
Abstract
The authors evaluated the neurobehavioral and neuropathologic sequelae after traumatic brain injury (TBI) in transgenic (TG) mice expressing truncated high molecular weight neurofilament (NF) protein fused to beta-galactosidase (NFH-LacZ), which develop Lewy body-like NF-rich inclusions throughout the CNS. TG mice and their wild-type (WT) littermates were subjected to controlled cortical impact brain injury (TG, n=19; WT, n=17) or served as uninjured controls (TG, n =11; WT, n =11). During a 3-week period, mice were evaluated with an array of neuromotor function tests including neuroscore, beam balance, and both fast and slow acceleration rotarod. Brain-injured WT and TG mice showed significant motor dysfunction until 15 days and 21 days post-injury, respectively (P < .025). Compared with brain-injured WT mice, brain-injured TG mice had significantly greater motor dysfunction as assessed by neuroscore (P < .01) up to and including 15 days post-injury. Similarly, brain-injured TG mice performed significantly worse than brain-injured WT mice on slow acceleration rotarod at 2, 8, and 15 days post-injury (P < .05), and beam balance over 2 weeks post-injury (P < .01). Histopathologic analysis showed significantly greater tissue loss in the injured hemisphere in TG mice at 4 weeks post-injury (P < .01). Together these data show that NFH-LacZ TG mice are more behaviorally and histologically vulnerable to TBI than WT mice, suggesting that the presence of NF-rich inclusions may exacerbate neuromotor dysfunction and cell death after TBI.
Neurofilaments (NF) are intermediate filament members of the neuronal cytoskeleton that are composed of three different polypeptide subunits, NF-light (NFL; 70 kd), NF-medium (NFM; 160 kd), and NF-heavy (NFH; 210 kd) (Hoffmann and Lasek, 1975). Although myelinated axons contain both NF and microtubules, NF represent the major cytoskeletal element, making up the bulk of the axonal volume. Recent studies have shown that inhibition of NF transport into axons through the expression of an NFH-LacZ fusion protein in transgenic (TG) mice resulted in severe inhibition of radial growth of large myelinated axons, suggesting a biological role for NF proteins in growth and maintenance of axonal caliber (Yamasaki et al., 1992; Ohara et al., 1993; Eyer and Peterson, 1994; Tu et al., 1995). In several human neurodegenerative diseases including Parkinson's disease (PD), diffuse Lewy body dementia (DLBD), or amyotrophic lateral sclerosis (ALS), accumulation of NF-rich inclusions as well as disruption of NF networks in selectively vulnerable neurons has been observed (Goldman and Yen, 1987; Itoh et al., 1992; Trojanowski and Lee, 1994; Tu et al., 1997b). Although the precise biological mechanisms underlying the formation of NF-rich inclusions and their functional consequences are still unclear (Pollanen et al., 1993; Trojanowski and Lee, 1994), increasing evidence suggests that inclusions could have deleterious effects on neuronal function and survival (Trojanowski et al., 1993; Coté et al., 1993; Collard et al., 1995; Galvin et al., 1997; Tu et al., 1997b).
Transgenic mice expressing excessive levels of NFL, NFM, or NFH proteins show massive NF accumulation in perikarya, dendrites, and axons of spinal motor neurons, indicating that altered NF metabolism may result in the formation of NF-rich inclusions (Lee et al., 1994; Collard et al., 1995; Tu et al., 1995). These mice eventually develop ALS-like neuronal cell loss. Recently, a TG mouse which expresses truncated mouse NFH protein fused to beta-galactosidase (LacZ) was shown to develop inclusions composed of all three endogenous NF proteins and the NFH-LacZ fusion protein in neurons throughout the CNS (Eyer and Peterson, 1994; Tu et al., 1997a). Loss of cerebellar Purkinje's cells observed in NFH-LacZ TG mice was delayed, similar to neuronal loss in age-dependent human neurodegenerative diseases such as PD and DLBD (Tu et al., 1997a). Although the numbers of affected neurons and size of inclusions became stable by 2 months of age, the onset of clinical phenotype (i.e., tremor, ataxia, or muscular weakness) was substantially delayed in NFH-LacZ TG mice, appearing only after 1 year of age (Tu et al., 1997a).
Since the formation of NF-rich inclusions has been thought to be involved in the pathogenesis of certain neurodegenerative diseases, we hypothesized that NF-rich inclusions and concomitant alterations in NF organization throughout the neurons may render affected neurons more vulnerable to insults or stress. Traumatic brain injury (TBI) in rats induces acute disturbances in somodendritic NF proteins in regions that later exhibit neuronal loss (Posmantur et al., 1994, 1996; Saatman et al., 1998). In addition, NF perturbation is a well-established characteristic of traumatically injured axons (Povlishock, 1993). Therefore, in the present study, controlled cortical impact (CCI) brain injury was induced in NFH-LacZ TG mice to examine the hypothesis that the pre-existing NF pathology in these mice influences their neurobehavioral and histopathologic response to trauma. To our knowledge, this is the first study to use mice with altered expression of a major cytoskeletal protein to explore links between traumatic and neurodegenerative processes.
MATERIALS AND METHODS
Mice
A breeding pair of NFH-LacZ transgenic mice was a generous gift from J. Eyer and A. Peterson. The NFH-LacZ construct and the production of NFH-LacZ TG mice have been described (Eyer and Peteron, 1994; Tu et al., 1997a). Briefly, the NFH-LacZ construct was microinjected into male pronuclei of B6C3 zygotes. The injected embryos were transferred to the oviducts of pseudopregnant B6C3 females. Heterozygous TG and age-matched wild-type (WT) littermate control mice were bred on a B6C3 background and a colony of these mice was maintained in the laboratories of one of the authors (JQT). The presence of the transgene was determined for each mouse used in the current study by the polymerase chain reaction on DNA extracted from tail biopsy specimens, using primers identical to that described in Eyer and Peterson (1994). The presence or absence of the NFH-LacZ transgene was also confirmed post-hoc by immunohistologic examination of brain sections. All NFH-LacZ mice had easily identifiable neurofilamentous inclusions in virtually all neurons, as has been described for these mice (Eyer and Peterson, 1994; Tu et al., 1997a).
