
Abstract
Increasing evidence indicates that glucocorticoids (GCs), produced in response to physical/emotional stressors, can exacerbate brain damage resulting from cerebral ischemia and severe seizure activity. However, much of the supporting evidence has come from studies employing nonphysiological paradigms in which adrenalectomized rats were compared with those exposed to constant GC concentrations in the upper physiological range. Cerebral ischemia and seizures can induce considerable GC secretion. We now present data from experiments using metyrapone (an 11-β-hydroxylase inhibitor of GC production), which demonstrate that the GC stress-response worsens subsequent brain damage induced by ischemia and seizures in rats. Three different paradigms of brain injury were employed: middle cerebral artery occlusion (MCAO) model of focal cerebral ischemia; four-vessel occlusion (4VO) model of transient global forebrain ischemia; and kainic acid (KA)-induced (seizure-mediated) excitotoxic damage to hippocampal CA3 and CA1 neurons. Metyrapone (200 mg/kg body wt) was administered systemically in a single i.p. bolus 30 min prior to each insult. In the MCAO model, metyrapone treatment significantly reduced infarct volume and also preserved cells within the infarct. In the 4VO model, neuronal loss in region CA1 of the hippocampus was significantly reduced in rats administered metyrapone. Seizure-induced damage to hippocampal pyramidal neurons (assessed by cell counts and immunochemical analyses of cytoskeletal alterations) was significantly reduced in rats administered metyrapone. Measurement of plasma levels of corticosterone (the species-typical GC of rats) after each insult showed that metyrapone significantly suppressed the injury-induced rise in levels of circulating corticosterone. These findings indicate that endogenous corticosterone contributes to the basal level of brain injury resulting from cerebral ischemia and excitotoxic seizure activity and suggest that drugs that suppress glucocorticoid production may be effective in reducing brain damage in stroke and epilepsy patients.
Keywords
Considerable evidence suggests that glucocorticoids (GCs), the adrenal steroids released during stress, play an important role in neurodegenerative processes. While some data indicate that chronic elevation of circulating levels of GCs alone can result in neuronal degeneration in the hippocampus, particularly in aged animals (Landfield et al., 1981; Sapolsky et al., 1990; Landfield, 1994), the most striking effects of GCs are observed when the brain is subjected to excitotoxic/metabolic insults. GCs have been shown to impair the capacity of neurons to contain cascades of glutamate and calcium during neurological insults, thus exacerbating cytoskeletal pathology and neuron death (Sapolsky, 1985; Landfield, 1987; Elliott et al., 1993; Stein-Behrens et al., 1994). In vivo studies have shown excitotoxic (Elliott et al., 1993) and ischemic (Sapolsky and Pulsinelli, 1985) injury to hippocampal neurons is greater in adrenalectomized rats exposed to constant GC concentrations (in the upper physiological range of 15–40 μg/dl) than in adrenalectomized controls. Moreover, elevated plasma GC levels in aged humans have been correlated with decreased cognitive performance (Lupien et al., 1994), an alteration consistent with hippocampal damage. Studies of hippocampal cell cultures have shown that GCs can (a) exacerbate hypoxic and hypoglycemic neuronal injury (Tombaugh et al., 1992); (b) reduce cellular glucose uptake (Horner et al., 1990); (c) exacerbate the toxicity of kainic acid (KA), metabolic poisons, and oxygen radical generators (Sapolsky et al., 1988); (d) accelerate ATP depletion following metabolic insults (Tombaugh and Sapolsky, 1992; Lawrence and Sapolsky, 1994); and (e) enhance excitatory amino acid–induced elevation of intracellular calcium levels (Elliott and Sapolsky, 1992).
The mechanism of ischemic brain injury involves a reduction in energy availability to neurons leading to a drop in ATP levels and promotion of excitotoxicity (Novelli et al., 1988; Siesjo, 1988; Beal, 1992). The ability of glutamate receptor antagonists to reduce neuronal injury in rodent models of focal cerebral ischemia and transient global forebrain ischemia (Grotta et al., 1990; Nellgård and Wieloch, 1992) attests to the importance of excitatory amino acids in ischemic brain injury. Overactivation of excitatory amino acid receptors induces membrane depolarization and calcium influx, which results in activation of proteases and lipases and accumulation of reactive oxygen species, which can damage membranes and proteins and kill neurons (Siman and Noszek, 1988; Lafon-Cazal et al., 1993; Verity et al., 1993). Taken together, the evidence suggests that a plausible mechanism whereby GCs increase neuronal vulnerability to excitotoxic and ischemic insults is by impairing glucose transport leading to ATP depletion and increased vulnerability to excitotoxicity (see Sapolsky, 1994 for review).
Early studies implicating GC “endangerment” of neurons were nonphysiological in that they compared adrenalectomized rats with those exposed to constant GC concentrations in the upper physiological range. Considerable evidence indicates that neurologic insults can induce significant endogenous GC secretion. For example, levels of circulating GCs were shown to be elevated in stroke patients (Feibel et al., 1977; Davalos et al., 1994; Fassbender et al., 1994) and in patients experiencing severe epileptic seizures (Pritchard, 1991; Calabrese et al., 1993), and increased levels and duration of GC elevation were correlated with worse outcome. These reports suggested that endogenous GC secretion at the time of the insult could add to the damage. This view is supported by a study showing that metyrapone, a potent and rapid 11-β-hydroxylase inhibitor of GC production, can decrease hippocampal damage due to local KA infusion (Stein and Sapolsky, 1988). In addition, a preliminary report indicated that metyrapone could reduce hippocampal damage induced by transient global forebrain ischemia in the gerbil (Morse and Davis, 1989). In the present study, we extended preliminary findings and provide data showing that the protective effects of metyrapone are more general, in that they protect against additional insults relevant to the pathogenesis of both stroke and severe epileptic seizures (systemic KA-induced hippocampal damage). In addition, we show that metyrapone reduces seizure-induced cytoskeletal damage to hippocampal neurons. When taken together with data showing that metyrapone suppressed circulating levels of corticosterone (CORT), the species-typical GC of rats, in the early postinjury period, the findings indicate that endogenous GCs contribute to ischemia and seizure-induced brain injury, and testify to the physiological and clinical relevance of GC neuroendangerment.
MATERIALS AND METHODS
Animals and experimental treatments
Male Wistar rats were used for the four-vessel occlusion (4VO) model of transient global forebrain ischemia, and males of a spontaneously hypertensive inbred strain of Wistar rat (250–350 g, purchased from Harlan, Indianapolis, IN, U.S.A.) were used for the middle cerebral artery occlusion (MCAO) model of focal cerebral ischemia. Male Sprague–Dawley rats (280–300 g) (Simonsen, Gilroy, CA, U.S.A. or Harlan) were used for KA lesion studies. Metyrapone (Sigma, St. Louis, MO, U.S.A.) was dissolved in sterile saline and injected either subcutaneously or intraperitoneally in a volume of 1.5 or 3 ml, respectively, (200 mg/kg body wt). For all experiments, metyrapone was administered 30 min prior to the ischemic insult or KA administration. KA (Sigma) was dissolved in sterile saline and injected either intraperitoneally or directly into the dorsal hippocampus. CORT (Sigma) was dissolved in peanut oil and injected subcutaneously in a volume of 1 ml (40 mg/kg body wt). Rats were maintained under conditions of controlled lighting (12:12 light/dark cycle with lights on at 0700) and temperature (22°C) and allowed free access to lab chow and tap water. All experiments were performed during “lights on” hours.
MCAO model of focal cerebral ischemia
The method established by our laboratory is a modification of that of Brint et al. (1988) and involves tandem occlusion of the middle cerebral artery (MCA) and ipsilateral common carotid artery (CCA). Animals were fasted overnight prior to surgical preparation for ischemia. Rats were anesthetized with 500 mg/kg chloral hydrate for isolation of MCA and CCA. A cannula was inserted into one femoral artery for sampling of arterial blood and measurement of systemic blood pressure. Thermistor probes were inserted into the rectum and temporalis muscles to monitor body and brain temperature, which was maintained at 36–37°C by external warming. The left CCA was isolated through an anterior incision in the neck. A second incision was made between the lateral canthus of the left eye and the ipsilateral external auditory canal to expose the underlying skull. Under direct visualization with a Zeiss operating microscope, the left MCA was exposed through a 2-mm burrhole drilled 2–3 mm rostral to the fusion of the zygomatic arch and the squamosal bone. The dura was opened with a sharp needle and an alloy wire (0.1 mm diameter) was inserted beneath the MCA just superior to the inferior cortical vein. The MCA was elevated from the cortical surface and cauterized by applying electrical current to the wire. The scalp incision was closed and 4–0 silk was used to tie off the CCA. After the neck incision was sutured, the rat was returned to its cage and given free access to water and rat chow. Twenty-four h following MCAO, rats were anesthetized with chloral hydrate (500 mg/kg body wt) and perfused transcardially with saline, followed by 2% triphenyltetrazolium chloride (TTC), using our modification of the technique described by Bederson et al. (1986). The stained sample was fixed for 24 h in 10% formalin at 4°C. The brain was cut into 1-mm coronal sections and infarct area was determined with a Macintosh II computer using National Institutes of Health (NIH) Image Analysis software (version 1.52). An optical density threshold taken from healthy gray matter in the unaffected right hemisphere was used to create a mask that excluded infarcted (nonstaining) brain tissue. The mask was superimposed over the corresponding video image, thereby permitting exclusion of white matter tracts from the area of infarction. All measurements of infarct volume were confined to cortex and were performed by a single operator blinded to treatment status. Animals were divided into three treatment groups: saline-treated control (n = 8), metyrapone-treated (n = 8), and metyrapone H-CORT-treated (n = 7) animals.
