
Abstract
In vitro studies suggest that adenosine may attenuate anoxic white matter damage as an intrinsic protective substance. The authors investigated ischemic alterations of purines in relation to tissue depolarization and extracellular calcium and amino acid concentrations in vivo using microdialysis and ion-selective electrodes in cortical gray and subcortical white matter of 10 cats during 120 minutes of global brain ischemia. Immediately on induction of ischemia, regional cerebral blood flow ceased in all cats in both gray and white matter. The direct current potential rapidly decreased, the decline being slower and shallower in white matter. Extracellular calcium levels decreased in gray matter. In contrast, they first increased in white matter and started to decrease below control levels only after approximately 30 minutes. Adenosine levels transiently increased in both tissue compartments; the peak was delayed by 30 minutes in white matter. Thereafter, levels declined faster in gray than in white matter and remained elevated in the latter tissue compartment. Inosine and hypoxanthine elevations were progressive in both regions but smaller in white matter. Levels of gamma-aminobutyric acid, another putatively protective agent, steadily increased, starting immediately in gray matter and delayed by almost 1 hour in white matter. The delayed and prolonged accumulation of adenosine correlates with a slower adenosine triphosphate breakdown in white matter ischemia and may result in protection of white matter by suspending cellular calcium influx.
Cerebral ischemia leads to consumption of energy stores by reducing the glucose and oxygen supply (Siesjö, 1978). Consequently, adenosine triphosphate (ATP) is metabolized to adenosine and further to inosine and hypoxanthine, which all accumulate in the extracellular space during ischemia (Hagberg et al., 1987). Subsequent to the breakdown of energy-rich phosphates, cellular ion homeostasis is impaired. The resulting cellular uptake of Ca2+ is thought to play a major role in the pathophysiologic sequence of events leading to irreversible cell damage (Hansen, 1985; Siesjö and Bengtsson, 1989; Stys, 1998).
Most studies on the mechanisms underlying ischemic brain damage have been conducted on gray matter (GM). Because cerebral white matter (WM) is affected in most human strokes, studies on the specific pathophysiology of WM ischemia are of utmost relevance for the development of effective strategies in stroke treatment. Taking into account the differences in structure, function, and metabolism of GM and WM, in particular the fact that WM consists of axons and glia but not synapses, one might assume that different mechanisms lead to ischemic dysfunction and damage. Evidence for this hypothesis is derived from in vitro work on the rat optic nerve preparation showing that anoxic injury in central WM depends on extracellular [Ca2+] and that it is, for example, modulated by inhibitors of the Na+/Ca2+ exchanger (Stys et al., 1992) and by both L-type and N-type Ca2+ channel antagonists (Fern et al., 1995). A recent in vivo study from this laboratory has shown Ca2+ extrusion from the extracellular space in WM ischemia to be delayed by about 30 minutes compared with GM ischemia (Kumura et al., 1999). This probably reflects a suspended ischemic Ca2+ influx and thereby a lower ischemic vulnerability of WM. The underlying mechanisms of this difference between the two central nervous system compartments remain to be clarified.
In vitro studies using the rat optic nerve as a model for a central WM tract have shown synergistic neuroprotective effects of gamma-aminobutyric acid (GABA) and adenosine (Fern et al., 1994), and it has been suggested that these effects are exerted by an attenuation of Ca2+ influx into cells. The authors have shown endogenous release of GABA to be delayed by 1 hour in WM versus GM ischemia (Shimada et al., 1993). Because the delay in Ca2+ extrusion in WM amounts to only 30 minutes, it is not sufficiently explained by the later GABA accumulation. In the current study the authors investigated alterations of in vivo extracellular concentrations of adenosine and its metabolites in relation to simultaneously measured extracellular GABA and Ca2+ concentrations, and the direct current potential in WM and GM ischemia.
MATERIALS AND METHODS
Adult male cats (n = 10) weighing 3.1 to 4.6 kg were fasted overnight and then anesthetized with ketamine hydrochloride (25 mg/kg given intramuscularly). After catheterization of the left femoral artery and vein, the animals were tracheotomized, immobilized with pancuronium bromide (0.2 mg/kg given intravenously, and ventilated artificially. Anesthesia was maintained with halothane (∼1% in a mixture of 70% nitrous oxide and 30% oxygen) during surgery. An intravenous infusion of 2 mL/kg per hour Ringer's solution containing 5 mg/kg per hour gallamine triethiodide for muscle relaxation was maintained throughout the experiment. Arterial blood pressure was monitored continuously and arterial blood gases were measured intermittently. Physiologic variables were controlled. Preparation and occlusion of both common carotid and subclavian arteries were performed as previously described (Kumura et al., 1999).