Controlled cortical impact brain injury
All animal use procedures were in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals (Publication No. 85-23, 1985) and were approved by the University of Pennsylvania Institutional Animal Care and Use Committee. Mice (n=58) were anesthetized with sodium pentobarbital (65 mg/kg, intraperitoneally) and placed in a stereotactic head holder. Ointment was applied to their eyes to prevent drying during surgery. The skull was exposed, and a 5-mm craniotomy was performed over the left parietotemporal cortex between the coronal and rhomboid sutures, keeping the dura mater intact. Forty-five minutes after administration of anesthesia, animals were either subjected to CCI of mild severity (n = 36) or sham (no impact, n = 22) treatment. Brain injury CCI was performed using a pneumatic impactor, a procedure that has been previously described in detail (Dixon et al., 1991). The impactor has a 3-mm diameter round tip driven at a velocity of 4.8 to 5.2 m/sec. The depth of the impact was 1.0 mm and the duration was kept constant at 200 milliseconds. The impactor rod was angled 20° from the vertical plane so that the impactor was perpendicular to the exposed dura mater. The upper rod was attached to a linear velocity displacement transducer (Shavitz Model 500HR). Animals remained in the stereotactic head holder during CCI injury and the dura was kept moist with saline at all times. After CCI, the craniotomy was covered with a 7-mm square of plastic and the scalp was closed with 4-0 silk suture. Core body temperature was recorded by insertion of a plastic coated thermocouple into the rectum, while temporalis muscle temperature was monitored through placement of a temperature needle probe into the temporalis muscle.
Behavioral training and testing
Sixteen female and 14 male NFH-LacZ TG mice and 15 female and 13 male WT littermate controls were used. All mice were from 3 to 6 months of age; WT were 4.1 ± 0.7 (mean ± SD) months old and TG mice were 4.2 ± 1.0 months old. Mice of this age were selected to assure that the number of inclusions had reached its plateau (after 2 months of age) but no age-dependent neurodegeneration had begun (approximately 12 months of age). Mice were acclimated to the rotarod test over a 2-day period before the injury, and to the beam balance 1 day before the injury. Post-injury neuromotor function was evaluated at 2, 8, 15, and 21 days post-injury using the composite neuroscore described below. The ability to maintain balance on an accelerating rotarod at two different rotational accelerations was evaluated at 2, 8, and 15 days post-injury. The ability to balance on a narrow beam (beam balance) was assessed every other day until 14 days post-injury. Because this study was the first in our laboratory to evaluate behavioral deficits in mice by the beam balance task, this test was evaluated more frequently than other tasks. All behavioral testing was performed by two trained observers who were unaware of both injury status and genotypes of the test subjects.
Composite neuroscore. This battery of motor tests was previously developed for the rat (McIntosh et al., 1989). and subsequently adapted for mice (Raghupathi et al., 1998; Murai et al., 1998). The mice were scored on a 4 (normal) to 0 (severely impaired) integral scale for (l) right forelimb flexion on suspension by the tail, and (2) decreased resistance to lateral pulsion toward the right. Because CCI was induced over the left parietal cortex, motor deficits exhibited by injured mice were markedly more profound on the right side. In addition, animals were tested for their ability to stand on an inclined plane (angle board) in each of four directions (face down, left, up, and right) for 5 seconds. The mice were scored according to the decrease in angle from their pre-injury level, where 0° = 4, 2.5° = 3, 5° = 2, 7.5° = 1, and 10° or more = O, and the scores for each of the 4 directions were averaged for inclusion in the composite scores. A composite neuroscore (0-12) was generated by combining the scores for each of these three tests.
Beam balance. This test of motor function was originally developed for the rat (Clifton et al., 1991) and was adapted for mice (Mikawa et al., 1996). Mice were placed on a suspended narrow wooden beam (0.7-cm width) and were rated on a modified six-point scale for their performance on the beam as follows: 5, balances with steady posture; 4, grasps the side of the beam and/or has unsteady movements; 3, hugs the beam without falling; 2, hangs off the beam without falling; 1, hangs on the beam and falls off; 0, does not attempt to balance on the beam. Animals were trained on the beam 24 hours before injury until they achieved a rating of 5.
Rotarod. The rotarod test described by Hamm, et al. (1994) was modified for use with mice (Raghupathi et al., 1998; Murai et al., 1998). The latency to balance on a 36-mm outer diameter rotating rod which has a rubber surface was measured at two different rotational accelerations (fast and slow). The sensitivity of this task for evaluating motor dysfunction in brain-injured rats was previously described by Hamm, et al. (1994). The initial velocity was 5 rpm for fast acceleration (17.3 rpm/10 sec) and for slow acceleration (5.0 rpm/10 sec). All animals were acclimated to the rotarod test paradigm 2 days before the injury. The rotarod tests were performed 24 hours before injury to score the baseline latencies for each animal. Each trial ended when the animal fell off the rotarod or gripped the rod and spun around the lowest point. Four trials for the fast acceleration rotarod were performed and the average of the two middle latencies was taken as baseline, while two trials were performed and the average of the latencies was used as baseline for the slow acceleration rotarod. Evaluation of rotarod tests after the injury was based on the individual scores relative to their baseline latencies.
Histopathologic analysis
At 4 weeks post-injury, a randomly selected subset of animals (10 brain-injured TG mice and 8 brain-injured WT mice) were reanesthethized (100 mg/kg sodium pentobarbital) and perfused transcardially with heparinized saline. Brains were removed and postfixed with 4% paraformaldehyde overnight. Paraffin-embedded coronal sections (6 µm in thickness) were obtained and collected onto slides consecutively. Sections were stained with hematoxylin and eosin or 1% cresyl violet (Nissl) every 120 µm to evaluate histopathologic changes. To provide a more accurate determination of the irregularly shaped lesion caused by CCI, we evaluated the brain tissue loss volumetrically. Tissue loss in brain-injured mice was measured using nine sections, distributed from bregma 0.14 to −3.6 mm. Percent of volumetric tissue loss was calculated for the injured hemisphere relative to the contralateral hemisphere according to the formula described below:
Percent Volumetric Tissue Loss =
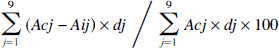
Statistical analysis
Composite neuroscore, beam balance, and rotarod score (percent baseline latencies) are nonparametric data, and are represented by a median score. Comparisons between multiple groups were made using the Kruskal-Wallis test followed by the Mann-Whitney U test for comparing pairs of groups. Tissue loss data are parametric and are represented by mean percentages. Comparisons between groups were made using unpaired student t-tests. A P value of less than .05 was considered statistically significant.
