
Abstract
In vitro studies suggest that ischemic injury of cerebral white matter is mediated by nonsynaptic cellular mechanisms, such as Ca2+ entry into axons through reversal of the Na+-Ca2+ exchanger. The authors investigated extracellular Ca2+ concentration in relation to tissue depolarization (direct current potential) in vivo using ion-selective electrodes in cortical gray and subcortical white matter of α-chloralose-anesthetized cats during 120 minutes of global cerebral ischemia. On induction of ischemia, regional CBF, as measured by hydrogen clearance, ceased. The direct current potential decreased rapidly within minutes in gray matter and with little time delay in white matter. Extracellular Ca2+ concentration decreased just as quickly in gray matter. In white matter, in contrast, extracellular Ca2+ increased in the first 20 to 30 minutes, and a delayed and much slower decline, compared with gray matter, was observed thereafter, reaching a minimal level only about 60 minutes after occlusion. Our results suggest that smaller and delayed transmembrane shifts of Ca2+ are correlates of delayed ischemic membrane dysfunction in central white matter tracts, which may be explained by a lack of synaptic mechanisms.
Keywords
Human stroke usually affects cerebral gray matter and white matter. Since these two major constituents of the brain differ functionally, metabolically, and structurally, mechanisms underlying ischemic dysfunction and damage also are suspected to differ. Ionic movements across neuronal membranes are critical for the understanding of anoxic or ischemic injury (Hansen, 1985; Stys, 1998). Among ion fluxes that occur in the early course of ischemia, Ca2+ entry into neurons is considered to be the most deleterious, since increases in intracellular [Ca2+] are not only related to neuronal transmission failure through its influence on transmitter release, but also to degeneration of neurons, possibly through activation of degredative enzymes (Choi, 1985, 1988; Rothman and Olney, 1986). Excitotoxin-mediated influx of Ca2+ into neurons has been assumed to be a crucial mechanism of the ischemic process (Benveniste et al., 1988; Rothman and Olney, 1986; Choi, 1985). Since white matter consists of axons and glia but not synapses, ischemic injury of white matter presumably is mediated by nonsynaptic cellular mechanisms. Ca2+ influx through reversal of the Na+/Ca2+ exchanger and possibly also through voltage-gated Ca2+ channels has been suggested to play such role (Stys, 1998). Evidence for this hypothesis derives from several in vitro studies of the rat optic nerve preparation showing that anoxic injury in central white matter depends on extracellular concentration of Ca2+ ([Ca2+]ₒ) and, that it is, for example, modulated by inhibitors of the Na+/Ca2+ exchanger (Stys et al., 1991) and by both
Here, we present simultaneous determinations of [Ca2+]ₒ, direct current (DC) potential and regional CBF measured in cortical gray and subcortical white matter during 120 minutes of global ischemia in cats.
MATERIALS AND METHODS
Five cats weighing 2.2 to 3.4 kg were anesthetized with ketamine hydrochloride (25 mg/kg intramuscularly). After canulating the left femoral artery and vein, the animals were tracheotomized and immobilized with pancuronium bromide (0.2 mg/kg intravenously). Artificial ventilation was initiated, and anesthesia was changed to halothane (0.6% to 1.2% in a 70% nitrous oxide/30% oxygen gas mixture) to avoid protective effects of ketamine against excitotoxic and ischemic damage (Lees 1989). Arterial blood pressure was monitored continuously. Arterial blood gases were measured intermittently, and Pa
To measure [Ca2+]ₒ, double-barreled ion-selective microelectrodes with tip diameters of 1.5 to 2 µm were manufactured using theta glass with thick septa. The ion-selective barrel was filled with Ca2+ ionophore (Fluka, Neu-Ulm, Germany) and 150 mmol/L CaCl2, and the reference barrel was filled with 150 mmol/L NaCl. Calibration at 37°C was performed in 0.03, 0.3, and 3.0 mmol/L CaCl2 solutions with constant background of 150 mmol/L NaCl and 3 mmol/L KCl. The reference barrel also served for DC potential and for electrocorticogram recordings. The DC potential was recorded against a calomel electrode placed at the nasal region. Etched platinum iridium electrodes (tip diameter less than 50 µm) were used to measure CBF.