Additional rats were prepared for each treatment group as described above for Nissl staining. Twenty-four h following MCAO, rats were anesthetized with sodium pentobarbital and perfused transcardially with saline, followed by cold 4% paraformaldehyde. The brain was removed, blocked, and cut with a freezing microtome into 40 μm sections, which stained with 0.1% cresyl violet. Sections were visualized with an inverted Nikon Diaphot microscope under bright field for qualitative assessment of the infarcted area of the cortex.
4VO model of transient global forebrain ischemia
Transient global forebrain ischemia was accomplished with the 4VO model (Pulsinelli and Brierley, 1979), which we adapted for modest hypotension produced by reversible exsanguination (Geddes et al., 1994; Smith-Swintosky et al., 1994). Briefly, fasted rats were anesthetized with chloral hydrate (500 mg/kg; i.p.), the common carotid arteries isolated with loose ligatures, and the vertebral arteries cauterized. Preischemic and 30 min post-ischemie blood glucose levels, blood gases and hematocrits were determined. Electroencephalographic (EEG) and electrocardiographic (ECG) activities were monitored with subdermal electrodes and recorded with a Gould Graphics Recorder. Body temperature and blood pressure were monitored continuously using the same physiological standards applied to MCAO animals. Ischemia was initiated by clamping the carotid arteries with surgical clips and withdrawing arterial blood to produce a mean systemic blood pressure of 80–85 mm Hg. After 20 min of isoelectric EEG, surgical clips were removed and the shed blood infused. This method results in highly reproducible delayed selective death of CA1 pyramidal neurons (Pulsinelli and Brierley, 1979; Smith-Swintosky et al., 1994). Seventy-two h following ischemia, rats were perfused transcardially with 4% paraformaldehyde. Brains were cut into 30 μm coronal sections, which were stained with cresyl violet. Cell counts were made from high power (40 ×) microscope fields of region CA1 of the hippocampus as previously described (Smith-Swintosky et al., 1994). Rats were divided into two treatment groups: control saline-treated (n = 8) and metyrapone-treated (n = 9) animals.
KA administration
KA was administered either systemically or by focal injection into the dorsal hippocampus as described previously (Stein and Sapolsky, 1989; Elliott et al., 1993; Stein-Behrens et al., 1994). For systemic administration, KA was injected intraperitoneally into unanesthetized rats in a single 1.5-ml bolus containing 15 mg KA/kg body wt. For focal KA injection into the dorsal hippocampus, rats were anesthetized with a ketamine/ropun cocktail, placed in a stereotaxic head holder, and the skull exposed along the midline. KA (0.07 μg in a volume of 1 μl) was injected unilaterally into dorsal hippocampus [stereotaxic coordinates: anterior/posterior (AP) = −3.0, medial/lateral (ML) = 2.1, dorsal/ventral (DV) = 4.0 mm from lambda]. As expected (Ben-Ari, 1985), essentially all animals administered KA (by either route) manifested seizures within the first h postinjection. In a previous study using the intrahippocampal KA administration paradigm, we recorded EEG and showed that metyrapone did not significantly affect epileptiform activity (Stein and Sapolsky, 1988). Four hours following administration of KA, rats were anesthetized with 20% urethane and perfused transcardially with a 0.1 M phosphate-buffered saline (PBS)/0.5% heparin solution followed by 4% paraformaldehyde. Coronal brain sections (30 μm) were cut on a freezing microtome and used for Nissl staining and immunohistochemistry. Neuronal damage in the hippocampus induced by KA administered by these two different routes was previously characterized in considerable detail (Elliott et al., 1993; Stein-Behrens et al., 1994). Neuronal damage was quantified as described in our previous studies (Elliott et al., 1993; Smith-Swintosky et al., 1994). Briefly, coronal brain sections were stained with cresyl violet and Nissl-positive neurons were counted in three high power 40 × microscope fields in hippocampal regions CA1, CA3, and hilus. Counts were made in four sections/brain. A mean number of neurons/hippocampal region was obtained for each animal, and determinations were made in five-to-eight rats/treatment group.
Immunohistochemistry and Western blot analysis
Coronal brain sections (30 μm) were cut on a freezing microtome, and free-floating sections were immunostained with tau or MAP2 antibodies using the biotin-avidin-peroxidase method as described previously (Elliott et al., 1993). The primary antibodies included a monoclonal antibody to MAP2 (1:1000, vol/vol (Sigma) and a monoclonal antibody (tau-1) that recognizes an epitope on tau in a dephosphorylated state. Sections were incubated overnight in primary antibody, followed by sequential incubations (with intervening PBS washes) in biotinylated secondary antibody, avidin-peroxidase complex, and diaminobenzidine tetrahydrochloride. To control for nonspecific staining, additional sections were subjected to the immunostaining procedure without the primary antibody.
Methods for Western blot analysis of tau and MAP2 were similar to those used in our previous studies (Mattson, 1992; 1995). Briefly, proteins were separated by SDS-PAGE (10% acrylamide), transferred to nitrocellulose, and then immunoreacted with primary antibodies followed by incubation with peroxidase-labeled secondary antibody (1:4,000, vol/vol).
Quantification of plasma corticosterone levels
Radioimmunoassay was used to quantify levels of CORT in plasma samples as described previously (Stein-Behrens et al., 1994). The procedure employed uses a highly specific CORT antibody (B3–163) (Endocrine Sciences, Tarzana, CA, U.S.A.) and 3H-CORT tracer (Gwosdow-Cohen et al., 1982). This assay detects CORT levels as low as 0.95 μg/dl and the coefficients of variation within and between assays was <10%. Blood samples were taken at the following times: MCAO paradigm: predrug, 30 min postdrug (preischemia), and 60 min postischemia; 4VO paradigm: predrug, 30 min postdrug (preischemia), and 30 min postischemia; kainate: 30 and 90-min postkainate.
Statistical analyses
Comparisons of infarct volumes (in the MCAO ischemia paradigm) were made using analysis of variance (ANOVA) with Scheffe's post hoc test for pairwise comparisons. Comparisons of numbers of surviving CA1 neurons (4VO ischemia paradigm) in saline-treated control rats and metyrapone-treated rats were made using the unpaired t-test (two-tailed). Comparisons of numbers of undamaged neurons in hippocampal regions CA1, CA3, and hilus (focal and systemic KA seizure paradigms) were made using ANOVA followed by Scheffe's post hoc test for pairwise comparisons of control and metyrapone-treated groups. Comparisons of plasma CORT levels in saline- and metyrapone-treated rats were made using ANOVA and Scheffe's post hoc test for pairwise comparisons.
RESULTS
Focal ischemic injury to cortex is reduced in rats administered metyrapone
Permanent occlusion of the MCA resulted in the formation of a major infarct of the frontal cortex within 24 h (Fig. 1). We found that administration of metyrapone 30 min prior to the onset of ischemia significantly reduced infarct volume by 54% compared to saline-treated control animals (p < 0.001) (Fig. 2). Moreover, infarcts in the metyrapone-treated animals contained a band of surviving cells (n = 7 of eight rats) that was never observed in saline-injected control ischemic rats (n = 8). Examination of Nissl-stained brain sections indicated that the cells protected by metyrapone were predominantly located within cortical layers 4–5 (data not shown). The average volume of the band of “protected cortex” in metyrapone-treated rats was 8 ± 3 mm3 (range = 0.0–17.7 mm3) (Fig. 1). An additional group of rats was given both metyrapone and CORT prior to the onset of ischemia. If metyrapone exerted its protective effects via reduction of circulating CORT levels, then the presence of exogenous CORT would attenuate or block the metyrapone protective effect. The mean infarct size in metyrapone + CORT rats was intermediate to infarct volumes in saline- and metyrapone-treated rats (Fig. 2), although the difference in values for metyrapone + CORT and metyrapone alone fell just short of statistical significance. Interestingly, none of the rats receiving metyrapone + CORT (n = 7) exhibited the band of surviving cells within the infarct that were seen in seven of eight rats receiving metyrapone alone (Fig. 1); this difference was highly significant (p < 0.001 by χ2 analysis). These data indicated that the protective effect of metyrapone against MCAO-induced cortical injury was at least partially due to its ability to suppress CORT production rather than to some other action (see Discussion).