Microdialysis probes (CMA/12, membrane 1 mm in length, 0.5 mm in diameter; Carnegie Medicin, Stockholm, Sweden) were perfused continuously with artificial cerebrospinal fluid (expressed in mmol/L: 1.2 CaCl2, 145 NaCl, 2.7 KCl, 1.0 MgCl2, adjusted to pH 7.4 with phosphate buffer) at a flow rate of 2 μL/min. Ten-minute samples of dialysate (20 μL) were divided into two 10-μL aliquots that were analyzed in two different high-performance liquid chromatography systems. Amino acids were determined by fluorescence detection (excitatory wavelength 330 nm, emission wavelength 480 nm; RF-535 Shimadzu, Kyoto, Japan) after precolumn derivatization with o-phtaldialdehyde and separation by a 5-μm C18-Nucleosil column (100 × 4.6 mm; Alltech, Munich, Germany), using a linear gradient elution profile (buffer A: sodium acetate 10 mmol/L, pH 5.7, buffer B: 10% to 60% methanol) at a flow rate of 2 mL/min (Shimada et al., 1993, with modifications). Purine catabolite concentrations were determined by ultraviolet detection (wavelength 254 nm; Knauer, Berlin, Germany) after separation by a 3-μm C18 column (10 × 4 mm, Knauer) using an elution of 10 mmol/L NH4HPO4/CH3OH (20:1 v/v; pH 5.5) with a flow rate of 1.2 mL/min (Matsumoto et al., 1992, with modifications). All substances were identified and quantified by comparing retention times and peak areas with those of external standards.
For measurement of extracellular calcium activity [Ca2+]o, double-barreled ion-selective microelectrodes with tip diameters of 1.5 to 2 μm were manufactured using theta glass with thick septa. The ion-selective barrel was filled with Ca2+ ionophore (Fluka, Neu-Ulm, Germany) and 150 mmol/L CaCl2, and the reference barrel was filled with 150 mmol/L NaCl. Calibration at 37°C was performed in 0.03, 0.3, and 3.0 mmol/L CaCl2 solutions with constant background of 150 mmol/L NaCl and 3 mmol/L KCl. The reference barrel served also for direct current potential and for electrocorticogram recordings. The direct current potential was recorded against a calomel electrode placed at the nasal region. Etched platinum iridium electrodes (tip diameter < 50 μm) were used to measure cerebral blood flow.
Bur holes were drilled according to stereotactic coordinates (Reinoso-Suárez, 1961) above the middle ectosylvian and the marginal gyrus. The dura mater was removed under microscopic control. A microdialysis probe, a Ca2+-selective electrode, and a platinum electrode were assembled with a tip-to-tip distance of 1.5 mm. The electrodes were inserted at the middle ectosylvian gyrus site 1.0 mm deep into cortex, and at the marginal gyrus site 9 to 11 mm deep into the corona radiata. Both bur holes were filled with Gelfoam soaked in cerebrospinal fluid. Body and brain temperatures were controlled separately at 37°C with two independent feedback temperature control systems.
Animal preparation was followed by a 2-hour stabilization period. In this period, general anesthesia was switched to α-chloralose by administering α-chloralose (60 mg/kg) intravenously at least 1 hour before arterial occlusion and turning off halothane thereafter. During the last 30 minutes of this period, control values for hydrogen clearance were measured after inhalation of hydrogen gas (<5%, added to the respiratory gas mixture). Cerebral blood flow (CBF) was calculated from the initial 2 minutes of the clearance curve using the formula: CBF (mL/100 g/min) = 69.3/T1/2. Shortly before occlusion, the microdialysis control sample was taken. Thereafter, global ischemia was initiated and kept for 2 hours by occluding the prepared extracranial arteries. Cerebral blood flow was measured 10 minutes after arterial occlusion and thereafter every 30 minutes. Microdialysate was sampled every 10 minutes after occlusion.