Burr holes were drilled according to stereotactic coordinates (Reinoso-Suárez, 1961) above the temporal auditory cortex (middle ectosylvian gyrus) and above the frontal cortex (marginal gyrus). The dura mater was removed under microscopic control. Assemblies of a Ca2+-selective electrode and a platinum electrode (electrode-to-electrode distance, about. 1.5 mm) were inserted at the middle ectosylvian gyrus site 1.0 mm deep into the cortex, and at the marginal gyrus site 9 mm deep into the corona radiata (14 mm anterior to the interauricular line, 9 mm lateral to midline). Thin thermocouples (diameter 250 µm) were inserted adjacent to the electrode assemblies at same depth and at a distance of about 2 mm. Both burr holes were filled with Gelfoam soaked in CSF. Body and brain temperatures were controlled separately at 37.0°C with two independent feedback temperature control systems. After animal preparation, halothane was turned off, α-chloralose (60 mg/kg) was intravenously administered at least 1 hour before arterial occlusion, and general anesthesia was maintained with artificial ventilation of 70% nitrous oxide/30% oxygen. During the last 30 minutes of this period, control values for hydrogen clearance were measured after inhalation of hydrogen gas (less than 5%, added to the respiratory gas mixture). The CBF was calculated from the initial 2 minutes of the clearance curve using the following formula: CBF (mL/100 g/min) = 69.3/T1/2. Thereafter, global ischemia was initiated and kept for 2 hours by occluding the prepared extracranial arteries. The CBF was measured 10 minutes after arterial occlusion and thereafter every 30 minutes.
All experimental data are expressed as means ± SD. The significance of differences at P < 0.05 was tested between sequential measurements and between the two locations using analysis of variance with multiple post hoc comparisons (Scheffé's test) (Statistica, StatSoft, Inc., Tulsa, OK, U.S.A.).
RESULTS
Regional cerebral blood flow, brain temperature, and physiologic variables
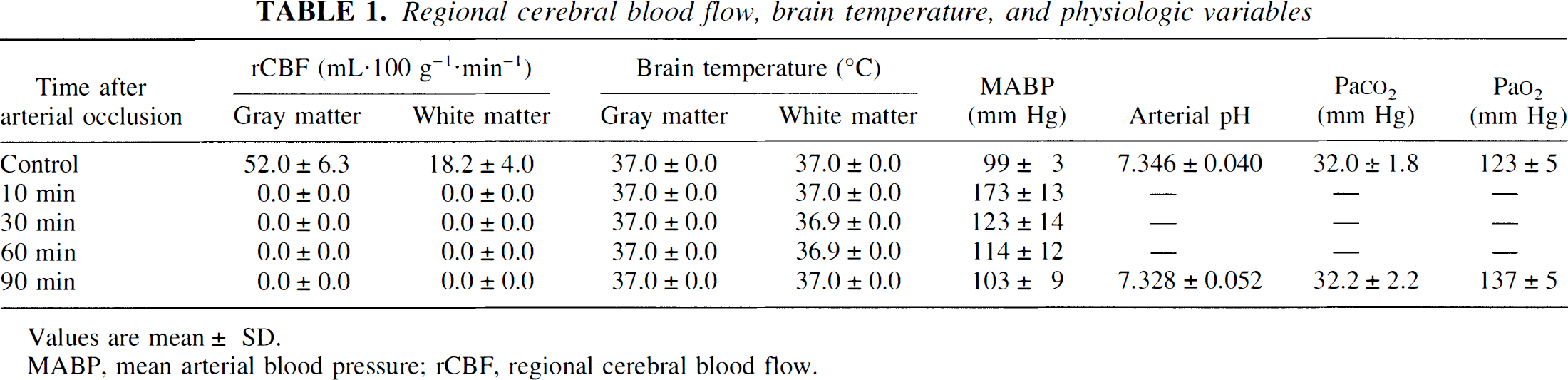
Values are mean ± SD.
MABP, mean arterial blood pressure; rCBF, regional cerebral blood flow.