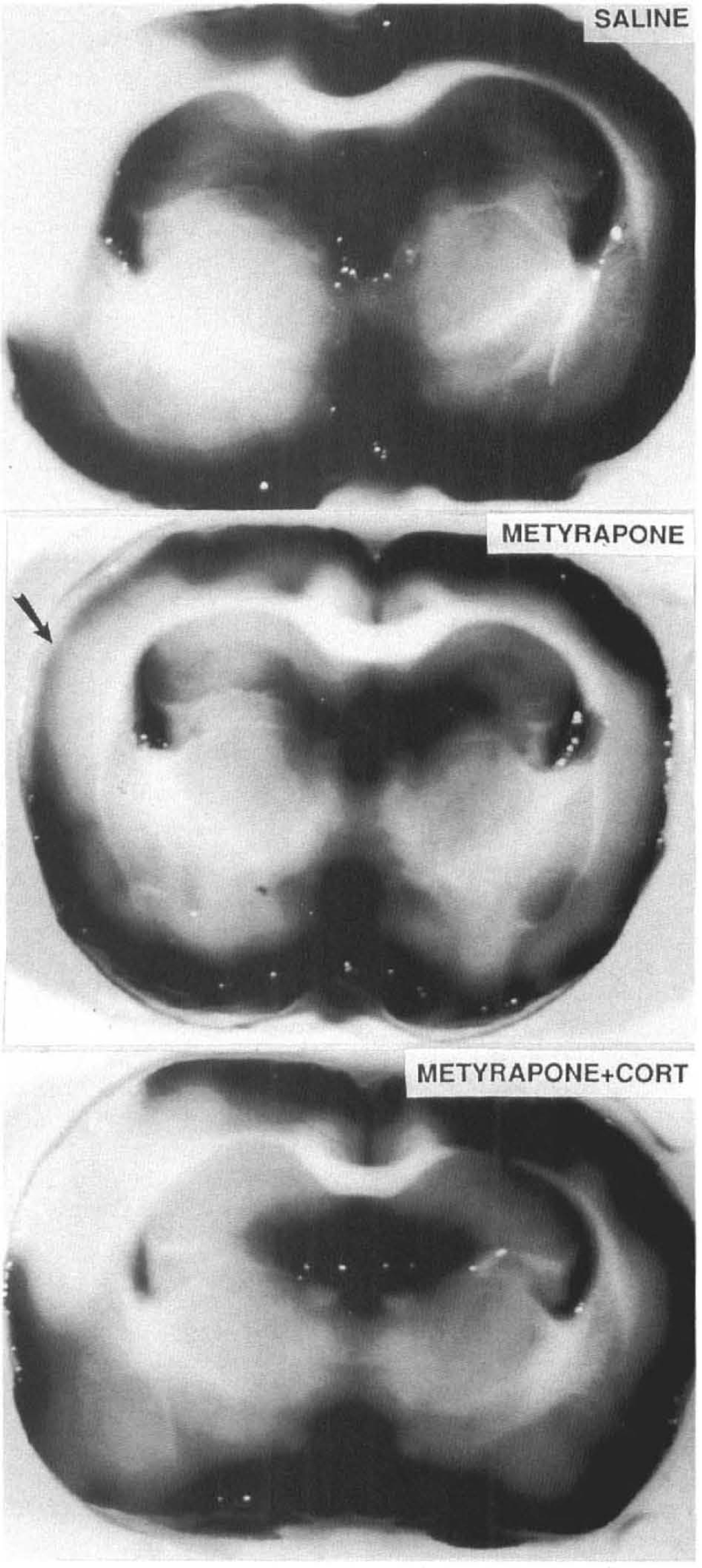
Metyrapone reduces cortical damage induced by middle cerebral artery occlusion. Photomicrographs of TTC stained coronal brain sections from rats receiving: saline (upper panel), metyrapone (middle panel), or metyrapone + CORT (lower panel). Saline-treated animals exhibited a large cortical infarct. Infarct size was reduced in metyrapone-treated animals with a characteristic band of preserved cells appearing within the infarcted area (arrow). Metyrapone + CORT treatment resulted in modest reduction in infarct size and loss of cells in all cortical layers.
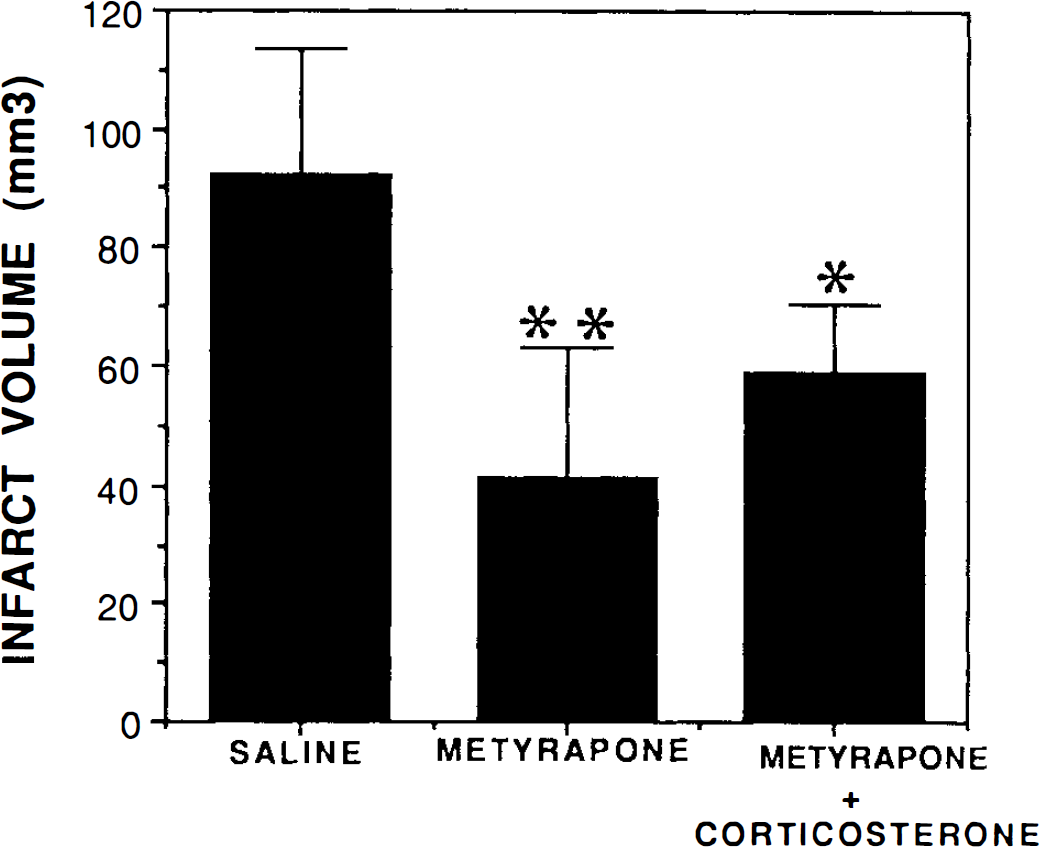
Metyrapone significantly reduces infarct volume in rats subjected to middle cerebral artery occlusion. Rats were pretreated with saline, metyrapone, or metyrapone + CORT for 30 min prior to permanent occlusion of the middle cerebral artery. Rats were killed 24 h later and brains were fixed and stained with TTC to determine infarct volume. Values represent the mean and SD (n = 7–8 rats/condition). *, p < 0.05 compared to saline-treated animals; **, p < 0.001 compared to saline-treated animals. ANOVA with Scheffe's post hoc test for pairwise comparisons.
Plasma levels of CORT following MCAO were significantly reduced in rats administered metyrapone, to ∼45% those of control rats administered saline (Fig. 3). This reduction in CORT corresponded with a significant decrease in infarct volume (Fig. 2). Plasma CORT levels in rats treated with metyrapone + CORT were 58 ± 6.8 μg/dl (n = 6), a level significantly greater than in rats administered metyrapone alone (p < 0.01). Based upon our previous studies (Sapolsky et al., 1995), basal CORT levels during the time of day at which experiments were performed in the present study ranged from 2 to 10 μg/dl, indicating that metyrapone reduces postinjury CORT levels to values somewhat higher than levels in unstressed rats.
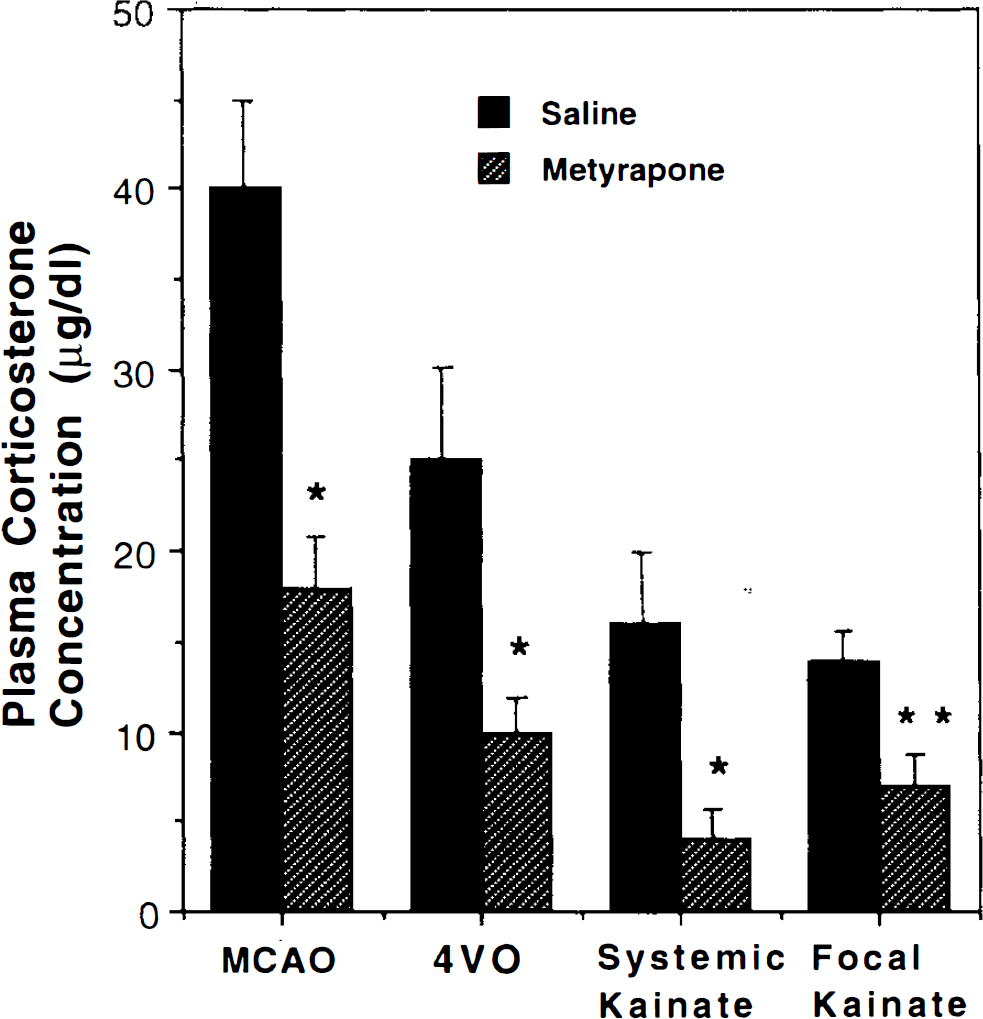
Effect of metyrapone on plasma CORT levels in rats subjected to cerebral ischemia and seizures. Plasma CORT levels were determined from blood samples taken 60 min following MCAO, 30 min following 4VO, or 90 min following administration of kainate (kainate was administered either systemically or by focal injection into the dorsal hippocampus). Values represent mean ± SD (n = 5–9 rats/condition). *, p < 0.01 compared to corresponding value for saline-treated rats. **, p < 0.05 compared to corresponding value for saline-treated rats. ANOVA with Scheffe's post hoc test for pairwise comparisons.