RESULTS
Neurobehavioral status before brain injury
Approximately 80% of WT mice and TG mice did not fully spread their hindlimbs when suspended by the tail, suggesting that both WT and TG mice have subtle hindlimb dysfunction before injury (data not shown). For this reason, we did not include hindlimb function in the composite neuroscore in the present study, as we have in previous studies of brain-injured mice (Murai et al., 1998). For the angle board task, no significant difference was observed in the sum of the baseline angles for four directions between WT mice (152 ± 10.8°; mean ± SD) and TG mice (151.0 ± US) (P = not significant by unpaired t-test). For the beam balance task at 24 hours before injury, all WT and TG mice quickly learned to balance on the narrow beam. However, the baseline latency of the fast acceleration rotarod test was 13.1 ± 3.5 seconds for WT mice compared with 10.7 ± 3.3 seconds for TG mice (P < .01 by unpaired t-test). This result shows that TG mice are not identical to the WT animals in regard to the pre-injury baseline performance of the rotarod task. Post-injury performance for rotarod was therefore reported as percent of baseline. Before brain injury, no difference was observed in core brain (temporalis muscle) temperature between WT mice (37.7°C) and TG mice (37.5°C).
Evaluation of neurobehavioral function after CCI
Composite neuroscore. Fifteen minutes after brain injury, core brain (temporalis muscle) temperature of injured WT mice (37°C) and injured TG mice (37.6°C) did not differ from pre-injury values or between groups. Both brain-injured WT mice and TG mice showed significant motor deficits relative to their respective uninjured controls at 2, 8, and 15 days post-injury (P < .05) (Fig. 1). By 21 days after injury, brain-injured WT mice had recovered to the extent that there was no significant difference compared with uninjured WT mice; however, brain-injured TG mice still showed a significant deficit at this time point compared with uninjured TG mice (P < .05). In addition, brain-injured TG mice had significantly lower composite neuroscores when compared with brain-injured WT mice at 2, 8, and 15 days post-injury (P < .025), but not 21 days (P = .053).
Composite neuroscores at 2, 8, 15, and 21 days post-injury for brain-injured (inj) and uninjured (sham) mice. The composite neuroscore reflects performance in forelimb flexion, lateral pulsion, and inclined planes tasks (total possible score=12). Bars represent median scores, while dots represent composite neuroscores for individual animals. Open bars are WT mice, and filled bars are TG mice. * = P < .001,** = P < .025, *** = P < .05.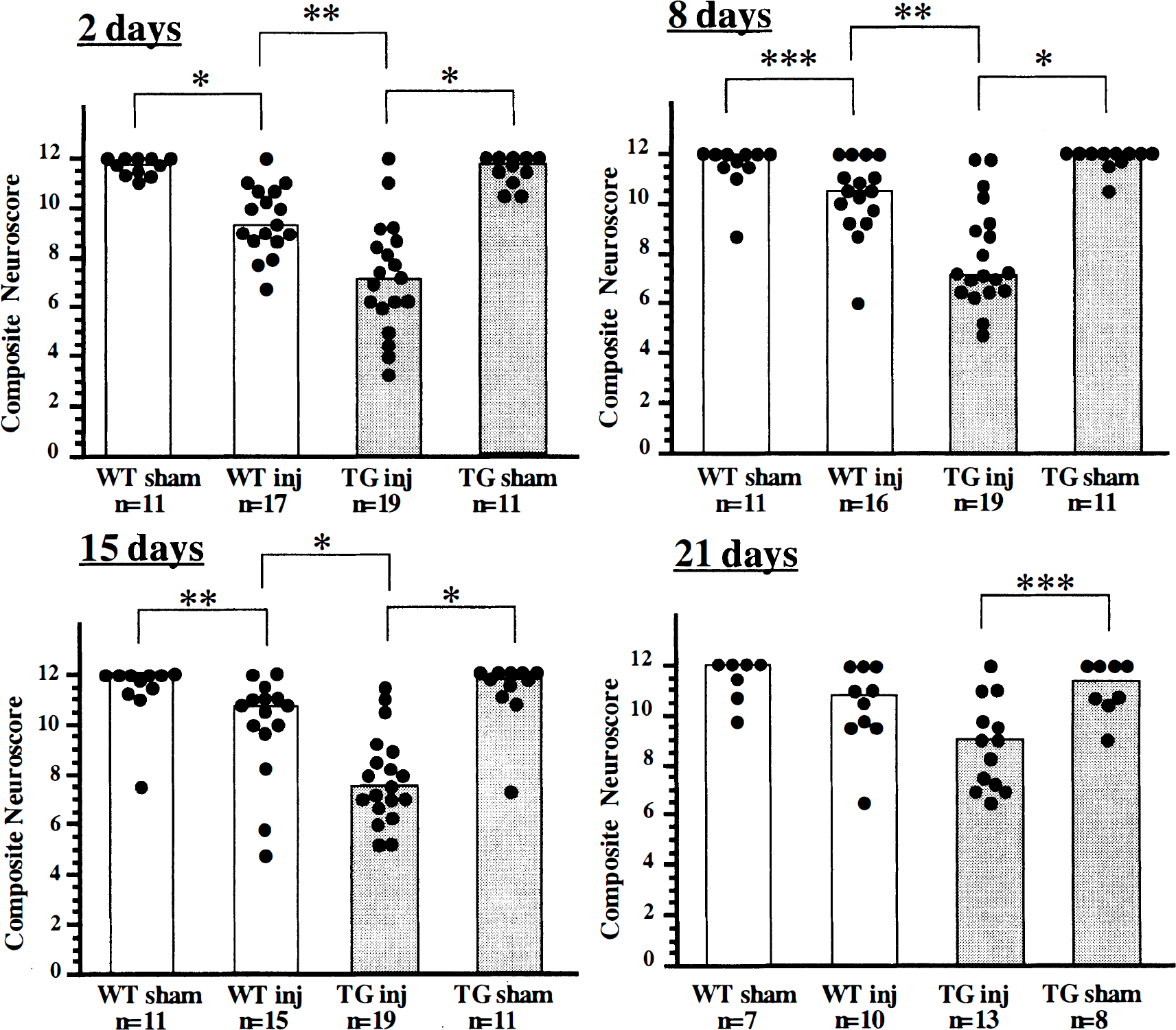
Beam balance. Beam balance performance was significantly decreased 2, 4, and 6 days after injury in both brain-injured WT and TG mice compared with uninjured controls (Fig. 2) (P < .025 and P < .01, respectively). Brain-injured WT mice gradually recovered to pre-injury level such that there were no significant deficits after 10 days post-injury. At 8 days post-injury, TG mice exhibited dysfunction, but their deficits did not reach statistical significance (P = .052). However, brain-injured TG mice showed significant deficits relative to uninjured mice which persisted from 10 to 14 days post-injury (P < .01). In addition, their scores were significantly lower than those of brain-injured WT mice up to 14 days post-injury (P < .01).