Figure 1 shows representative [Ca2+]ₒ and DC potential recordings in gray matter and white matter before and after multiple extracranial arterial occlusion. In gray matter, [Ca2+]ₒ increased for a brief period immediately after arterial occlusion and decreased thereafter in two steps: a first, steep fall about 3 minutes after occlusion was interrupted by a gradual decline, and then again was followed by a steep decrease, until a final level of less than 0.2 mmol/L was reached after about 15 minutes. A negative shift of the DC potential preceded the fall in [Ca2+]ₒ and reached its final level earlier than [Ca2+]ₒ, almost at the time when [Ca2+]ₒ reached its first plateau. During the remaining course of the experiment, the DC potential recovered partially. In white matter, in contrast, [Ca2+]ₒ increased during the initial part of the experiment and decreased below baseline, only about 30 minutes after occlusion. Thereafter, it decreased gradually to about 0.3 mmol/L.
Examples of direct current (DC) potential and extracellular concentration of Ca2+ ([Ca2+])ₒ recordings in cortical gray matter and subcortical white matter of the same animal before and during global ischemia.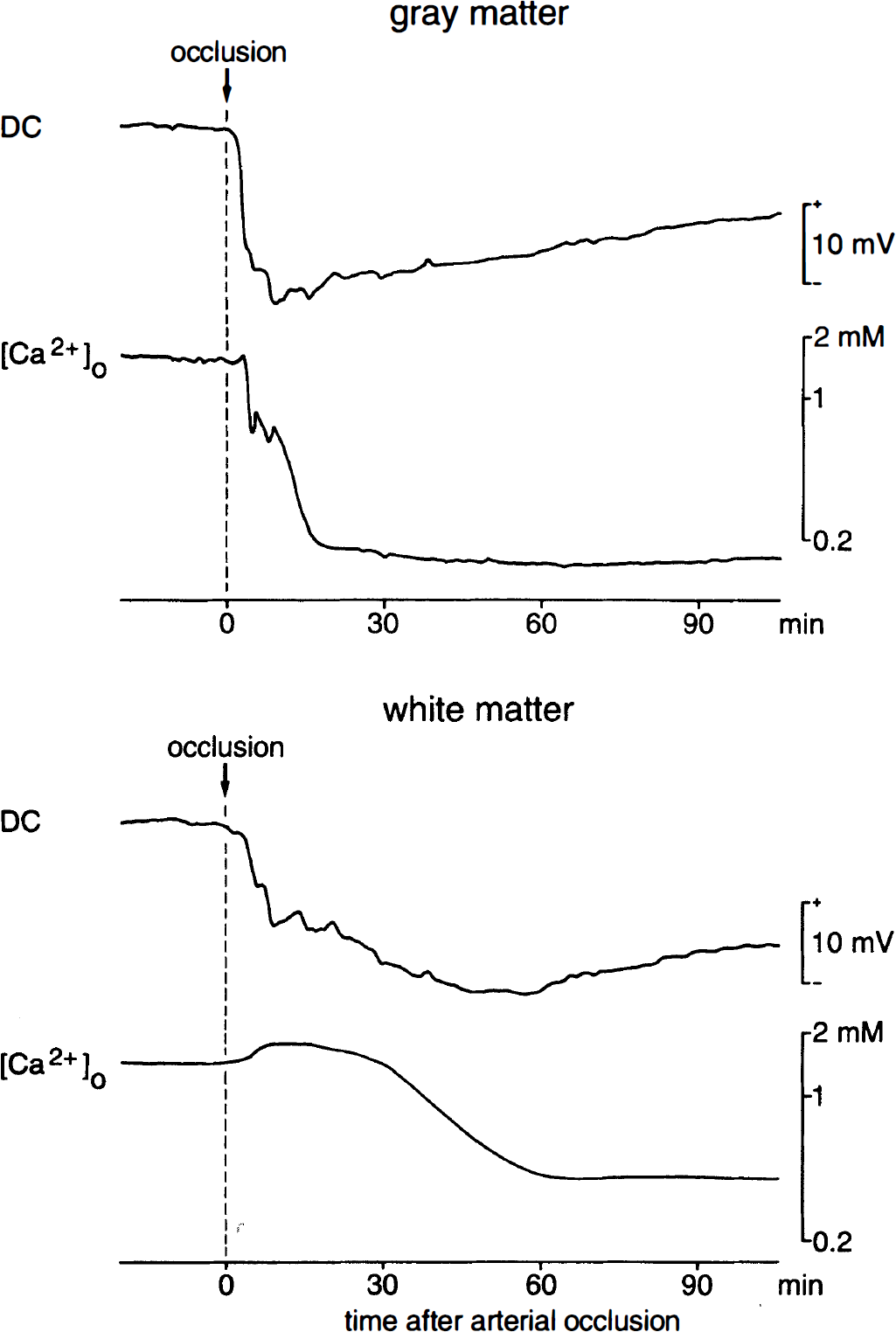
Figure 2 summarizes [Ca2+]o and DC recordings in gray matter and white matter regions. Preischemic values of [Ca2+]ₒ in gray matter and white matter were 1.22 ± 0.05 mmol/L (mean ± SD) and 1.22 ± 0.04 mmol/L, respectively. During the first 20 minutes after occlusion, [Ca2+]ₒ decreased in gray matter to about 10% (0.19 ± 0.07 mmol/L) of its preischemic control. In white matter, in contrast, [Ca2+]ₒ increased during the early period, reaching maximal values about 10 minutes after occlusion (1.62 ± 0.13 mmol/L) and slowly fell thereafter until it reached its final level of about 20% of preischemic control after about 60 minutes (0.28 ± 0.04 mmol/L). Although the negative shift of the DC potential after arterial occlusion also was slower in white matter than in gray matter (Fig. 1), the overall comparison of DC potential changes revealed similar alterations in the two regions during the first 20 minutes after occlusion, resulting in minimal levels of −12.05 ± 5.60 mV in gray matter and −13.03 ± 4.65 mV in white matter (Fig. 2). Thereafter, the DC potential tended to shift back to positive values in gray matter, reaching −6.41 ± 4.10 mV at the end of the observation period 120 minutes after occlusion, whereas in white matter, low values of the DC potential persisted throughout the experimental session (–12.21 ± 3.67 mV at the end of the experiment).