All experimental data are expressed as means ± standard deviation. The significance of differences at P < 0.05 was tested between sequential measurements and between the two locations using analysis of variance with multiple post hoc comparisons (Newman–Keuls test and critical ranges) (Statistica, StatSoft, Tulsa, U.S.A.).
RESULTS
During control, regional CBF was 52.0 ± 6.3 mL/100 g per minute in GM and 18.2 ± 4.0 mL/100 g per minute in WM and rapidly decreased on arterial occlusion to 0 mL/100 g per minute in both WM and GM regions. This value was kept throughout the experimental period. The electrocorticogram flattened on occlusion and all animals showed fully dilated pupils with no light reflex. During the initial 20 minutes after arterial occlusion, arterial blood pressure increased significantly from 103 ± 6 mm Hg to 170 ± 16 mm Hg and returned thereafter to preischemic levels.
Alterations of the direct current potential and of [Ca2+]o in the course of ischemia are shown in Fig. 1. After the onset of ischemia, the direct current potential rapidly decreased in GM, reaching −17 mV after 5 minutes and a minimum of −19 mV after 10 minutes of ischemia. Thereafter, the direct current potential tended to shift back to higher values, reaching −5.7 mV at the end of the observation period. In WM, the negative shift of the direct current potential was slower and smaller, reaching a minimum of −11 mV after about 45 minutes of ischemia. Interestingly, a remarkable change in the rate of depolarization from fast to slow was observed in WM 10 minutes after the onset of ischemia. After reaching the minimum, the direct current potential in WM tended to shift back, too, but values remained lower than in GM. Control determinations of [Ca2+]o yielded comparable values of about 1.2 mmol/L in both GM and WM. [Ca2+]o steeply declined in GM during the first minutes of ischemia, reaching minimal levels of 0.19 mmol/L by 15 minutes after occlusion. In contrast, [Ca2+]o in WM increased during the early period of ischemia, peaking at approximately 1.8 mmol/L 10 minutes after occlusion. It decreased slowly thereafter and fell below control values only 30 minutes after occlusion, reaching its final level of 0.28 mmol/L after about 60 minutes.
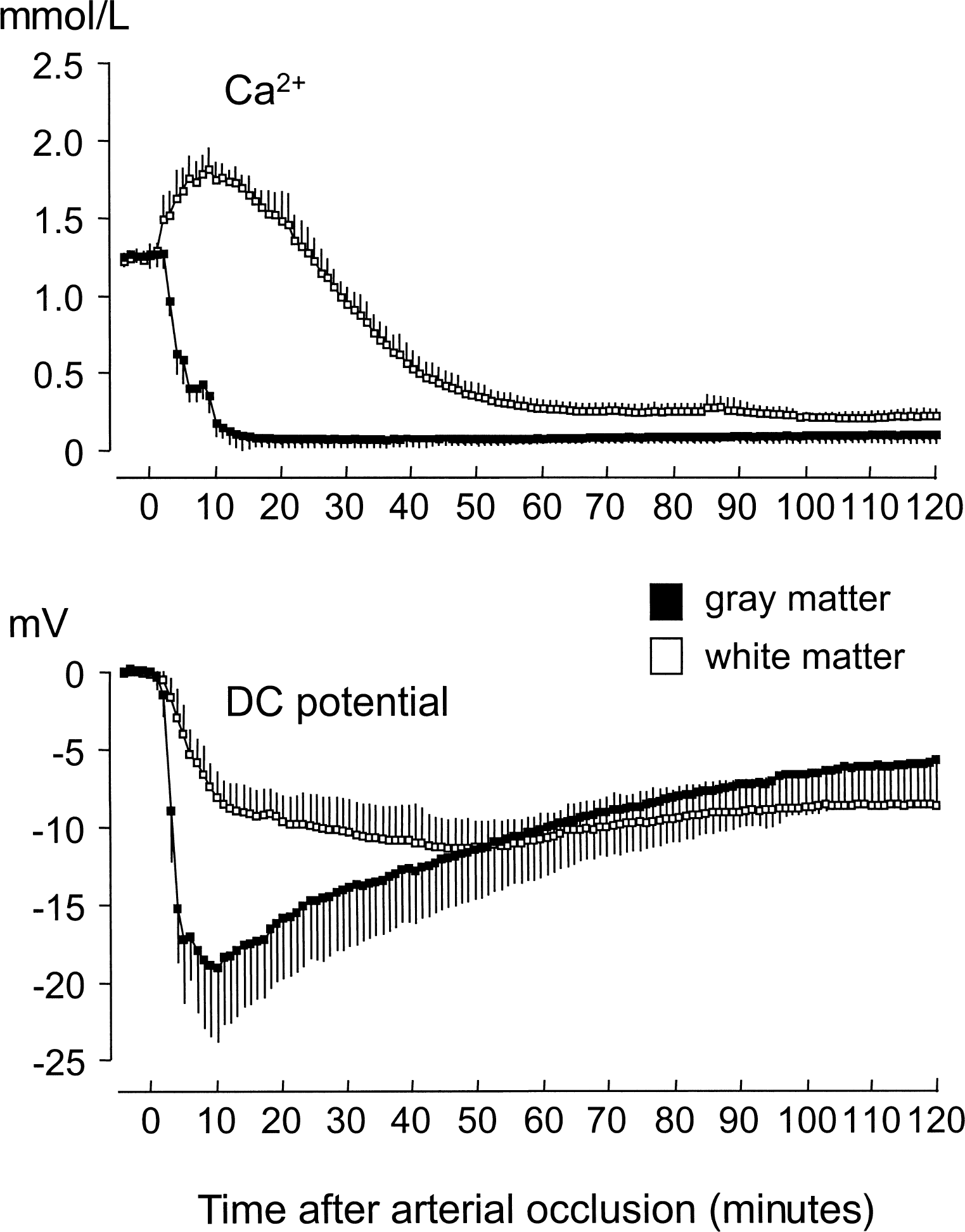