Beam balance performance evaluated every second day for 14 days post-injury. Open circles and closed circles represent median scores for brain-injured (inj) wild-type (WT) and transgenic (TG) mice, respectively, at each time point, while open squares and closed squares represent those of uninjured WT and TG mice, respectively. * = P <.01, ** = P < .025 comparison between brain-injured WT or TG mice and their respective sham group; # = P < .001, ## = P < .01 comparison between WT and TG brain-injured mice. Note that the numbers of animals tested are not the same for all time points (WT sham, n=7-11; WT inj, n=9-17; TG sham, n=7-11; TG inj, n=11-19). (○) WT injured. (●) TG injured. (□) WT sham. (■) TG sham.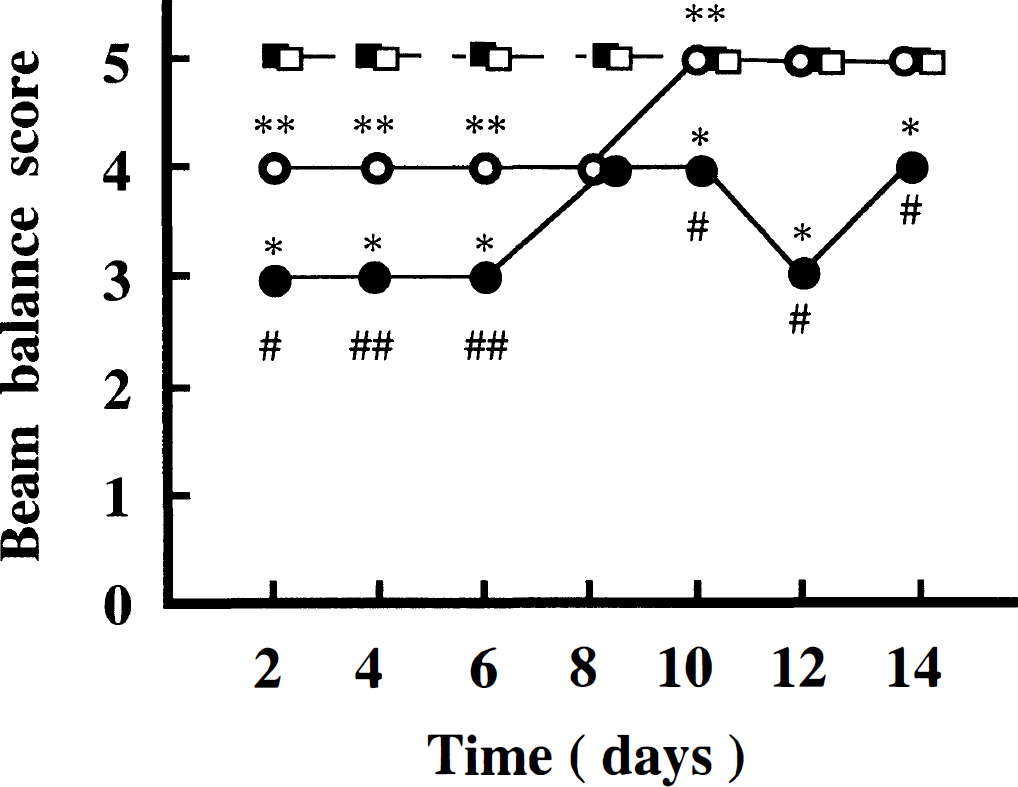
Rotarod test. Using the fast acceleration paradigm at 2 days post-injury, both brain-injured WT and TG mice showed significant deficits compared with uninjured controls (P < .001) (Fig. 3A). At 8 and 15 days postinjury, brain-injured WT mice had recovered so that there was no significant difference compared with uninjured controls. Brain-injured TG mice exhibited significantly lower scores when compared with brain-injured WT mice at 8 and 15 days post-injury (P < .025). Using the slow acceleration paradigm, brain-injured WT mice performed at a level comparable to uninjured WT control at all times (Fig. 3B). In contrast, brain-injured TG mice showed significant dysfunction up to 15 days post-injury (P < .05). Brain-injured TG mice were found to have significantly lower scores when compared with brain-injured WT mice at each time point (P < .025).