Metyrapone reduces damage to CA1 hippocampal neurons induced by transient global forebrain ischemia
Twenty min of transient global ischemia resulted in the selective degeneration of CA1 pyramidal neurons by 72 h postischemia; <300 undamaged neurons were present in CA1 of ischemic rats receiving saline (Figs. 4 and 5). Our previous studies established that the normal numbers of neurons in CA1 sections from nonischemic sham-operated controls ranged from 850–950 neurons (Smith-Swintosky et al., 1994). A 30-min treatment with metyrapone prior to the onset of the ischemic episode significantly increased the number of CA1 neurons surviving 72 h following the insult (p < 0.001) (Fig. 5). There was a threefold increase in CA1 cell survival in the metyrapone-treated group compared with the saline-treated group. Saline-treated rats exhibited severe damage of CA1 pyramidal neurons indicated by their pyknotic and irregularly shaped appearance. Metyrapone-treated animals displayed many healthy CA1 neurons indicated by their round soma and distinct nuclei (Fig. 4). Rats in the metyrapone-treated group had a significantly lower level of plasma CORT 30 min following the ischemic insult compared to rats in the saline-treated group (Fig. 3).
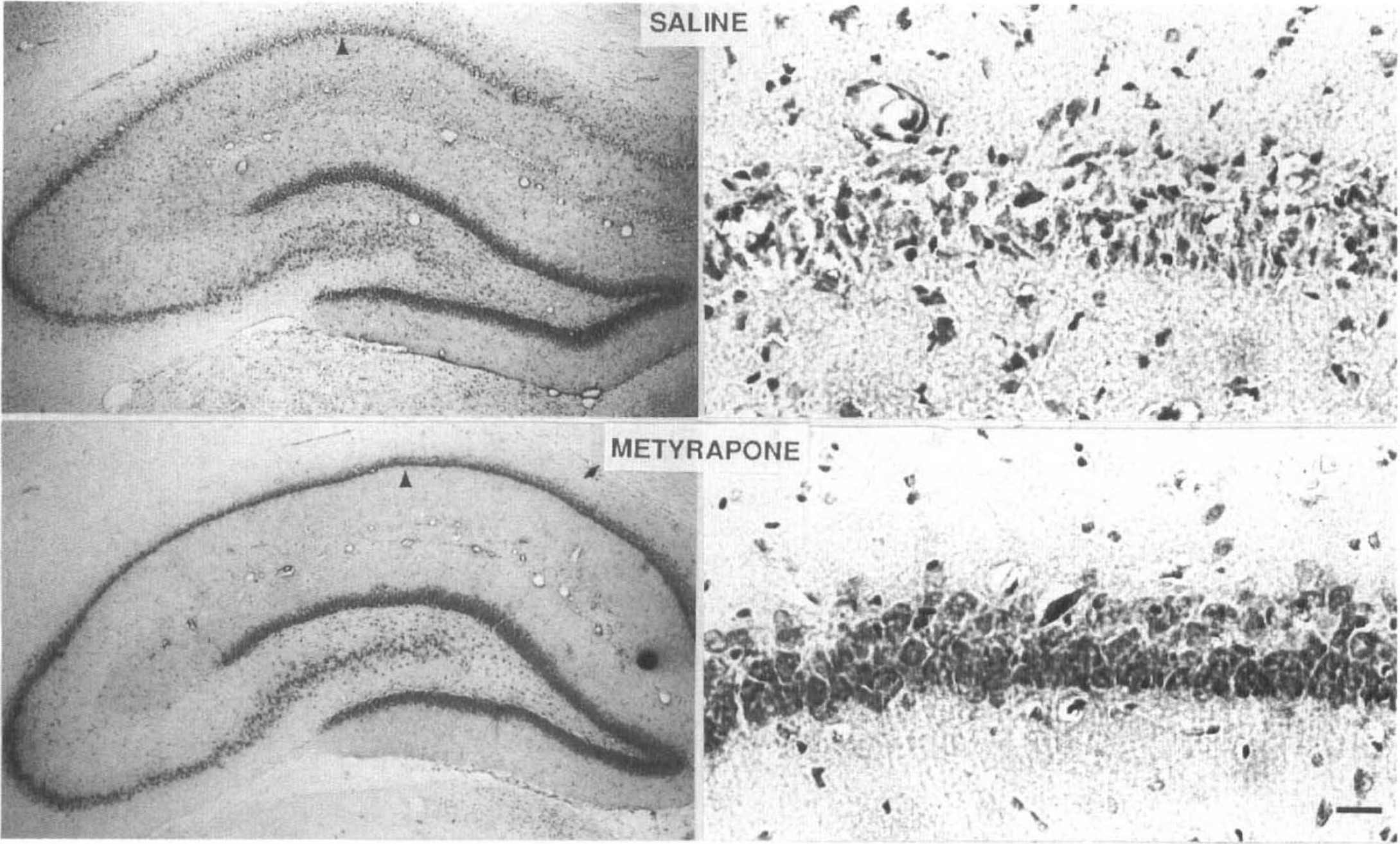
Damage to hippocampal CA1 pyramidal neurons induced by transient global forebrain ischemia is reduced in rats administered metyrapone. Photomicrographs of Nissl-stained coronal brain sections containing the hippocampus. Both 4× and 40× magnifications were made to illustrate the extent of neuronal damage observed after transient global ischemia in both saline-treated and metyrapone-treated rats. More neurons survived the ischemic insult in rats administered metyrapone. Scale bar, 25μm.
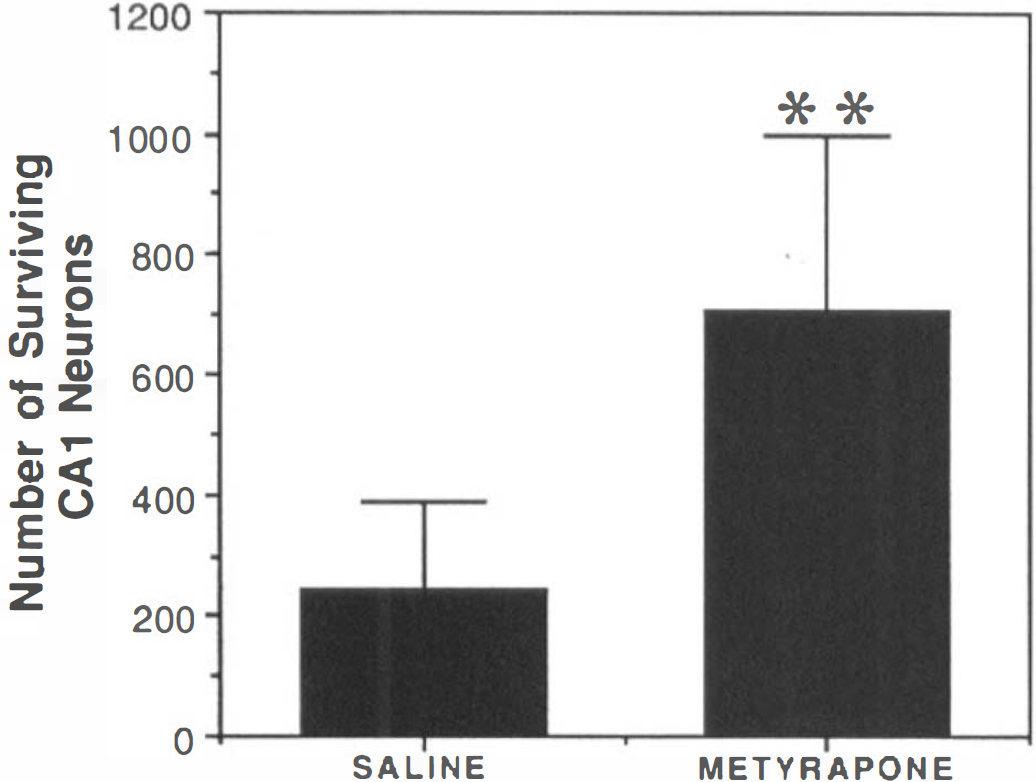
Metyrapone significantly reduces ischemia-induced loss of CA1 hippocampal neurons. Rats were pretreated with saline (control) or metyrapone for 30 min prior to 20 min of 4VO ischemia. Rats were killed 72 h later and coronal brain sections were stained with cresyl violet. Cell counts were made in six fields in the CA1 region of both hippocampi. Values represent mean and SD (n = 8–9 rats/condition). **, p < 0.001 compared to the saline-treated rats (Student's t-test).