Changes in direct current (DC) potential and extracellular concentration of Ca2+ measured in cortical gray matter (n = 11) and subcortical white matter (n = 9) during 120 minutes of global ischemia. Data (mean ± SD) were sampled every minute from 5 minutes before (control) to 120 minutes after induction of ischemia. A negative value for the DC potential indicates a decrease from baseline. Ischemic Ca2+ concentrations tested at time points 10, 30, 60, and 120 minutes of ischemia significantly differed from control values in both gray matter and white matter. Differences between the 2 regions reached statistical significance at 10, 30, 60, and 120 minutes of ischemia; control values in the 2 regions did not differ significantly (repeated measures analysis of variance with post hoc comparison). The DC potential was significantly different in both compartments from preischemic controls after 10 minutes of ischemia and remained altered throughout the measurement. Significant differences between gray matter and white matter were reached at the 10-and 30-minute measurements after the onset of ischemia (P < 0.05; repeated measures analysis of variance with post hoc comparison), respectively.
Figure 2 shows changes of adenosine, inosine, and hypoxanthine in GM and WM during the course of ischemia. Basal levels of these purines did not differ significantly between GM and WM. In GM, adenosine levels increased immediately after induction of ischemia and reached a maximum concentration of 1.88 μmol/L after 10 minutes of ischemia. After this transient increase, adenosine levels returned roughly to control levels during the next 2 hours of ischemia. In WM, adenosine started to increase promptly after the onset of ischemia, too, but the slope of this increase was not as steep. Adenosine concentrations at 10 minutes after ischemia were significantly less in WM than in GM. A maximum concentration of 1.88 μmol/L was reached only in the 30-minute sample. After this peak, the concentration slowly declined, staying well above GM values until the end of the observation period. In comparison with the transient rise in adenosine, inosine and hypoxanthine concentrations increased continuously in GM and WM throughout ischemia, the rise in WM being somewhat smaller. Inosine levels seemed to reach a plateau in GM after approximately 1 hour, whereas inosine levels in WM and hypoxanthine levels in GM and WM peaked at the end of the 2-hour ischemia period.