Performance of the fast acceleration 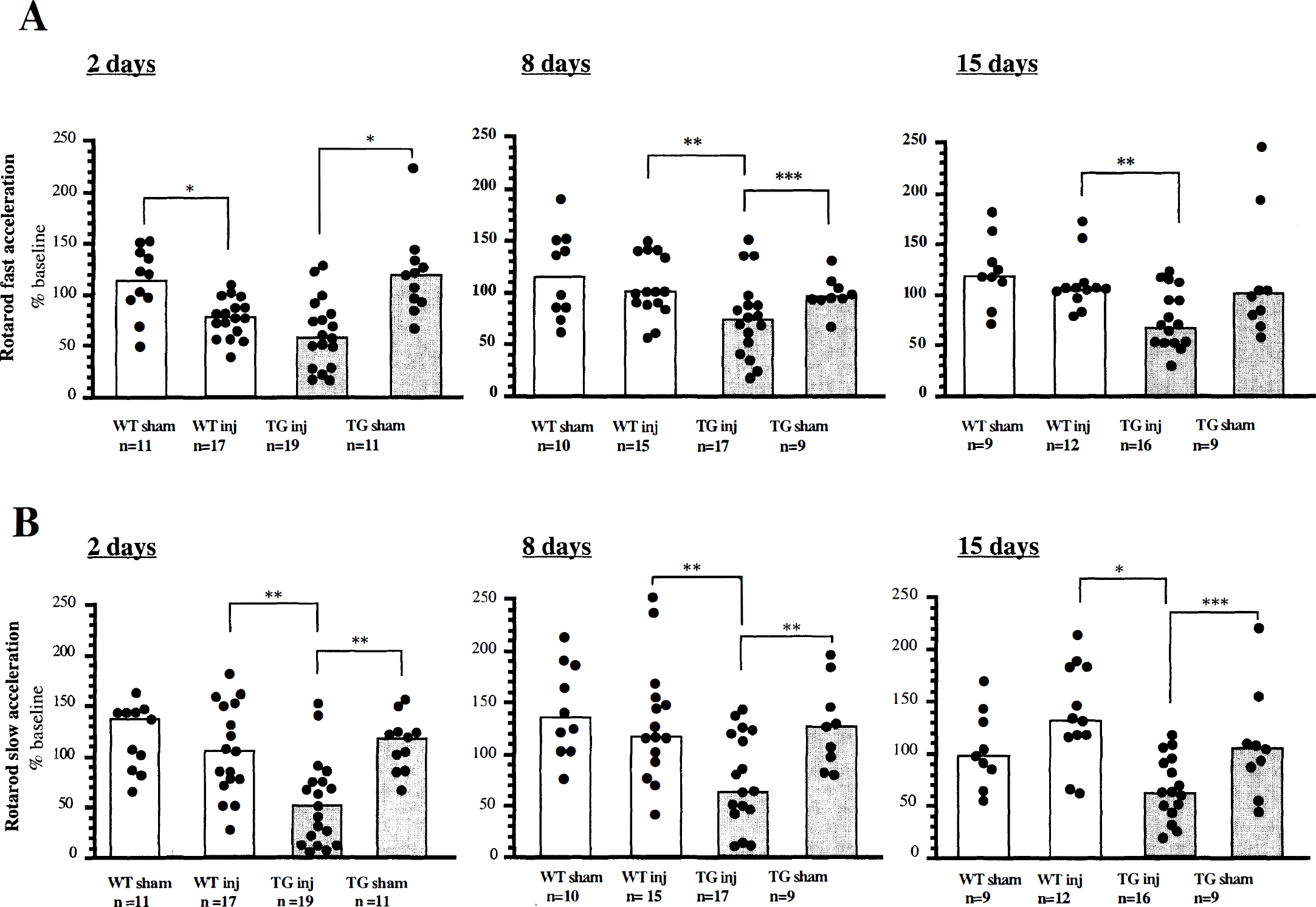
Histopathologic analysis
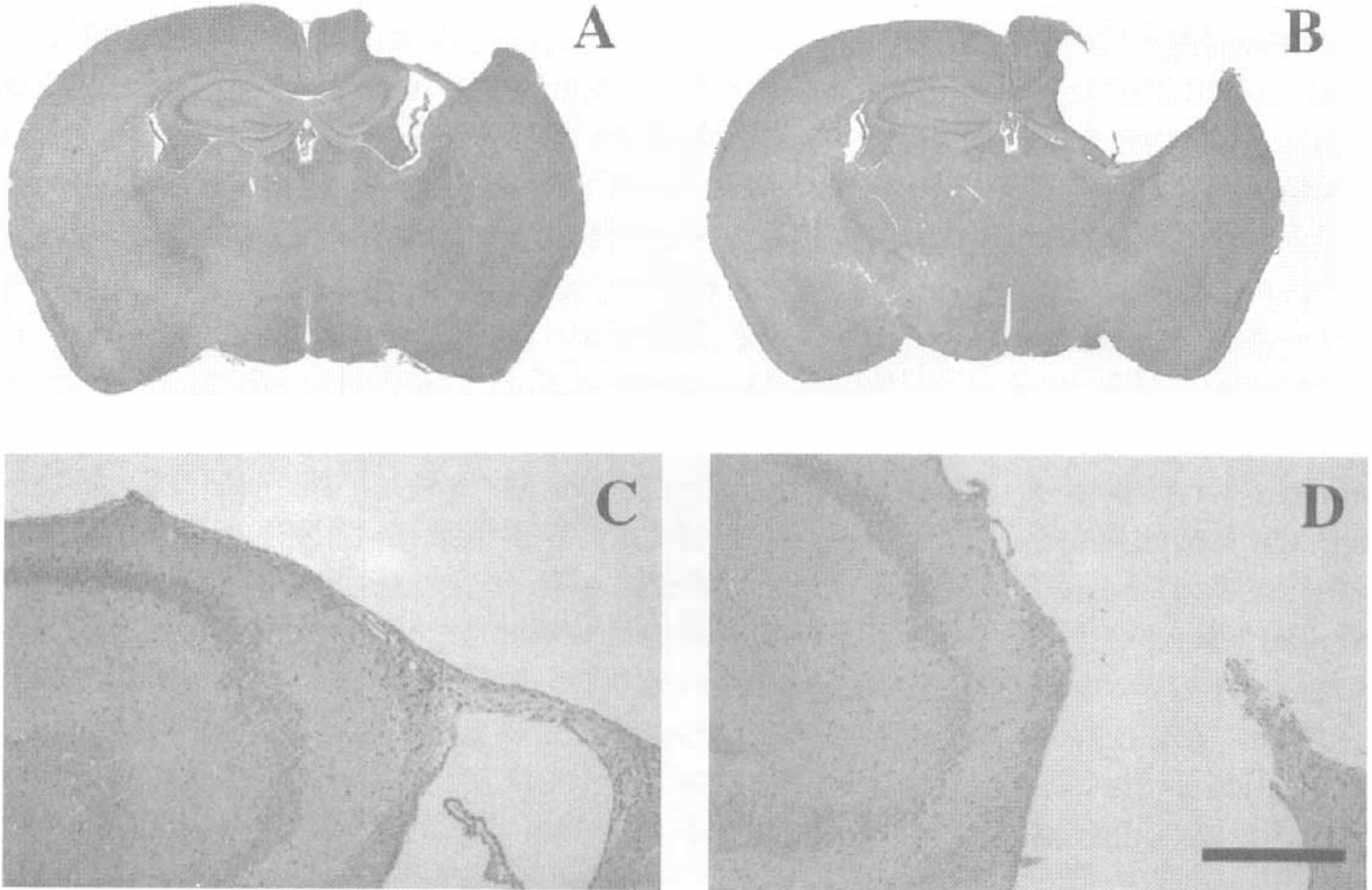
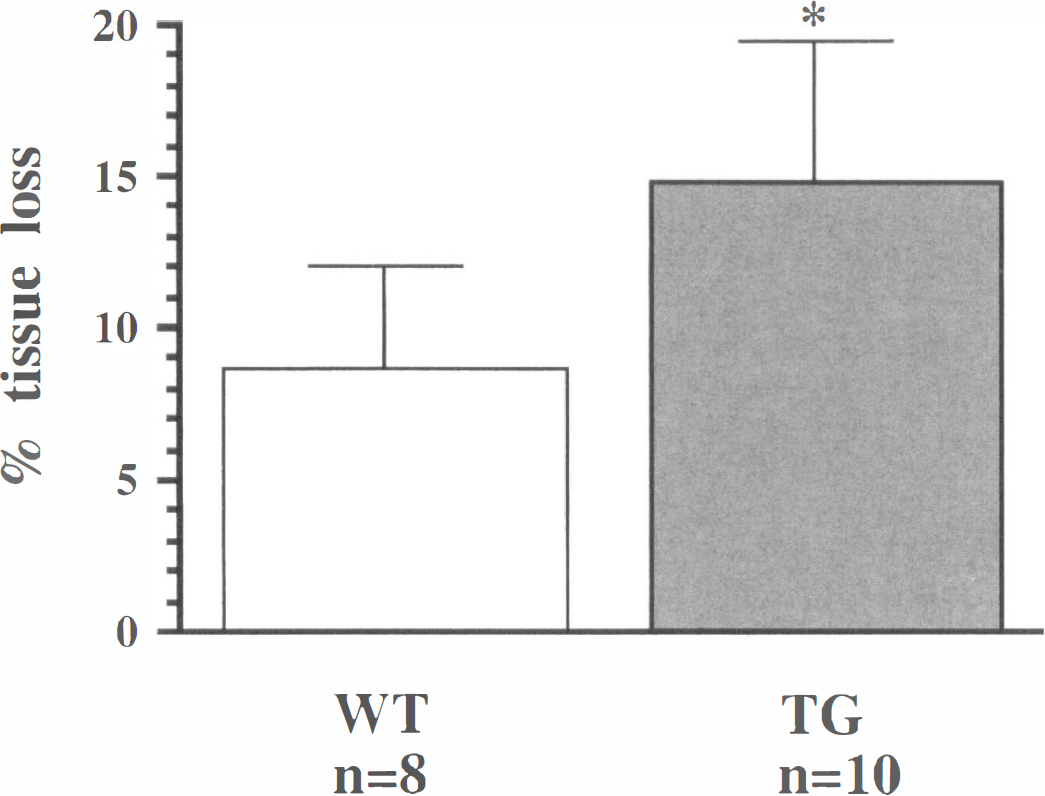
As has been described in previous reports (Eyer and Peterson, 1994; Tu et al., 1997a), NFH-LacZ mice exhibit neurofilamentous inclusions within neurons throughout the brain that are clearly detected using NF immunohistochemistry (Fig. 4). By 4 weeks post-injury, both brain-injured WT and TG mice had developed atrophy in the injured hemisphere and most had a well-demarcated necrotic cavity localized to the impact site. The necrotic cavities were always limited to the cortex in brain-injured WT mice (Fig. 5A, C). However, in most of the brain-injured TG mice, the necrotic cavities reached to the white matter, often communicating with the ventricle (Fig. 5B, D). Atrophy of the injured hemisphere was also observed caudal and rostral to the boundaries of the necrotic cavity in both WT and TG mice. Hippocampal atrophy and/or distortion with associated CA2/CA3 pyramidal neuron loss, commonly observed at this chronic postinjury timepoint, appeared more pronounced in the TG mice. No overt morphologic changes were observed in the contralateral hemisphere in either WT or TG mice. The percentage of tissue lost in the injured hemisphere was significantly greater in brain-injured TG mice (14.8 ± 4.8%; mean and SD) when compared with brain-injured WT mice (8.7±3.4%, P < .01) (Fig. 6).
Distribution of neurofilament-rich inclusions throughout the CNS of NFH-LacZ transgenic mice. Inclusions are stained with an antibody against neurofilament-light and are observed in the cortex (particularly levels 3 and 5), hippocampus (Hipp), thalamus (Thai), and cerebellum (Cbl). All panels are at the same magnification and the scale bar = 100 µm. Photomicrographs of representative 6µm coronal sections of injured wild-type and NFH-LacZ mouse brain stained with hematoxylin and eosin. Sections obtained at approximately bregma − 1.70 mm at 4 weeks post-injury