Physiological data in the ischemia paradigms
Physiological variables were measured in all animals subjected to MCAO (Table 1) or 4VO (Table 2). There were no significant differences in baseline physiological variables measured prior to treatment with saline vehicle or metyrapone in either the MCAO or the 4VO paradigm. Measurements made 30 min following administration of metyrapone (immediately prior to MCAO or 4VO ischemia) revealed that the drug had no significant effect on any of the physiological variables. The same data were reacquired 60 min after reversal of carotid occlusion and infusion of shed blood in the 4VO animals to verify that there was no postischemic compromise of respiratory control in the medulla. We found no significant difference between saline or metyrapone-treated 4VO groups after ischemia. Table 2 includes temperature data to show that neither global forebrain ischemia nor metyrapone treatment affected rectal temperature. Temporalis muscle temperatures were matched to rectal measurements in all animals. Table 2 also contains hematocrit data to show that the effects of reversible exsanguination during 4VO ischemia were uniform for both vehicle -and metyrapone-treated rats. Postischemic blood sampling was unnecessary in MCAO animals because of their rapid recovery from anesthesia after CCA-MCA occlusion.
Physiological parameters in control and metyrapone-treated MCAO rats
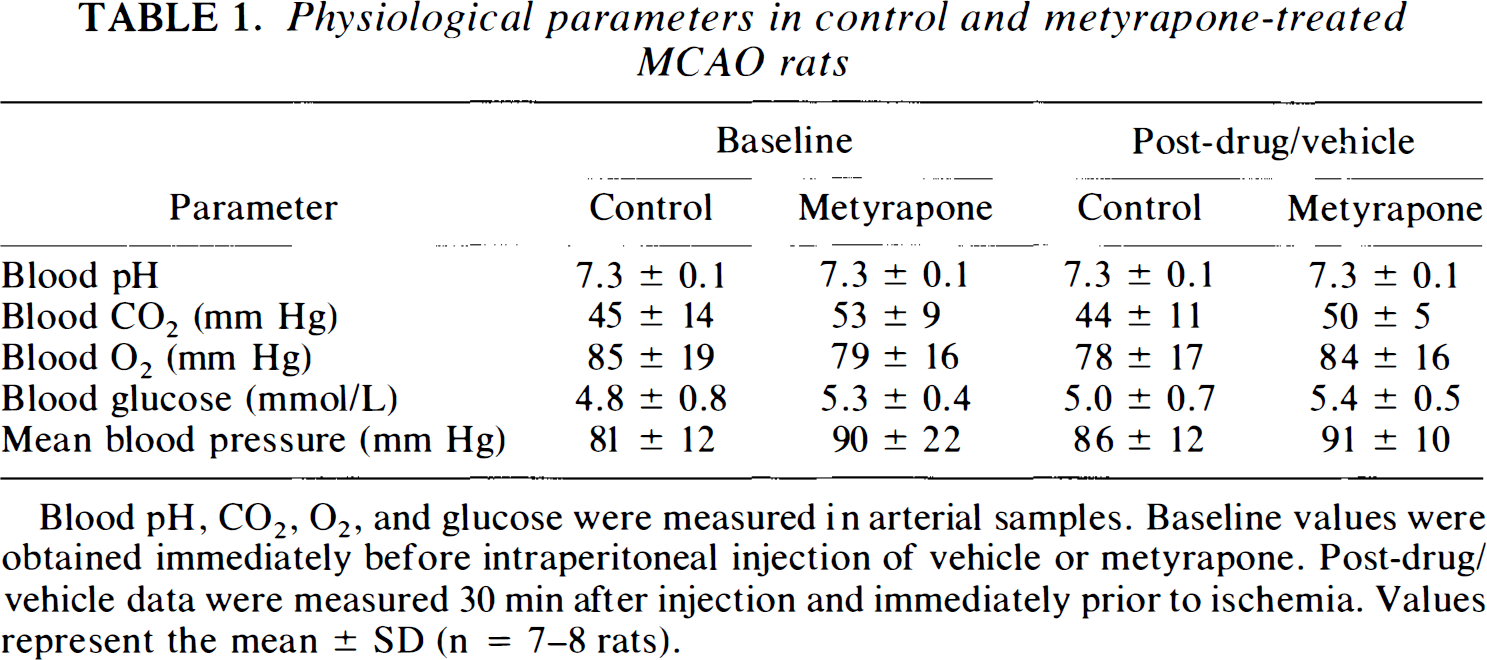
Blood pH, CO2, O2, and glucose were measured in arterial samples. Baseline values were obtained immediately before intraperitoneal injection of vehicle or metyrapone. Post-drug/vehicle data were measured 30 min after injection and immediately prior to ischemia. Values represent the mean ± SD (n = 7–8 rats).
Physiological parameters in control and metyrapone-treated 4VO rats
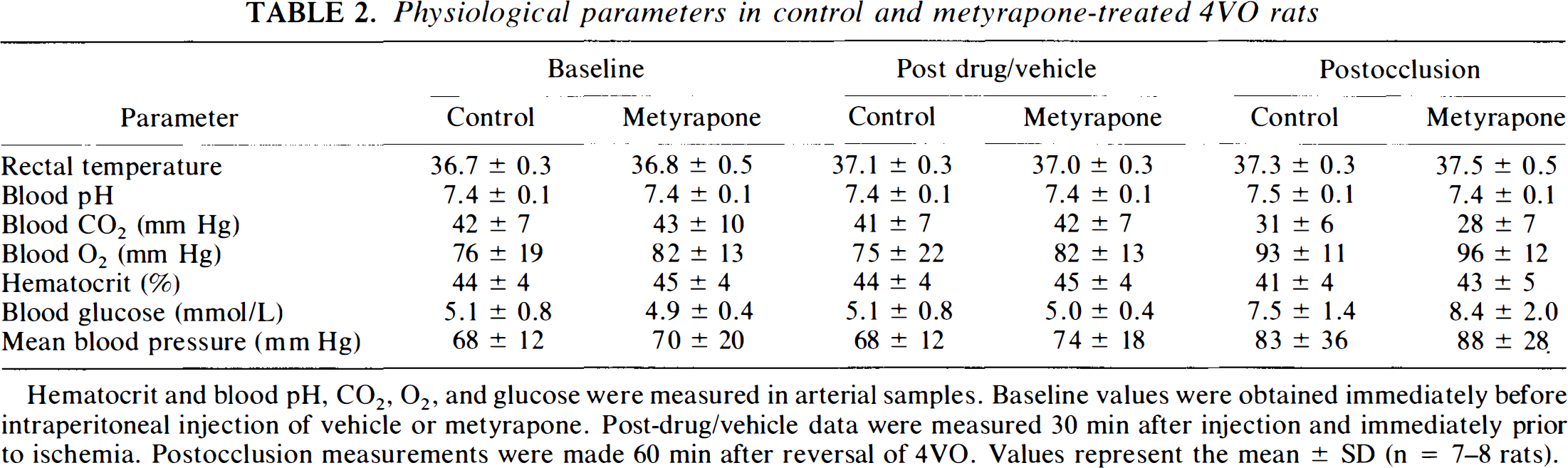
Hematocrit and blood pH, CO2, O2, and glucose were measured in arterial samples. Baseline values were obtained immediately before intraperitoneal injection of vehicle or metyrapone. Post-drug/vehicle data were measured 30 min after injection and immediately prior to ischemia. Postocclusion measurements were made 60 min after reversal of 4VO. Values represent the mean ± SD (n = 7–8 rats).
Seizure-induced damage to hippocampus is reduced in rats administered metyrapone
Adult male Sprague–Dawley rats were administered convulsant doses of KA either systemically (15 mg/kg body wt) or by stereotaxic injection of 0.7 μg of KA into one hippocampus. In previous studies, we characterized neuronal damage to hippocampal neurons in these paradigms (Elliott et al., 1993; Stein-Behrens et al., 1994). Systemic KA administration resulted in damage and death of pyramidal neurons in regions CA3 and CA1, and hilar neurons (Stein and Sapolsky, 1989). Focal injection of KA into the dorsal hippocampus resulted in damage and death of CA3 and hilar neurons (Elliott et al., 1993). In both cases, damage occurs relatively rapidly, within 3–12 h of administration of KA. In the present study, rats were killed 4 h following KA injection, and 30 μm coronal brain sections were stained with cresyl violet. In control rats not receiving KA, there were no signs of injured cells in any regions of the hippocampus; values for neuronal numbers in regions CA1, CA3, and hilus in uninjured control rats are presented in Fig. 6A. In animals receiving systemic KA, the numbers of undamaged neurons in regions CA1, CA3, and hilus were significantly reduced (Figs. 6 and 7). Whereas normal healthy pyramidal neurons had relatively round somata with nuclei and nucleoli often visible, neurons damaged by KA were shrunken, with irregular shapes and nuclear structures not visible (Fig. 7). In rats administered metyrapone (200 mg/kg body wt, s.c.) 30 min prior to exposure to KA, seizure-induced neuronal damage was significantly reduced in CA1, CA3, and hilar regions of the hippocampus (Figs. 6A and 7). Damage to CA3 and hilar neurons induced by focal injection of KA into the dorsal hippocampus was also significantly reduced in rats pretreated with metyrapone (Fig. 6B). Quantification of blood levels of CORT 90 min following KA administration (in both systemic and focal KA paradigms) showed that levels were markedly reduced in rats administered metyrapone compared to saline-injected control rats (Fig. 3).