Changes in direct current (DC) potential and extracellular concentration of Ca2+([Ca2+]ₒ) measured in cortical gray matter (n = 5) and subcortical white matter (n = 5) during 120 minutes of global ischemia. Data (means ± SD) are plotted every 10 minutes for the initial 30 minutes and every 30 minutes afterward. A negative value for the DC potential indicates a decrease from baseline. * P < 0.01: significantly different from preischemic control; ° P < 0.01: significantly different from gray matter. ANOVA and post hoc comparisons.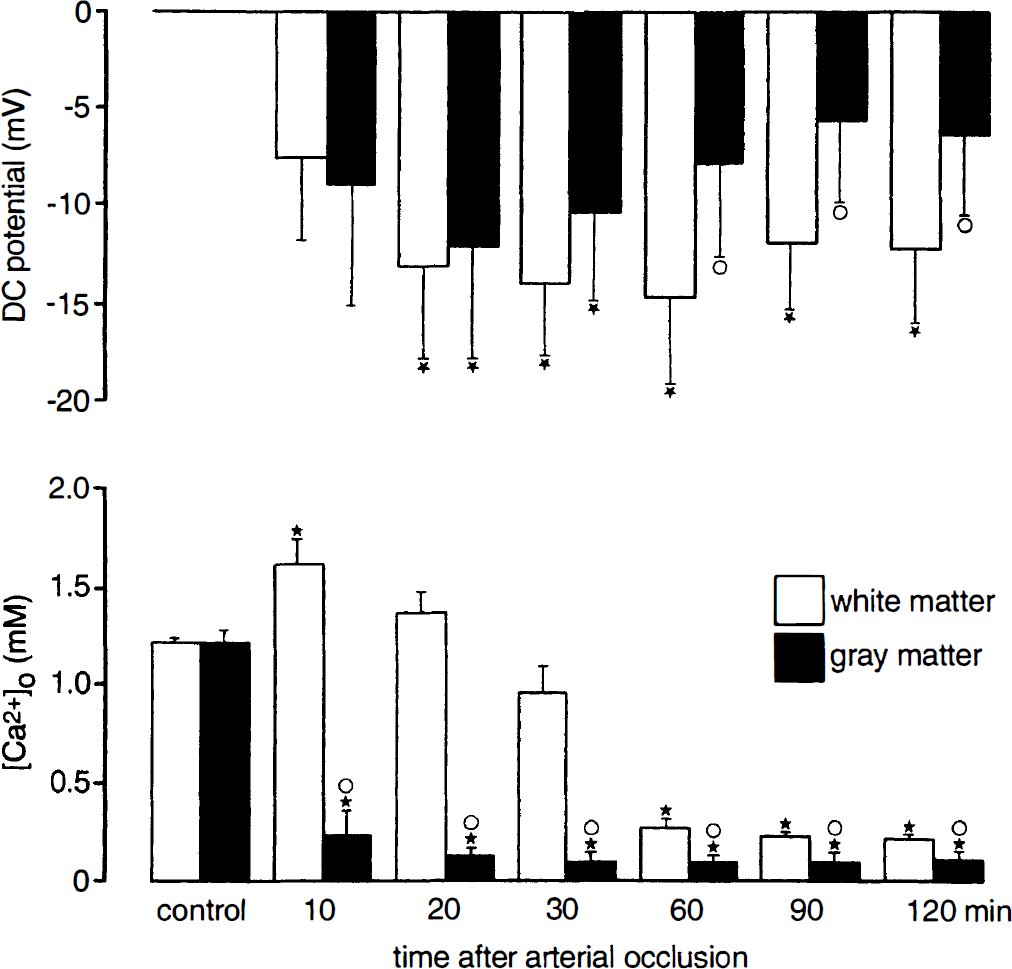
DISCUSSION
Our study demonstrates that during the early phase of global cerebral ischemia, alterations of [Ca2+]ₒ differ tremendously among gray matter and white matter regions. The decline of [Ca2+]ₒ after arterial occlusion is delayed in white matter by about 30 minutes, and it is not as steep as that in gray matter. Furthermore, the [Ca2+]ₒ level reached after 30 to 60 minutes remains significantly higher in white matter than in gray matter. In contrast, differences among gray matter and white matter regions concerning ischemic DC potential shifts are not as pronounced. The deepest values below −10 mV are reached in both regions after about 20 minutes. Thereafter, a partial repolarization above −10 mV is observed in gray matter, whereas in white matter, the DC potential remains well below −10 mV. The current results provide evidence for a substantially different ischemic pathophysiologic mechanism of cerebral white matter compared with gray matter.
Cerebral blood flow
The same degree of complete blood flow reduction in gray matter and white matter was essential for the direct comparison between the two CNS components. It was confirmed by local hydrogen clearance determinations of cerebral perfusion in the vicinity of DC and ion-selective electrodes. Hydrogen clearance provides a quantitative measure of tissue blood flow (Aukland et al., 1964). Additionally, this method allows differentiation between gray matter and white matter perfusion already in the control state, since the slope of clearance curves in gray matter is considerably steeper than that in white matter (Pasztor et al., 1973). It was therefore used during the stereotactically guided insertion of electrodes (Reinoso-Suárez, 1961) as an additional means to verify the site of measurement.