show the lesser extent of tissue loss in wild-type Percent of volumetric tissue loss in the ipsilateral hemisphere of injured mice at 4 weeks post-injury. Sections stained with hematoxylin and eosin were chosen over the region from bregma 0.14 to − 3.6 mm. Bars represent means and SD. Open bar is wild-type mice and filled bar is transgenic mice. * = P <.01.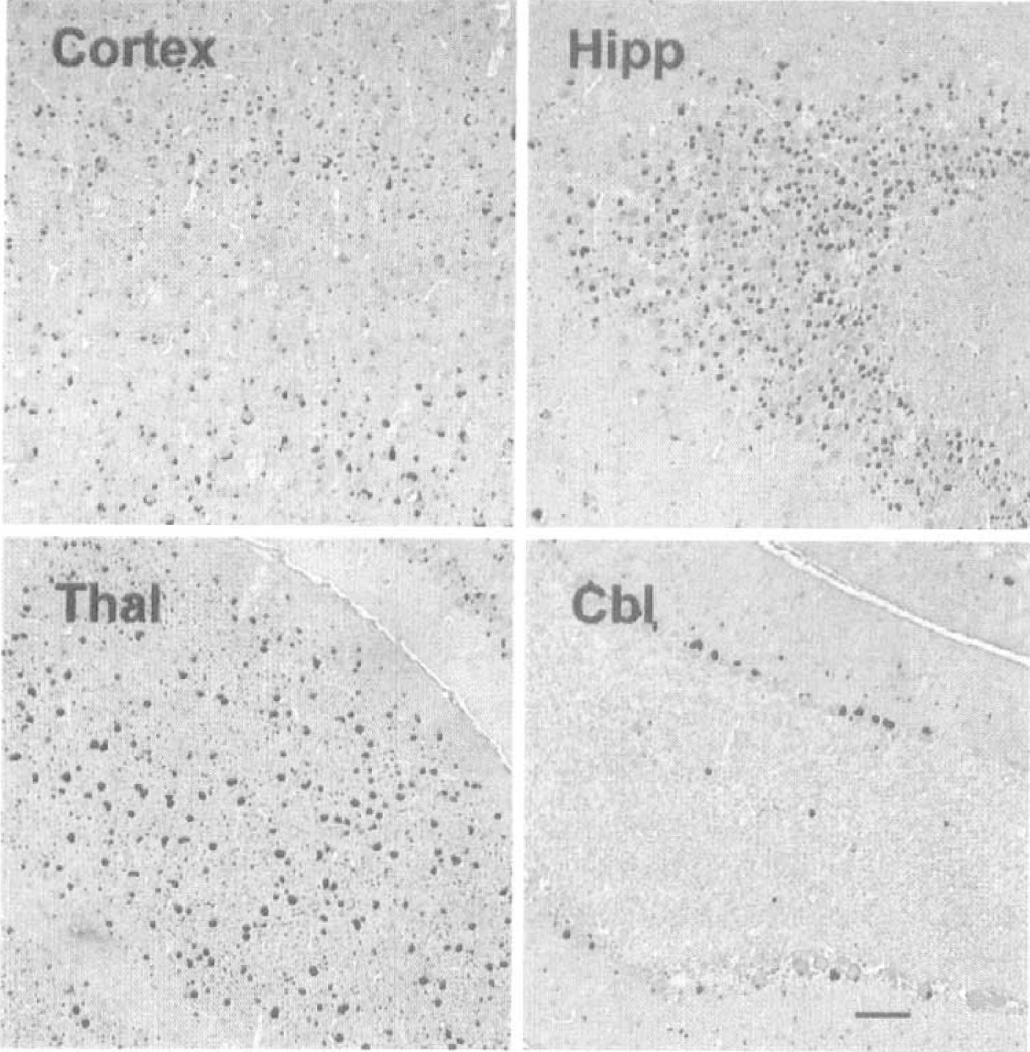
DISCUSSION
In the present study, we observed a marked exacerbation of post-traumatic neurobehavioral dysfunction and enhanced loss of brain tissue in brain-injured NFH-LacZ TG mice when compared with their WT littermates. After brain injury of this severity, neurologic motor deficits in brain-injured WT mice were modest and relatively short-lived when evaluated by neuroscore and beam balance. The performance of brain-injured WT mice on the slow acceleration rotarod task was similar to uninjured mice. However, brain-injured NFH-LacZ TG mice showed more profound and more prolonged neurobehavioral deficits in neuroscore, beam balance, and slow acceleration rotarod tests. The behavioral data were supported by histopathology that showed significantly greater tissue loss in the injured hemisphere in brain-injured NFH-LacZ TG mice than in WT mice at 4 weeks post-injury, suggesting that neuronal cell death was enhanced in NFH-LacZ TG mice after brain injury. The increased vulnerability of NFH-LacZ TG mice to TBI may be due in part to the aberrant NF distribution in nearly all neurons throughout the brain.
NF are thought to be major determinants of the caliber of large-diameter axons (Hoffmann et al., 1987), which occur in close association with other cytoskeletal elements such as microtubules and actin filaments (Tokutake, 1990), and provide mechanical support for axons (Landmesser and Swain, 1992). The core of a NF is composed of NFL protein, wheras the highly phosphorylated NFM and NFH proteins form side arms that can connect adjacent filaments (Willard and Simon, 1981), Imbalances in the expression of these three NF proteins are thought to lead to abnormal NF assembly with the subsequent formation of NF-rich inclusions in neurons (Côté et al., 1993; Xu et al., 1993; Tu et al., 1995).