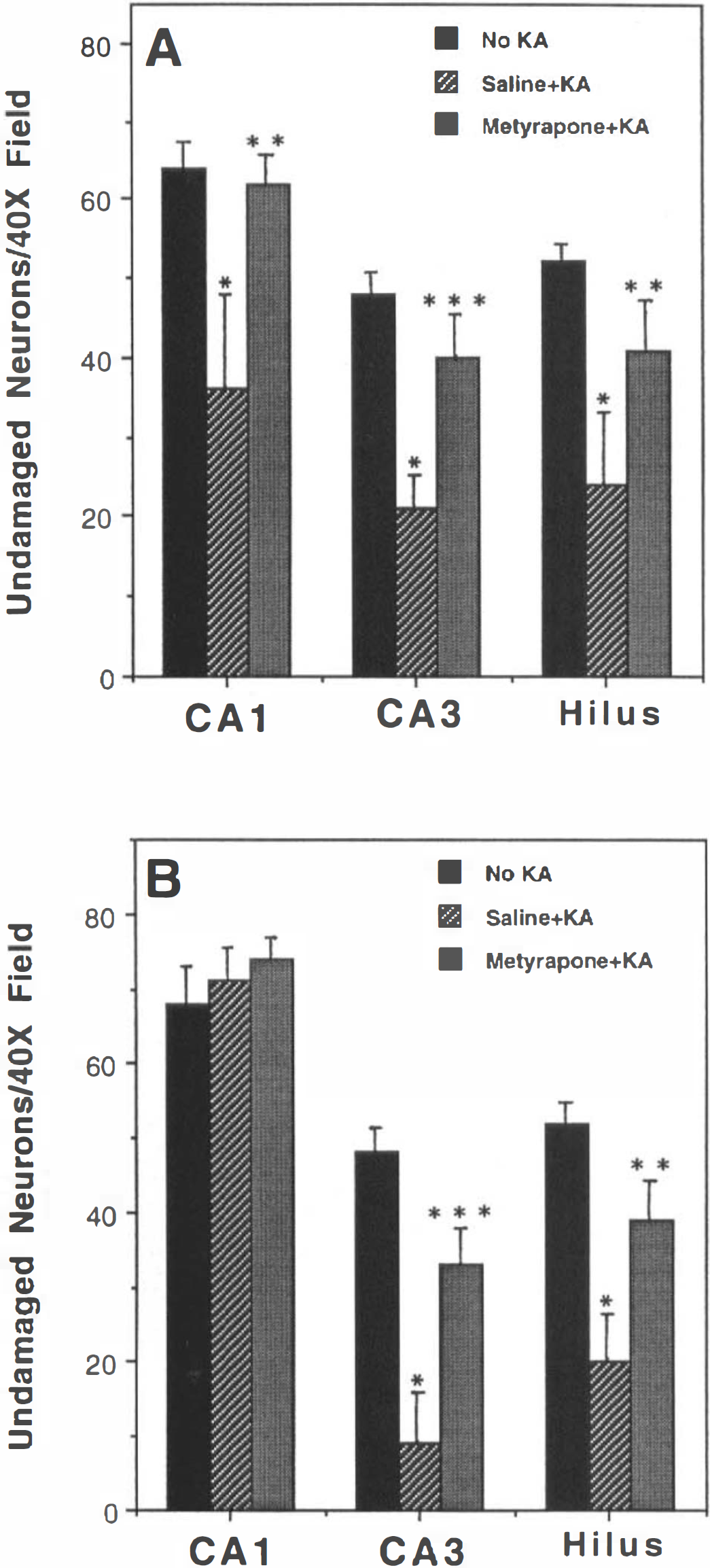
Metyrapone reduces seizure-induced neuronal damage in hippocampus. Rats pretreated with saline (control) or metyrapone were administered KA systemically (
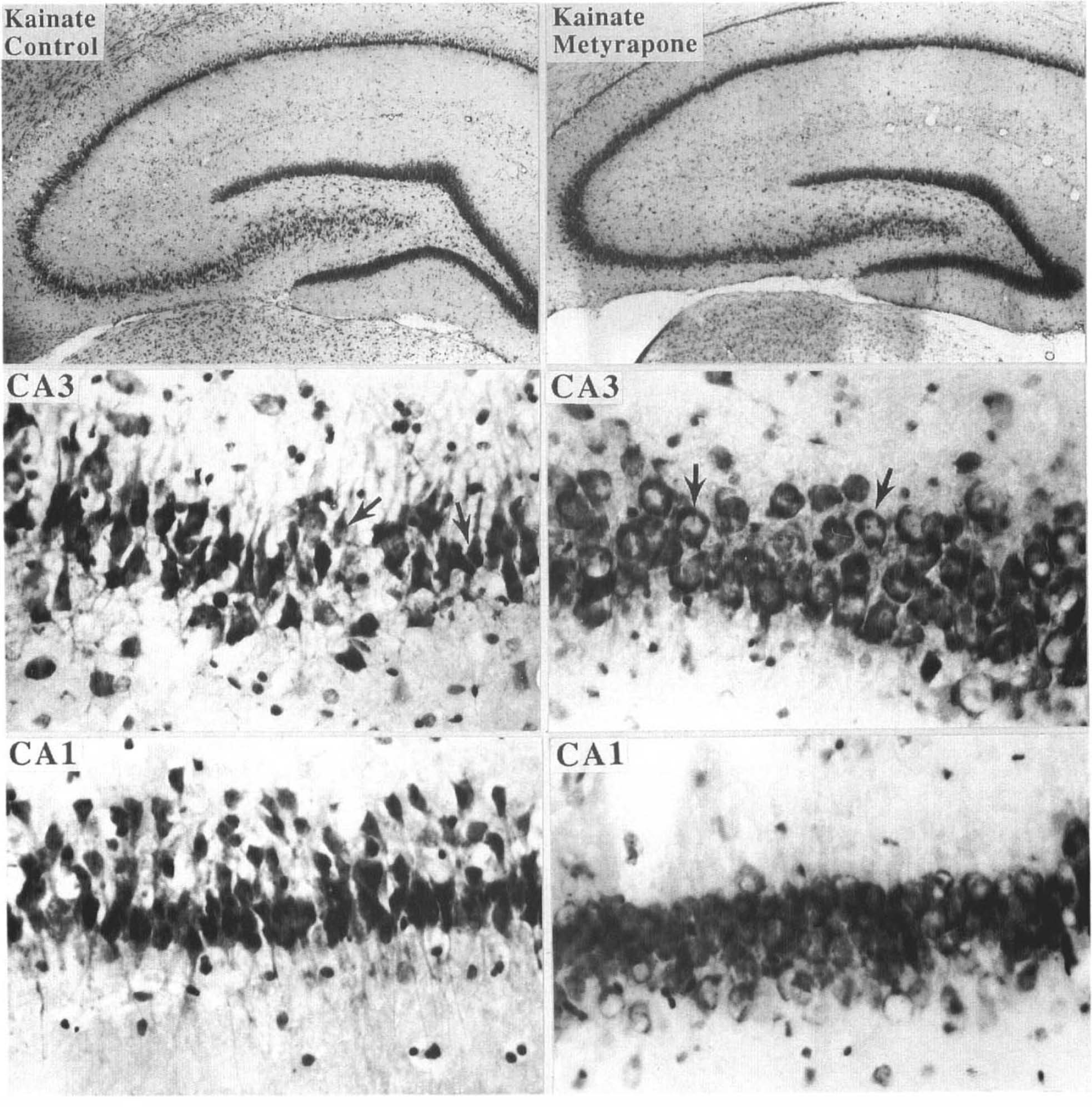
Seizure-induced damage to hippocampal CA3 and CA1 neurons is reduced in rats administered metyrapone. Nissl-stained coronal brain sections 4 h following systemic administration of KA from rats pretreated with saline (control; left panels) or metyrapone (right panels). Higher magnification micrographs of regions CA3 and CA1 are from the same sections shown at lower magnification in the upper micrographs. Note severe damage to CA3 and CA1 neurons in the control rat, and reduced damage in the rat receiving metyrapone. Arrows point to damaged neurons in CA3 of the control rat and to undamaged neurons in region CA3 of the rat receiving metyrapone.
Using the same seizure paradigms employed in the present study, we previously characterized the effects of KA on different components of the neuronal cytoskeleton, including the microtubule-associated proteins tau and MAP2, and spectrin (Elliott et al., 1993; Stein-Behrens et al., 1994). MAP2 is normally localized to dendrites throughout the hippocampus. Systemic administration of KA resulted in a loss of MAP2 immunoreactivity that was particularly obvious in the molecular layers of CA1 (Fig. 8). This reduction in MAP2 immunoreactivity was largely eliminated in rats administered metyrapone prior to KA. Tau is normally localized to axons in the different molecular layers of the hippocampus. As expected, systemic KA caused a loss of tau immunoreactivity (antibody tau-1) from axons in the molecular layers (particularly obvious in the CA1 region) and accumulation of tau-1 immunoreactivity in region CA3 (Fig. 9, upper panel). We previously reported that, in addition to causing a shift in tau localization (i.e., from axons to cell bodies), excitatory amino acids can alter phosphorylation of tau, as indicated by a reduction of a higher molecular weight tau band on Western blots (Mattson, 1995). Western blot analysis of hippocampi from KA-injected control and metyrapone-treated rats demonstrated that metyrapone largely prevented dephosphorylation of tau, as indicated by retention of the highest molecular weight tau-1 immunoreactive band, which was largely lost in KA-injected control rats (Fig. 9, lower panel). Western blot analysis showed that levels of MAP2 following KA administration were greater in metyrapone-treated rats than in rats receiving saline, indicating that metyrapone reduced KA-induced proteolysis of MAP2 (Fig. 9, lower panel). Taken together, these data indicate that suppression of endogenous GC production by metyrapone greatly reduces seizure-induced injury to hippocampal neurons.
