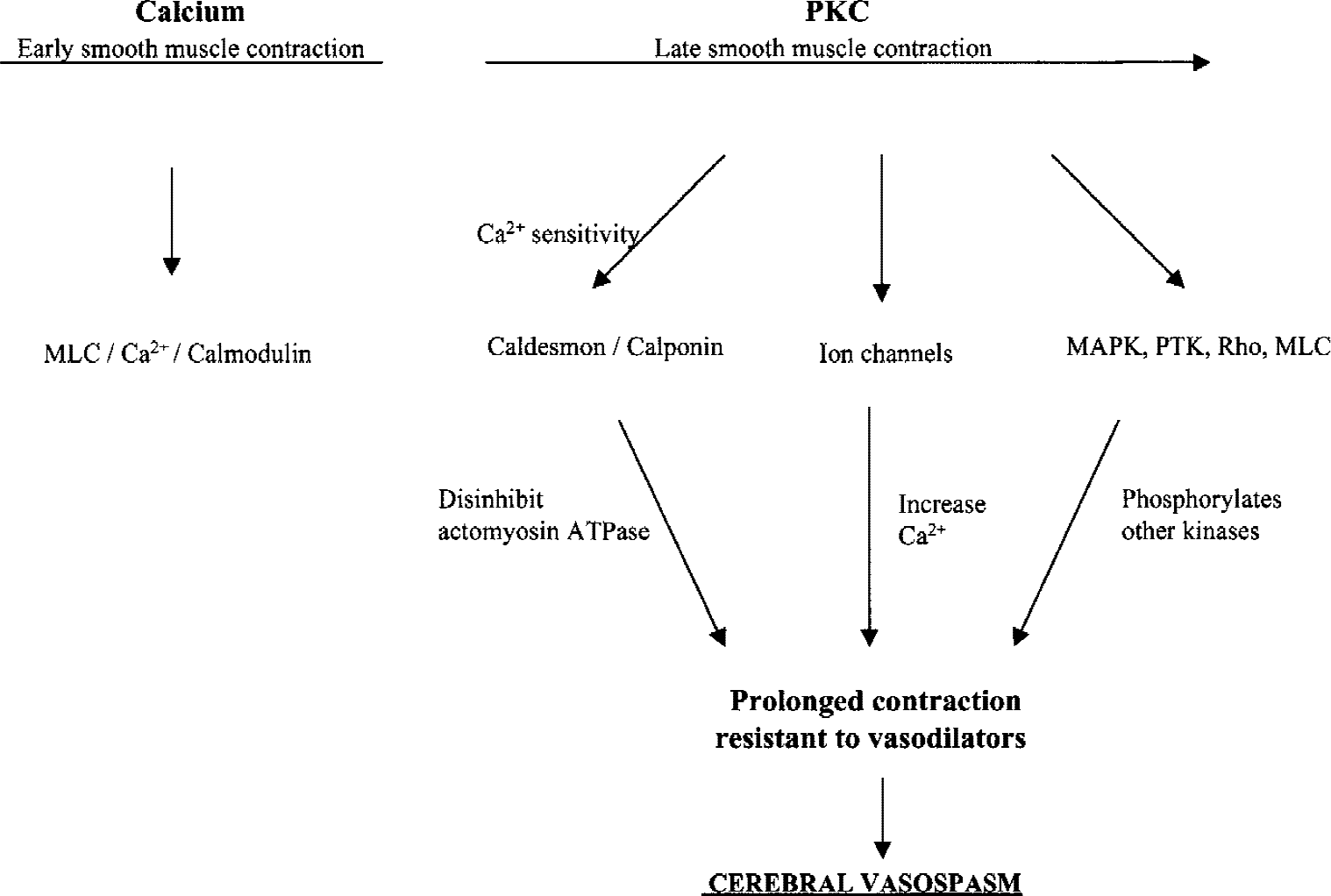
Putative scheme of cerebral vasospasm. The early phase of subarachnoid hemorrhage (SAH) is associated with enhanced Ca2+ permeability and Ca2+ -calmodulin activation of myosin light chain (MLC). During the later stages of SAH, activation of protein kinase C (PKC) occurs in response to a number of hemolytic products. Activated PKC enhances vascular smooth muscle sensitivity to Ca2+ by phosphorylating caldesmon and calponin, leading to disinhibition of actomyosin ATPase and thus enhancing vascular tone without further increases in cellular Ca2+. PKC also phosphorylates a number of ion channels (for example, Ca2+ and K+), channels ultimately causing greater entry of Ca2+ into the cell. Downstream targets of PKC are activation of other intracellular Ca2+ -sensitizing pathways (mitogen-activated protein kinase (MAPK), protein tyrosine kinase (PTH), Rho) that in turn phosphorylate other kinases in the production of cerebral vasospasm.
Footnotes
Acknowledgments:
The authors are grateful to Drs. Regent Laporte, Steve Pelech, and Chun Seow for their comments and suggestions.