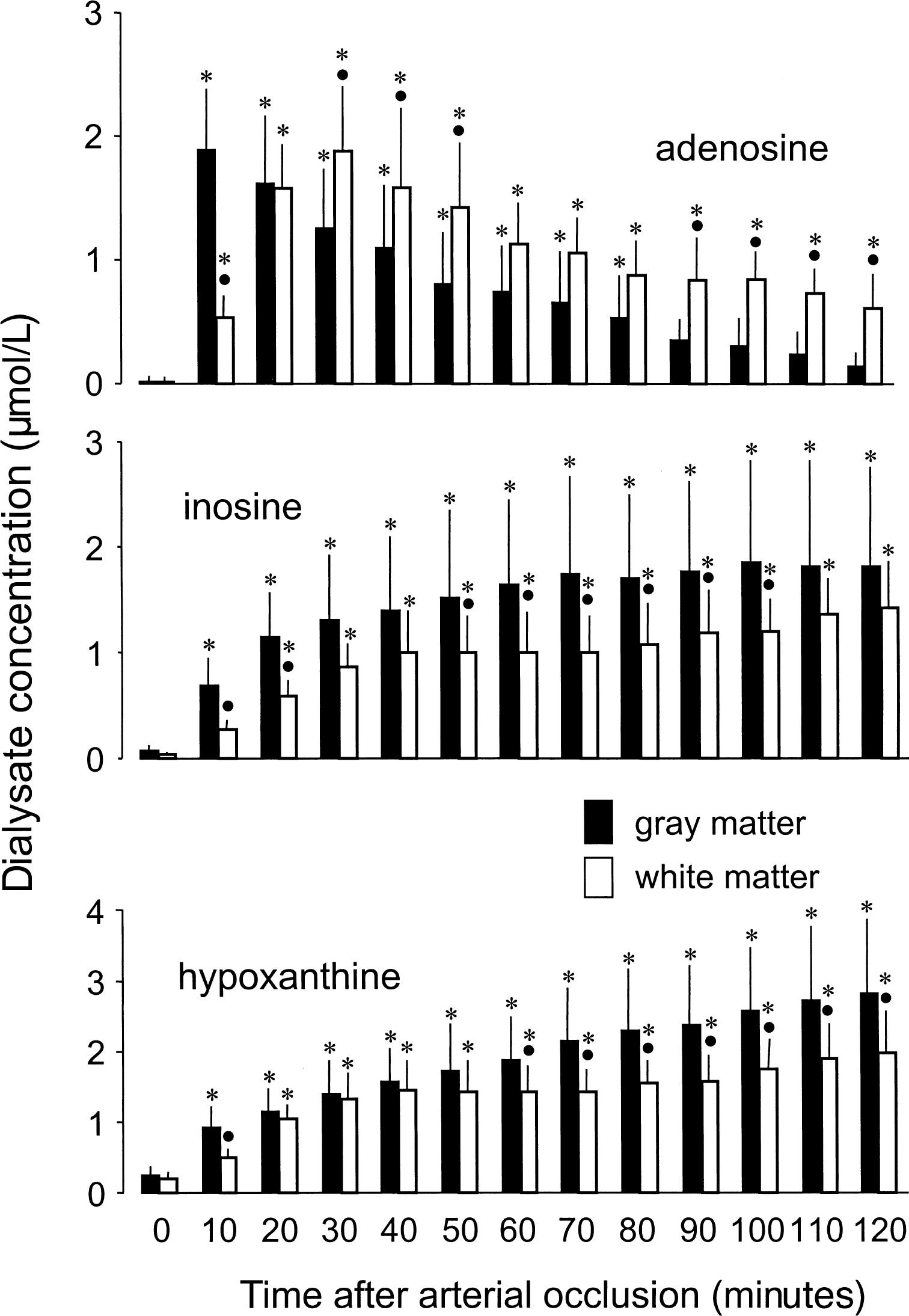
Changes in extracellular adenosine, inosine, and hypoxanthine concentrations measured in cortical gray matter (n = 10) and subcortical white matter (n = 8) during 120 minutes of global ischemia. Dialysate was sampled for 10 minutes preceding the times indicated in the figure. Data are expressed as mean ± SD. * P < 0.05, significantly different from preischemic control. •P < 0.05, significantly different from gray matter (repeated measures analysis of variance with post hoc comparison).
Temporal changes of neurotransmitter amino acids GABA and glutamate are illustrated in Fig. 3. In general, ischemic extracellular accumulation of both amino acids was immediate in GM and delayed by almost 1 hour in WM. The slope of the glutamate increase was steeper in the early phase of ischemia and reached a plateau almost 1 hour after the induction of ischemia, whereas the accumulation rate of glutamate in WM and of GABA in both GM and WM was continuous until the end of the observation period.