Direct current potential
Negative deflections of the DC potential are likely to be accompanied by anoxic or ischemic membrane depolarization of neurons and glia in brain tissue (Hansen, 1985). In the initial phase after arterial occlusion, cortical DC potential shifts obtained in our study followed a similar time course and fell to similar levels as described by other studies during complete or severe ischemia (Nicholson et al., 1977; Hansen and Zeuthen, 1981; Xie et al., 1995; Ohta et al., 1998). In vivo measurements of the DC potential in cerebral white matter ischemia are scarce. To our knowledge, only one study has reported anoxic negative DC potential shifts in subcortical white matter, which were induced by cardiac arrest in adult rats (Voříšek and Syková, 1997). In the mentioned study, DC shifts followed a time course similar to alterations in cortical gray matter. Minima were reached after 5 and 6 minutes in gray matter and white matter, respectively. Similarly, our results in adult cats did not reveal significant time course differences of the DC potential between gray matter and white matter in the early phase of ischemia, although in single cases, considerably slower decays were observed in white matter (Fig. 1). Analysis of means reveals relatively high variations of the DC potential, possibly reflecting that it is a gross potential undergoing multiple influences from potential changes at neuronal and glial membranes surrounding an extracellular electrode (Hansen, 1985). Minima of the DC potential were reached later than those in the rat cardiac arrest model. In particular, the minimum in white matter was reached after only about 1 hour (Fig. 2). In the second hour after occlusion, the DC potential in white matter remained significantly lower than that in gray matter. Partial recovery of the DC potential observed in gray matter during the second hour after occlusion probably reflects further degradation of membrane integrity, possibly resulting in progressive loss of compartmentalization between intracellular and extracellular space and thus one of the basic elements for polarization of nervous tissue. Following this rationale, persistent deep depolarization found in white matter would be a correlate of delayed, slower ischemic deterioration in this CNS tissue. In ischemia of longer duration, the hypothesis would have to be tested as to whether a secondary rise of the DC potential also is characteristic for white matter.