Accumulation of NF-rich inclusions in selectively vulnerable neurons is one common feature of late onset neurodegenerative diseases such as PD, DLBD, and ALS (Pollanen et al., 1993; Trojanowski et al., 1993; Liberini et al., 1996; Galvin et al., 1997). To evaluate the biological role of NF proteins and to investigate their possible role in the pathogenesis of neurodegenerative diseases, a number of transgenic mice overexpressing NFH, NFM, or NFL have been generated (Côté et al., 1993; Xu et al., 1993; Lee et al., 1994; Vickers et al., 1994; Eyer and Peterson, 1994; Collard et al., 1995; Tu et al., 1995). In mice overexpressing human NFH, Côté et al., (1993) reported abnormal accumulations of NF in the perikarya and proximal axons of anterior horn motorneurons, and these mice later underwent neuronal cell loss mimicking human ALS. Collard et al., (1995) postulated that cell death in NFH-overexpressing TG mice might be due to axonal transport blockade resulting from accumulation of NF-rich inclusions in the proximal axons, which might then lead to retrograde degeneration of the affected neurons. Although an impairment of axonal transport has been proposed as a mechanism of age-related neuronal degeneration in these NF protein overexpressing TG mice, recently Tu et al., (1997a) suggested another possible mechanism. In mice expressing truncated NFH-LacZ fusion protein, only those neurons containing inclusions in which cytoplasmic organelles such as mitochondria, endoplasmic reticulum, polyribosomes, and lysosomes (but never Golgi apparatus) were entrapped within the filamentous network of the inclusion underwent degeneration and death. From this observation, they suggested that entrapment of cytoplasmic organelles might initiate neuronal cell death by isolating the organelles, thereby impairing their function (Tu et al., 1997a).