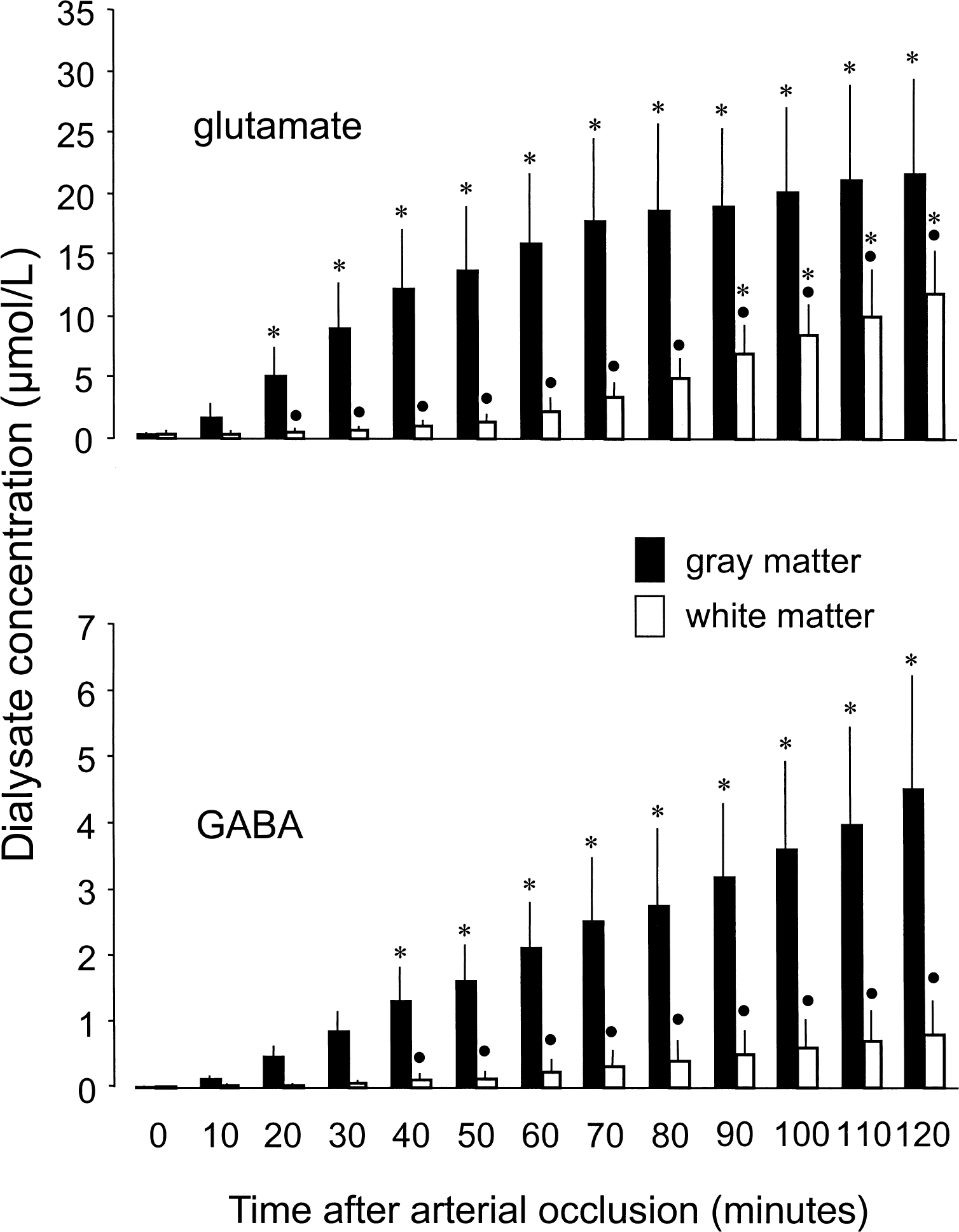
Changes in extracellular gamma-aminobutyric acid (GABA) and glutamate concentrations measured in cortical gray matter (n = 8) and subcortical white matter (n = 7) during 120 minutes of global ischemia. Data are expressed as mean ± SD. Dialysate was sampled for 10 minutes preceding the times indicated in the figure. * P < 0.05, significantly different from preischemic control. •P < 0.05, significantly different from gray matter (repeated measures analysis of variance with post hoc comparison).
DISCUSSION
To the authors' knowledge, this is the first study addressing changes in the levels of adenosine and its metabolites in WM ischemia. The most important finding is a marked difference between GM and WM in the course of ischemic adenosine accumulation. The peak of adenosine is delayed by about 20 minutes in WM; in contrast to GM, adenosine levels remain elevated throughout the ischemic period. Most significantly, the prolonged adenosine elevation in WM temporally correlates with the delayed extrusion of [Ca2+]o from the extracellular space. The measurements of the direct current potential and of extracellular GABA and glutamate revealed steady but smaller and slower alterations in WM than in GM.