Extracellular calcium activity
For the study of extracellular ion activity changes, consideration of both ion and water displacements is essential. Incongruous ischemic alterations of diffusion parameters among gray matter and white matter regions have been reported in young rats during development, including a decrease in volume fraction and an increase in tortuoisity of the extracellular space. In contrast, diffusion parameters seem to be homogeneous in the adult brain (Voříšek and Syková, 1997). When the finding of Vřríšek and Syková, (1997) is taken as a basis for our extracellular Ca2+ activity measurements in adult cats, the observed differences in [Ca2+]ₒ alterations among gray matter and white matter are largely attributable to differences in the development of Ca2+ conductances rather than variations in extracellular volume fraction. A first, brief increase of [Ca2+]ₒ in cortical gray matter probably was attributable to shrinkage of extracellular space taking place in the phase immediately after arterial occlusion (Hansen, 1985; Voříšek and Syková, 1997). The steep decline of [Ca2+]ₒ in cortical gray matter usually was preceded by the negative DC potential shift (Fig. 1), suggesting that Ca2+ influx was triggered by transmembrane depolarization and opening of voltage-gated and N-methyl-
The early rise in [Ca2+]ₒ in white matter may have to be explained similarly by shrinkage of the extracellular space in this tissue constituent (Voříšek and Syková, 1997), with yet little or no change of Ca2+ conductance, so that this phase with an elevated [Ca2+]ₒ lasts almost 30 minutes. The delayed [Ca2+]ₒ decrease in white matter ischemia seems to be at variance with observations of LoPachin and Stys (1995) who describe a continuous increase of intracellular [Ca2+] during 60 minutes of in vitro white matter anoxia using electron probe x-ray microanalysis. Careful consideration of their data, however, provides evidence for a biphasic intracellular [Ca2+] rise with a first slight increase immediately after onset of anoxia and a second steeper increase starting about 40 minutes later. The first, more gradual rise in intracellular [Ca2+] is difficult to explain by our data, since it occurs in the absence of a decay of [Ca2+]ₒ. The time of the second increase corresponds well with the time of the delayed decrease of [Ca2+]ₒ in our study. The importance of delayed Ca2+ influx into central white matter axons 30 to 60 minutes after arterial occlusion also is supported by in vitro observations during anoxia, showing that axonal damage can be prevented by removing Ca2+ from the bath solution at these later time points (Stys et al., 1990b). The mechanisms by which Ca2+ can enter central axons during ischemia are different from those in neurons. Myelinated axons have been described to lack voltage-activated and glutamate-gated Ca2+ channels (Foster et al., 1982; Ransom et al., 1990). Investigations in the rat optic nerve preparation did not disclose any protection using nonspecific Ca2+ channel blockers such as Co2+ and MN2+, or using more specific blockade of
CONCLUSION
The comparison between gray matter and white matter ischemia revealed slow, delayed decay of extracellular Ca2+ in white matter to be the most significant finding. Because of the central role of this ion as a “final common pathway” to necrotic cell death (Siesjö, 1986), this result seems to be relevant for lower ischemic vulnerability of central white matter, since it has been proposed by older studies for both functional and structural alterations during cerebral ischemia (Jones et al., 1981; Marcoux et al., 1982; Graf et al., 1990, Pantoni et al., 1996).