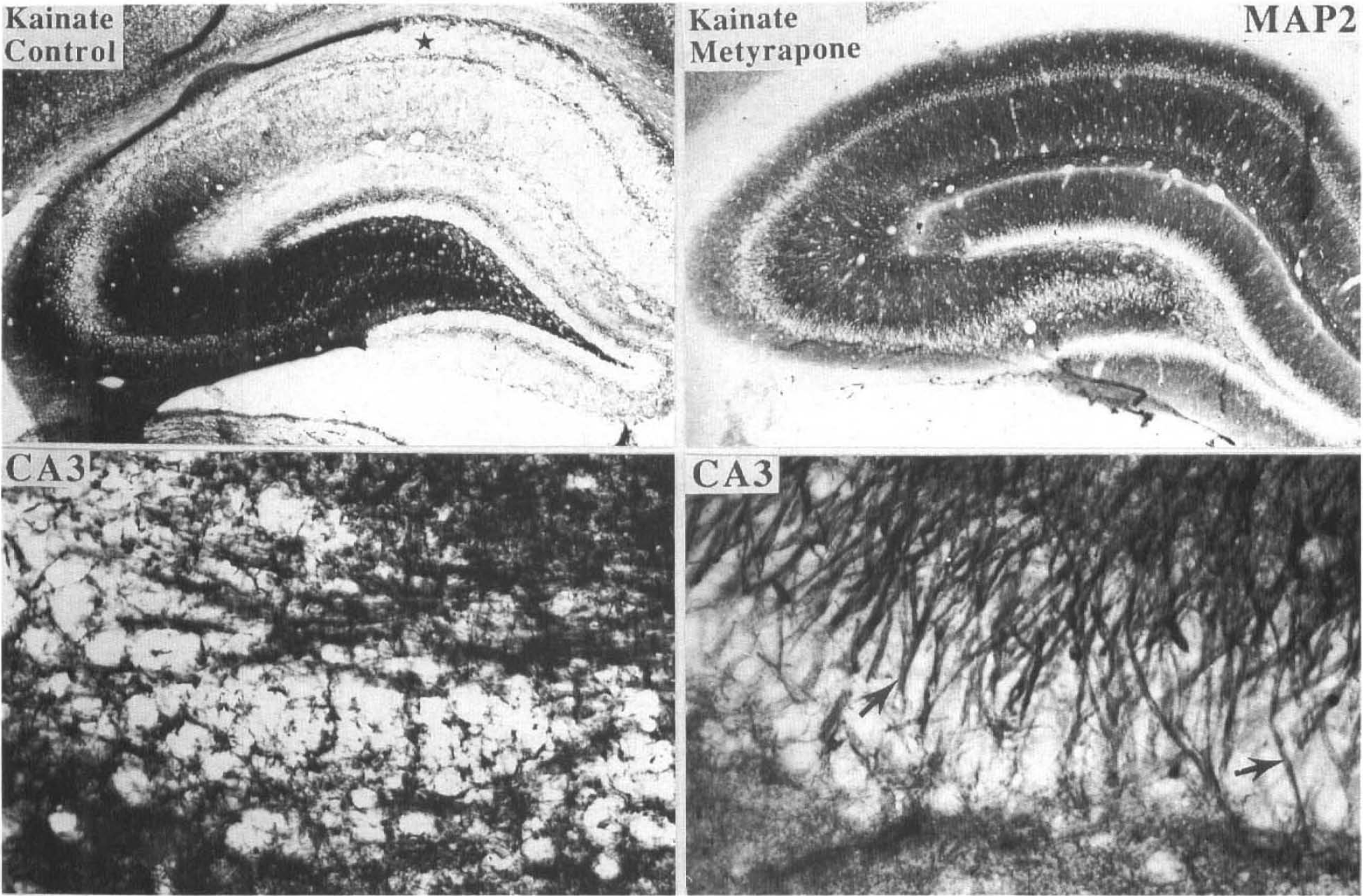
Metyrapone reduces KA-induced alterations in MAP2 immunoreactivity in hippocampus. Coronal brain sections 4 h following systemic administration of KA from rats pretreated with saline (control; left panels) or metyrapone (right panels). Sections were immunostained with MAP2 antibody. Lower panels are high magnification micrographs of a region of CA3 from the same brain sections shown in the upper panels. KA caused a loss of MAP2 immunoreactivity from molecular layers in region CA1 (star symbol), which was largely prevented by metyrapone.
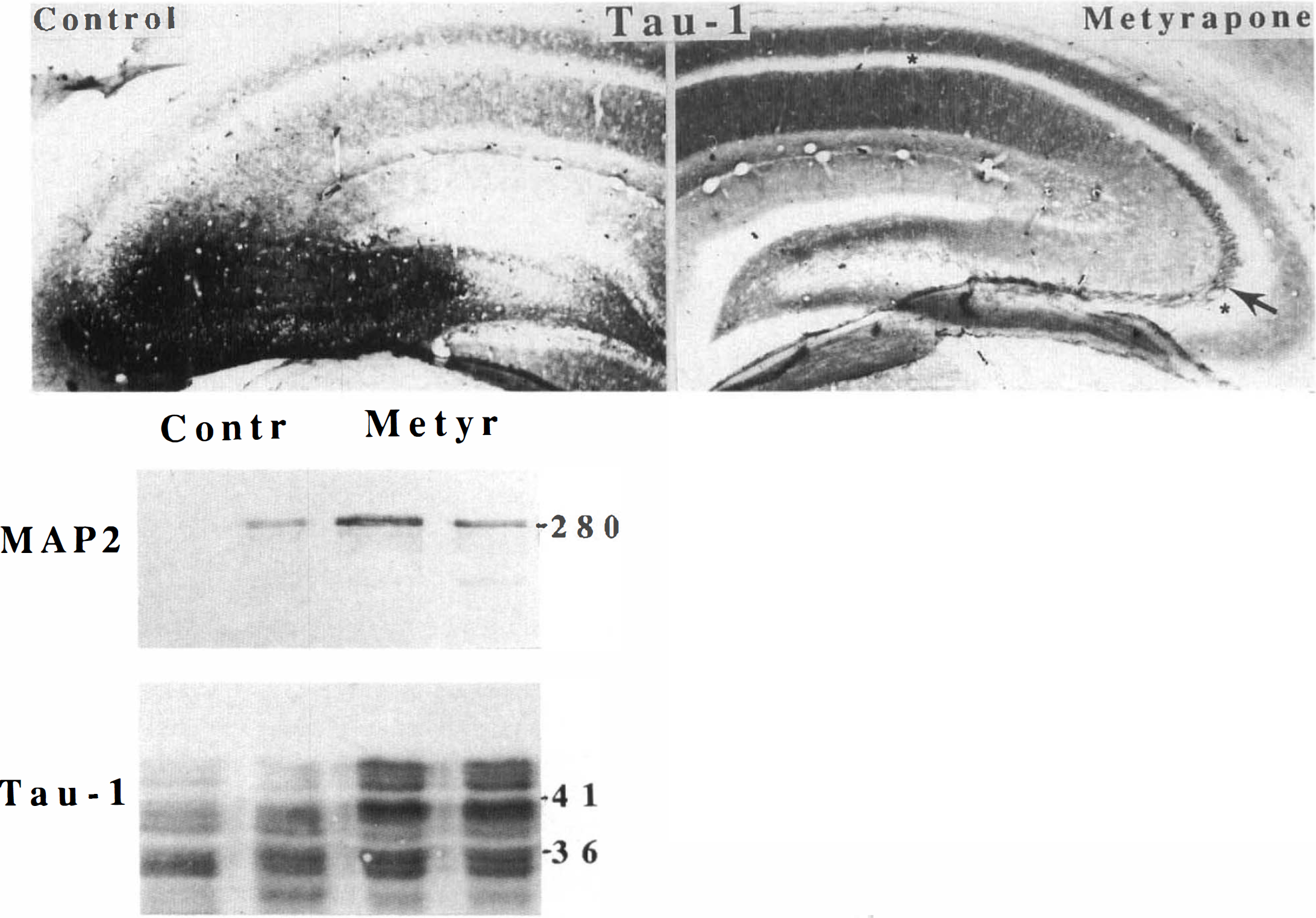
Metyrapone reduces KA-induced alterations in tau and MAP2 in hippocampus.
DISCUSSION
GCs have been shown to exacerbate toxicities of various neurological insults, presumably by their disruptive effects upon energetics in brain tissue (see introductory section). By inhibiting glucose uptake, GCs leave neurons metabolically vulnerable and less capable of the costly task of containing damaging cascades of glutamate and calcium (reviewed in Sapolsky, 1994). Our data add MCAO focal ischemia and systemic KA-induced seizures to the growing list of neurological insults that are exacerbated by GCs. Importantly, our data suggest that ongoing production of CORT in the postinjury period contributes to the severity of ischemia- and seizure-induced brain damage. We administered metyrapone, an inhibitor of GC synthesis, to a group of rats in each injury condition. As expected, metyrapone significantly attenuated the rise in plasma CORT after either ischemia- or seizure-induced injury. This suppression of plasma CORT paralleled a highly significant increase in cell survival such that infarct size was reduced in the MCAO focal ischemia paradigm, CA1 pyramidal neurons were preserved after transient global ischemia, and hippocampal neurons were significantly protected against seizure damage induced by either local or systemic KA administration. In two seizure paradigms with different characteristic patterns of hippocampal damage (convulsions induced by systemic KA resulting in damage to CA1, CA3, and hilar neurons; and local seizures induced by focal injection of KA resulting in damage to CA3 and hilar neurons only), metyrapone significantly reduced neuronal degeneration.