Adenosine may indicate slower ATP breakdown in white matter
Under ischemic conditions, adenosine accumulates in the tissue and particularly in the extracellular space as a result of massive and rapid ATP catabolism (Hagberg et al., 1987; Kleihues et al., 1974; Phillis et al., 1996; Siesjö, 1978). Adenosine is mainly formed intracellularly and transported out of the cell via a nucleoside transporter. Extracellularly, adenosine deaminase may account for the conversion of adenosine to inosine and hypoxanthine (Meghji et al., 1988), and perhaps for the extracellular accumulation of these substances in ischemic tissue (Hagberg et al., 1987). An intracellular conversion after cellular reuptake of adenosine by means of either facilitated diffusion or Na+-dependent active transport (Meghji and Newby, 1990) probably plays a minor role under ischemic conditions. Uptake by facilitated diffusion depends on a concentration gradient, and it is unlikely that the intracellular levels of adenosine were lower than those in the extracellular space. Na+-dependent active transport requires an appropriate Na+ concentration gradient, which is presumably absent in the ischemic brain. In comparison to GM structures, WM has long been recognized to possess much lower regional perfusion as well as glucose and oxygen utilization (Heiss et al., 1995; Sakurada et al., 1978; Sokoloff et al., 1977). Even if there is no simple a priori relation between metabolic rate and amount of energy-rich phosphates, one would expect a lower content of high-energy compounds and a slower ischemic consumption of them in WM, considering the close relationship between functional activity and metabolic rate. The results of studies on the content of energy stores in white matter are, however, contradictory. Ex vivo results from rapid-frozen brains of different species (Müller et al., 1970; Whittingham et al., 1995) show a lower amount of phosphocreatine and ATP in WM, if values are expressed on a dry-weight basis. Taking dry-weight rather than wet-weight values seems to be reasonable because the water content differs between the two central nervous system compartments (Müller et al., 1970). On a lipid-free dry-weight basis, differences in energy-rich substrates between GM and WM were not as pronounced (Folbergrováet al., 1970; Gatfield et al., 1966). In vivo determinations derived from magnetic resonance spectroscopy revealed a wide variety of results ranging from higher phosphocreatine concentrations (Wang and Li, 1998) and higher ratios of phosphocreatine to nucleoside triphosphates in GM (Mason et al., 1998; Wang and Li, 1998) to an equal distribution between the two regions (Buchli et al., 1994) and finally higher concentrations of energy-rich compounds and ratios in WM (Holtzman et al., 1996; Tsuji et al., 1996).
Referring to the unique although rather old study of Müller et al. (1970), in ischemic conditions the breakdown of energy-rich phosphates is slower in WM than in GM. Consequently, one would expect a slower ischemic elevation of adenosine in the extracellular space of WM regions that would reflect the slower loss of energy reserves, and perhaps also a smaller total amount of accumulated adenosine. The authors' data indeed show a delayed peak of adenosine in WM, but surprisingly the maximal accumulation was the same in WM and GM. Extracellular concentrations assessed by microdialysis depends not only on altered transmembrane fluxes but also on changes of the extracellular space, which is known to shrink during ischemia. Comparative data on this subject are scarce, but in a first approximation one would assume that shrinkage of the extracellular space would be of the same order of magnitude in the two compartments (Voríŝek and Sykova, 1997). Observations from studies using diffusion-weighted magnetic resonance imaging (Kuroiwa et al., 1998) suggest an even smaller reduction in extracellular space volume in WM than in GM during early ischemia. Therefore, the unexpectedly high concentrations of adenosine in WM cannot be attributed to a larger decrease of the extracellular space and may even be underestimated. Particularly with regard to the magnitude of the determined adenosine release, discussions on the autoprotective properties of adenosine become relevant.