These observations suggest that alterations of NF metabolism and subsequent formation of NF-rich inclusions may be related to neuronal degeneration. In NFH-LacZ TG mice, NF-rich inclusions plateau in number by 2 months of age and are widespread throughout the CNS (Tu et al., 1997a). However, neuronal loss is delayed until 1 year of age and is restricted to cerebellar Purkinje's cells, which contain organelle-entrapping inclusions (termed type II), as opposed to hippocampal cells that contain inclusions which did not entrap organelles (termed type I). Therefore, it was unclear whether the alteration of NF metabolism and subsequent formation of NF-rich inclusions perturbed neuronal function and led to neurodegeneration. We postulated that aberrant NF organization may lead to subtle functional impairment which could be revealed by stressing or challenging the neurons. Therefore, in the present study, we evaluated the response of NFH-LacZ TG mice to TBI, hypothesizing that neurobehavioral dysfunction and cell death after TBI may be accelerated in NFH-LacZ TG mice even when trauma is induced before neurons begin to degenerate.
Evaluated with a series of simple reflex and complex motor tests (composite neuroscore) as well as the more complicated beam balance task, brain-injured WT mice showed modest, relatively short-lived neurobehavioral deficits. Brain-injured NFH-LacZ TG mice, however, had greater initial deficits and took longer to recover from the injury. Previously, no phenotype such as tremor, ataxia, or muscle weakness was reported in NFH-LacZ TG mice until 1 year of age (Tu et al., 1997a). However, our extensive examination of motor function revealed a small but significant impairment in motor function (baseline latency for rotarod performance) in NFH-LacZ TG mice at only 3 to 6 months of age. After correcting for this pre-injury difference between the groups by evaluating individual post-injury latencies as percent of baseline, the slow acceleration rotarod task also revealed greater and longer lived neuromotor dysfunction in brain-injured NFH-LacZ TG mice. Although neuronal cell loss had not been reported in uninjured mice except in the cerebellar Purkinje's cells (Eyer and Peterson, 1994; Tu et al., 1997a), we observed greater tissue loss in the injured cerebral hemisphere (especially in the cortex) in NFH-LacZ TG mice compared with WT mice after injury.
Taken together, these data suggest that NFH-LacZ TG mice have increased vulnerability to TBI. However, the precise mechanisms underlying the exacerbation of the neurobehavioral dysfunction and histopathologic changes remain speculative. TBI has been shown to in duce a loss of NF proteins and disruptions in the neurofilamentous structure of both dendrites and axons (Yaghmai and Povlishock, 1992; Posmantur et al., 1994, 1996; Maxwell et al., 1997). Axonal injury, detected by increased immunoreactivity and apparent accumulation of NF, has been observed after TBI both in the clinical situation and in experimental models including CCI (Yaghmai and Povlishock, 1992). Traumatic axonal injury can lead to secondary axotomy. Subsequent anterograde degeneration and loss of synaptic terminals is thought to disconnect these axons functionally from their target neuronal population, thereby directly contributing to neuronal dysfunction (Povlishock, 1993). Since disruption of NF networks, reduction of endogenous NFM and NFL, and decreased levels of phosphorylation in endogenous NFH were observed in NFH-LacZ TG mice (Eyer and Peterson, 1994; Tu et al., 1997a), their axons may be structurally different from those of normal mice. Perhaps the preferential vulnerability of the transgenic animals to brain injury therefore involves both neuronal and/or axonal failure. Eyer and Peterson (1994) reported decreased axonal caliber of large myelinated axons in the sciatic nerves of NFH-LacZ TG mice. Structural differences caused by aberrant NF organization may confer vulnerability to mechanical stresses and result in more axonal damage, thereby leading to exacerbation of dysfunction.
Another possible explanation may involve potentially protective responses of neurons after TBI. Neurotrophic factors such as acidic or basic fibroblast growth factor, nerve growth factor, and brain-derived neurotrophic factor are known to have neuroprotective roles in various paradigms of neuronal stress (Koketsu et al., 1994; Hefti, 1986; Williams et al., 1986; Sinson et al., 1995; McDermott et al., 1997; Schäbitz et al., 1997). The expression of endogenous neurotrophic factors, such as basic fibroblast growth factor, is upregulated after brain injury (Finklestein et al., 1988; Frautschy et al., 1991; Kiyota et al., 1991). Administration of nerve growth factor or basic fibroblast growth factor after fluid percussion brain injury in rats attenuates neurobehavioral cognitive deficits or cell loss (Sinson et al., 1995; Dietrich et al., 1996; McDermott et al., 1997). Because axonal transport is indispensable for these neurotrophic factors to reach their targeted area (Gonzalez et al., 1994; Connor et al. 1997), it is possible that disturbances in the axonal transport due to inclusions located in proximal axons led to decreased neurotrophic factor delivery, thereby augmenting neuronal dysfunction after brain injury. Stress-induced proteins such as heat shock protein and glucose-regulated protein, and the proto-oncogene protein bcl-2 are also thought to have protective roles after brain injury (Lindquist, 1986; Raghupathi et al., 1995; Pelham, 1986; Raghupathi et al., 1998; Clark et al., 1997; Kane et al., 1995). These proteins are mainly localized in cytoplasmic organelles such as mitochondria or polyribosomes and are upregulated after brain injury (Morimoto, 1991; Kluck et al., 1997). Approximately 50% of neurons in the cerebral cortex of NFH-LacZ TG mice are reported to contain NF-rich inclusions that entrap cytoplasmic organelles (type II inclusions) (Tu et al., 1997a). Diminished function of entrapped organelles and their associated proteins might potentiate neuronal dysfunction and cell death after brain injury. Finally, we cannot exclude the possibility that TG animals may have had abnormalities in cerebral circulation and perfusion, and the possibility that the observed lesion expansion may be due to reduced (ischemic) blood flow in the penumbra of the contusion, which was not assessed in the present study.
In this report, we have provided the first evidence of vulnerability of NFH-LacZ TG mice to TBI both with respect to neurobehavioral function and histopathologic change, suggesting that disruption of the subcellular distribution of NF, evident by the formation of NF-rich inclusions, may be deleterious to neuronal function and survival after TBI. One could speculate that, in a similar manner, altered NF metabolism or structure might also confer an increased vulnerability to environmental risk factors such as neurotoxins, heavy metals, excitotoxicity, or growth factor deficiency which have been implicated in the pathogenesis of certain neurodegenerative diseases such as ALS (Tu et al., 1997b). Future studies that make use of targeted alterations in NF proteins should provide further insight into mechanistic and functional commonalities between trauma-induced pathology and neurodegenerative diseases, expanding on the findings described in this initial study.