Levels of plasma CORT following each insult were elevated into a range suggesting a robust stress response. These levels are similar to those we have observed previously following KA microinfusion (Stein and Sapolsky, 1988) and hypoxia-ischemia (R.M.S., unpublished data) in rats. Plasma CORT levels postinjury were markedly reduced in rats administered metyrapone to levels (∼1–5 μg/dl) in the unstressed to moderate stress range (Sapolsky et al., 1985). We found that damage to the cerebral cortex induced by MCAO was significantly reduced in rats administered metyrapone. This is interesting in that much of the previous “GC endangerment” literature has focused on the hippocampus. However, two previous studies, in which adrenalectomized rats were administered exogenous GC, demonstrated increased damage to the cortex induced by ischemic insults (Sapolsky and Pulsinelli, 1985; Koide et al., 1986). The present study provides several lines of evidence for a pathophysiological role for endogenous CORT in focal ischemic damage to neocortex. First, metyrapone significantly reduced plasma levels of CORT in the postinjury period. Second, infarct volume was reduced and preserved tissue within the infarct increased in rats administered metyrapone. Third, co-administration of metyrapone and CORT resulted in plasma CORT levels higher and infarct volume greater than in rats administered metyrapone alone. More striking was the absence of preserved tissue within the infarct of rats administered metyrapone plus CORT in contrast to a highly reproducible preservation of a band of tissue corresponding to layers 4 and 5 of the cortex in rats administered metyrapone alone.
In addition to exacerbating damage to neurons in different brain regions, several other data sets attest to the “generalizability” of the neuroendangering activity of endogenous CORT. The time course of neurodegeneration for each of the insults in the present study was distinct. KA caused a relatively rapid neurodegeneration of hippocampal neurons occurring within 4 h (cf. Elliott et al., 1993). MCAO led to a well-defined cortical infarct within 24 h (cf. Brint et al., 1988). Global ischemia resulted in delayed death of CA1 pyramidal neurons clearly seen by 72 h after the ischemic episode (cf. Pulsinelli and Brierley, 1979). Our results, therefore, indicate that metyrapone has a powerful effect on neurodegeneration resulting from both rapid excitotoxicity and more protracted ischemic insults. The ability of metyrapone to reduce plasma CORT levels and protect against brain injury in three different injury paradigms strongly suggests that endogenous stress-induced levels of CORT exacerbate ischemia- and seizure-induced brain injury. The fact that metyrapone was effective in two different strains of rat further supports a generalized neuroprotective activity of this compound. We observed that postinjury levels of plasma CORT in saline-injected control rats were different in the three different injury paradigms: CORT levels were highest in the MCAO paradigm, intermediate in the 4VO paradigm, and lowest in the KA paradigm (Fig. 3). These differences are likely due to the differences in the magnitude of the stress response induced by the different insults. However, we did not perform a complete characterization of the time course of changes in plasma CORT levels (before and following the insult) and, therefore, did not establish the complete profile of plasma CORT levels which may be quite different with each insult. Due to the limitations of the animal models we employed (e.g., keeping animals anesthetized and alive after severe neurological insult for extended periods of time), we were only able to monitor plasma levels of CORT up to 90 min postinsult and, therefore, do not know how long CORT levels remained suppressed in the rats administered metyrapone. Metyrapone is rapidly absorbed and can significantly lower plasma CORT levels within 10 min. Its half-life is ∼1–2.5 h, such that it takes ∼24 h to clear >99% of the drug from the body. Therefore, it is expected that plasma CORT concentrations would be reduced for many hours following a single injection of metyrapone. Finally, differences in the strain of rat could have also contributed to the observed differences in postinjury CORT levels because the highest levels were in the Wistar strain (MCAO and 4VO) and the lowest levels were in the Sprague–Dawley strain (KA). The latter possibility is supported by previous evidence indicating significant differences in basal plasma CORT levels and stress response plasma CORT levels for different strains of rats (Dhabhar et al., 1993).
Hippocampal neuronal populations vulnerable to ischemic and excitotoxic insults express high levels of CORT receptors. Indeed, CA1 and CA3 pyramidal neurons express the highest levels of type II (low affinity) GC receptors of, perhaps, any other neuronal population. Previous data indicated that it is the type II glucocorticoid receptor that mediates increased vulnerability of neurons to excitotoxic/metabolic insults (Koide et al., 1986; Packan and Sapolsky, 1990). Interestingly, the neocortical region with the highest level of type II GC receptors (layers 4 and 5) (Van Eekelen et al., 1987) was most markedly spared in rats administered metyrapone in the MCAO focal ischemia paradigm (Fig. 1). These observations are consistent with the possibility that activation of GC receptors in layers 4 and 5 cortical neurons contributes to their demise following MCAO. Our data are also consistent with those of previous studies of GC endangerment that provided evidence that the mechanism involves activation of type II receptors leading to suppression of glucose uptake and increased vulnerability to excitotoxic damage (see Sapolsky, 1994 for review). On the other hand, some of the available data are not consistent with a classic receptor-mediated mechanism of glucocorticoid endangerment. Nongenomic actions of CORT should be considered in light of the fact that metyrapone protected CA3 neurons against the rapid (<4 h) toxicity of KA. Such rapid actions of CORT could be mediated by membrane-associated steroid receptors, such as those described in brain synaptosomes (Orchinik et al., 1991), or by more direct effects on membrane properties. Indirect actions of GCs should also be considered because GCs can induce release of cytokines and fatty acids, both of which are known to induce membrane lipid peroxidation (Ahima et al., 1992; Herman, 1993). Moreover, there is evidence that high concentrations of CORT or GC receptor agonists (>30 nM) increase calcium action potentials and voltage-sensitive calcium currents in hippocampal neurons leading to increased levels of [Ca2+]i (Kerr et al., 1991; Kerr et al., 1992).
The present studies cast light on the ways in which metyrapone is neuroprotective via its capacity to decrease CORT secretion. In addition, metyrapone has some direct effects within the nervous system. For example, intracerebroventricular administration of metyrapone suppresses feeding in rats (Jain et al., 1993). Furthermore, metyrapone can inhibit cytochrome P450 (Fantuzzi et al., 1993)—a mechanism that gives it some antioxidant properties (Montoliu et al., 1994). Finally, metyrapone suppressed injury-induced production of tumor necrosis factor (Fantuzzi et al., 1993), a cytokine that can protect hippocampal and cortical neurons against excitotoxic and metabolic insults (Cheng et al., 1994). Thus, it is important to consider that perhaps the neuroprotective effects of systemic metyrapone, as documented in the present study, resulted solely from such central actions. Our data suggest that this is not the case. Co-administration of exogenous CORT with metyrapone in the MCAO paradigm resulted in plasma CORT levels equal to or greater than levels in control rats administered saline, and resulted in significantly greater cortical cell damage compared to metyrapone-treated animals. These data suggest that the neuroprotective actions of metyrapone were at least partially due to its effect in suppressing plasma CORT levels.
When taken together with previous findings, our data suggest that metyrapone reduces aberrant elevation of [Ca2+]i resulting from ischemic and excitotoxic insults. The ability of GCs to worsen the elevation of [Ca2+]i induced by excitotoxic/metabolic insults, documented in previous studies (Elliott and Sapolsky, 1992; Elliott et al., 1993), suggests that the reduction of plasma CORT levels, effected by metyrapone, likely resulted in stabilization of neuronal calcium homeostasis. Evidence supporting the latter possibility includes the ability of metyrapone to attenuate seizure-induced MAP2 proteolysis and alterations in tau immunoreactivity. We previously showed that administration of exogenous CORT to adrenalectomized rats (Elliott et al., 1993) and physiological stress in intact rats (Stein-Behrens et al., 1994) exacerbated KA-induced MAP2 and spectrin proteolysis, and resulted in redistribution of tau in the hippocampus. Each of these cytoskeletal alterations is believed to be mediated by calcium influx (Siman et al., 1984; Siman and Noszek, 1988; Mattson, 1990; Geddes et al., 1994; Roberts-Lewis and Siman, 1994; Mattson, 1995). The present data suggest that the stress of a seizure itself provokes sufficient GC secretion to augment calcium accumulation and resulting cytoskeletal alterations.
Metyrapone was given 30 min prior to the onset of each of the three insults described above and significantly protected against each. Additional studies are currently in progress to determine whether administration of metyrapone after ischemic insult or administration of KA also reduces subsequent neuronal injury and death. Such studies should reveal the potential clinical utility of metyrapone. This compound is one of several biochemically and physiologically well-characterized inhibitors of GC synthesis and secretion. Several compounds, including metyrapone, are used clinically in humans to test adrenal reserve in diagnostic endocrine tests. They are used therapeutically for months to inhibit GC secretion in Cushing's syndrome (Haynes and Murad, 1985; Miller and Crapo, 1993; Avgerinos et al., 1994) and, in recent experimental studies, have been used as a novel class of antidepressants (Rupprecht et al., 1991). Sustained GC exposure resulting from prolonged stress can be neurotoxic to the primate hippocampus (Sapolsky et al., 1990). Stress induces atrophy of hippocampal pyramidal neuron dendrites in rats (Watanabe et al., 1992), and sustained exposure to GCs in Cushingoid humans is associated with selective hippocampal atrophy (Starkman et al., 1992). If, as predicted from our data, endogenous GCs exacerbate ischemic and excitotoxic insults to the hippocampus and cortex in humans, then compounds such as metyrapone might prove protective in allaying brain damage associated with stroke and severe epileptic seizures.
Footnotes
Acknowledgment:
We thank S. Bose and L. Letson for technical assistance. This research was supported by grants to M.P.M. from the National Institutes of Health (NIH) (NS01640 and NS30583), and a Research Career Development Award the Alzheimer's Association (Evelyn T. Stone Fund), and the Metropolitan Life Foundation; fellowships to V.L.S. from the American Heart Association and the French Foundation for Alzheimer's Research; grants to L.C.P. from the NIH (NS01505 and NS33773) and a Clinical Investigator Development Award, the Kentucky Affiliate of the American Heart Association, and the Research and Development Office, Medical Research Service, Department of Veterans Affairs; and a grant to R.M.S. from the NIH (AG06633).