Adenosine may indicate intrinsic protection of white matter
Numerous in vitro and in vivo studies have documented neuroprotective effects of adenosine in GM structures (Fredholm, 1997; Rudolphi et al., 1992; Schubert and Kreutzberg, 1993). Mechanisms underlying these effects include blockade of pre-and postsynaptic Ca2+ channels, thereby counteracting the release of excitotoxic neurotransmitters and inhibiting or delaying excessive membrane depolarization by increasing the postsynaptic conductance of K+ and Cl− ions. These effects are presumably mediated by adenosine receptor activation coupled to activation of protein kinase C by a guanosine-triphosphate (GTP) binding membrane protein. Evidence exists that adenosine plays a protective role in WM ischemia, too, even though synaptic structures do not exist in this compartment (Fern et al., 1994, 1995). The results of in vitro studies using the rat optic nerve as a model for central white matter tracts have suggested that the protective effect of adenosine is also mediated at least partially by activation of protein kinase C that, in turn, is linked to the downregulation of Na+ channels and of membrane transporters such as the Na+-Ca2+ exchanger (Mené et al., 1992), thereby reducing ischemic Ca2+ influx.
The putative role of slower and extended adenosine elevation in WM ischemia may be related to smaller and slower alterations of other variables such as tissue depolarization and transmembrane shifts of various ions and neuromodulators. In the cascade-like pathophysiologic sequelae of a perfusional disturbance of the same magnitude, metabolic derangement is presumably slower in WM than in GM. Tissue depolarization and thereby processes linked to depolarization such as influx of Na+ and Ca2+ into cells by means of voltage-gated ion channels would be slowed. Preliminary determinations of extracelluar Na+ in the same model have revealed such slower extrusion of Na+ from the extracellular space in WM, paralleling the course of the direct current potential in this compartment (Kumura et al., 1999). Transmembrane ion gradients would remain in normal ranges for longer periods, leaving, in a positive feedback-like fashion, ATPase to some extent preserved. A system like the Na+-Ca2+ exchanger, which is very susceptible to changes of Na+ ion gradients, would remain intact for longer periods. These ameliorative effects of slower ATP depletion would be enhanced by sustained generation of intermediate products, in particular adenosine, which would exert its influence on protein kinase C (PKC) activation as described above. A result of this multifactorial ameliorative process is perhaps, in a final step, the prolonged maintenance of transmembrane Ca2+ gradients in WM as was found in the current study and also in vitro under anoxic conditions (Brown et al., 1998).
Interestingly, the peak of adenosine in WM after 30 minutes of ischemia and its subsequent decrease correspond well with the beginning of Ca2+ influx after 30 minutes of ischemia. The interpretation that this moment marks a turning point in the development of ischemic damage in WM is supported by the observation that the temporal threshold for morphologic damage in WM has been described to be in the range of 30 minutes (Pantoni et al., 1996). Even though interpretations on the basis of rather descriptive results obtained in in vivo experiments are speculative, the current authors suggest that the delayed and sustained accumulation of adenosine in the extracellular space of WM provides an autoprotective mechanism that, compared with GM, may help to hold up ischemic tissue depolarization and Ca2+ influx into the cells.
Whether GABA accumulation in the subsequent phase of ischemia provides another correlate of intrinsic neuroprotection seems more obscure, given that elevation of GABA levels was not only delayed but was less pronounced. GABA has been shown in vitro to possess such properties (Fern et al., 1994), and it has been suggested that the effects of GABA may be achieved by similar intracellular pathways, as have been described for adenosine. In view of the in vitro results, one could assume sequential or even synergistic effects of the two substances in the later phase of ischemia. In this context, the findings of a former study in a focal ischemia model may be relevant, describing partial recovery of cortical dysfunction attributable to WM ischemia to occur even after 1 hour of transient arterial occlusion (Graf et al., 1990). Evidence for a longer-lasting ischemic tolerance of WM, as provided by the current study and by earlier studies, implies that the specific pathophysiologic characteristics of WM ischemia should be taken into account in the search for more effective strategies in the treatment of brain ischemia.
Conclusion
The comparison between GM and WM ischemia revealed a slower and delayed extracellular accumulation of adenosine in WM versus GM. This delay corresponds well with the delayed [Ca2+]o decrease in WM. The authors propose that the slower accumulation of adenosine in WM reflects a slower ATP breakdown, and that a delayed but persistent adenosine accumulation provides an autoprotective mechanism, possibly by delaying cellular Ca2+ loading. These observations might help to explain the higher ischemic tolerance of central WM.
Footnotes
Acknowledgments:
The authors thank Mrs. P. Gabel and Dipl. Ing. B. Radermacher for valuable technical assistance.