
Abstract
The authors examined the protein expression of apurinic/apyrimidinic endonuclease (APE/Ref-1), a multifunctional protein in the DNA base excision repair pathway, before and after transient focal ischemia in mice. Immunohistochemistry showed the nuclear expression of APE/Ref-1 in the entire region of the control brains. Nuclear immunoreactivity was decreased as early as 5 minutes after 60 minutes of ischemia in the ischemic core, which was followed by a significant reduction of APE/Ref-1-positive cells in the entire middle cerebral artery territory, Western blot analysis of the sample from the nonischemic brain showed a characteristic 37-kDa band, which was reduced after ischemia. A significant amount of DNA fragmentation was observed at 24 hours, but not at 4 hours, after ischemia. The authors' data provide the first evidence that APE/Ref-1 rapidly decreases after transient focal ischemia, and that this reduction precedes the peak of DNA fragmentation in the brain regions that are destined to show necrosis and apoptosis. Although further examination is necessary to elucidate the direct relationship between the APE/Ref-1 decrease and ischemic necrosis and apoptosis, our results suggest the possibility that rapid decrease of APE/Ref-1 and the failure of the DNA repair mechanism may contribute to necrosis or apoptosis after transient focal ischemia.
The DNA repair enzyme apurinic/apyrimidinic endonuclease (APE/Ref-1) is a multifunctional protein in the DNA base excision repair pathway that is responsible for repairing apurinic/apyrimidinic sites in DNA (Bennett et al., 1997). DNA base excision repair is assumed to require two types of enzymes, such as DNA glycosylases and APE (Doetsch and Cunningham, 1990; Lindahl, 1990; Demple and Harrison, 1994). DNA glycosylases remove a damaged base, which could be caused by various kinds of insults including oxidative stress, creating an apurinic/apyrimidinic site in the DNA that is then acted on by an APE (Doetsch and Cunningham, 1990; Demple and Harrison, 1994). Then the repair is completed by insertion of an abasic residue followed by synthesis of a new base by DNA polymerase and ligation. Incomplete repair of apurinic/apyrimidinic sites is reported to cause mutagenesis and genetic instability (Loeb and Preston, 1986). Although little is known about the relationship of the DNA base excision repair pathway to necrosis or apoptosis in neuronal cells, recent evidence suggests that downregulation of APE expression is associated with apoptosis in cells of the myeloid lineage (Robertson et al., 1997). Also, in vivo it was shown that loss of APE/Ref-1 protein preceded the occurrence of DNA fragmentation in the CA1 neurons after global ischemia (Gillardon et. al., 1997).
Increasing evidence suggests that DNA damage, from the viewpoint of programmed cell death, may contribute to the death of neurons after focal ischemia (Tominaga et al., 1993; Li et al., 1995; Hara et al., 1997; Namura et al., 1998). Fragmented DNA is shown by both agarose gel electrophoresis (Tominaga et al., 1993) and terminal deoxynucleotidyl transferase-mediated uridine 5‘-triphosphate-biotin nick end labeling (TUNEL) staining (Li et al., 1995) after focal ischemia. Furthermore, we have shown that reactive oxygen species are an important factor for the development of DNA damage after transient focal ischemia (Chan, 1996; Kondo et al., 1997). However, from the standpoint of mutagenesis, Liu et al. (1996) have suggested that free radicals could attack the nuclear genes and cause genetic instability after mouse forebrain ischemia. Furthermore, nuclear translocation of a DNA repair enzyme, such as poly (ADP)- ribose polymerase (PARP), from the cytosol has been shown after transient cerebral ischemia (Endres et al., 1997), suggesting the role of these enzymes in repairing DNA damage after ischemia-reperfusion. In the present study, we sought to clarify this point by examining the expression of the DNA repair enzyme APE/Ref-1 protein before and after transient focal ischemia in which programmed cell death is assumed to play a role (Tominaga et al., 1993; Li et al., 1995; Hara et al., 1997; Namura et al., 1998). Also in the present study, a significant amount of DNA fragmentation was detected by DNA gel electrophoresis at 24 hours after ischemia. Using immunohistochemistry and Western blotting on the same ischemia-reperfusion model, we examined the APE/Ref-1 expression after transient ischemia. The results show that the APE/Ref-1 protein rapidly decreases as early as 5 minutes after 60 minutes of middle cerebral artery (MCA) occlusion, followed by a significant reduction of the protein in the entire MCA area 4 hours after ischemia. These data provide evidence that APE/Ref-1 rapidly decreases after transient ischemia, and this reduction precedes the peak of DNA fragmentation. Although further examination is necessary to elucidate the direct relationship between the APE/Ref-1 decrease and ischemic infarction or apoptosis, our results suggest the possibility that the rapid decrease of APE/Ref-1 and the failure of the DNA repair mechanism may contribute to neuronal cell death after transient focal cerebral ischemia.
MATERIALS AND METHODS
Focal cerebral ischemia
Adult male CD-1 mice (35 to 40 g) were subjected to transient focal ischemia by intraluminal MCA blockade with a nylon suture as previously described (Yang et al., 1994). The mice were anesthetized with 2.0% isoflurane in 30% oxygen and 70% nitrous oxide using a face mask. The rectal temperature was controlled at 37°C with a homeothermic blanket. Cannulation of a femoral artery allowed the monitoring of blood pressure and arterial blood gases (Table 1), samples for analysis being taken immediately after cannulation, 10 minutes after occlusion, and 10 minutes after reperfusion. After the midline skin incision, the left external carotid artery was exposed, and its branches were electrocoagulated. An 11.0-mm-long 5-0 surgical monofilament nylon suture, blunted at the end, was introduced into the left internal carotid artery through the external carotid artery stump. After 60 minutes of MCA occlusion, blood flow was restored by withdrawal of the nylon suture.
Summary of APE/Ref-1 expression after 60 minutes MCA occlusion

APE/Ref-1, apurinic/apyrimidinic endonuclease.
normal constitutive expression
moderate reduction of nuclear immunoreactivity
marked decrease of the number of APE/Ref-1-positive cells.
Histologic assessment
The experimental animals were killed at 5 minutes, 30 minutes, 4 hours, and 24 hours after 60 minutes of MCA occlusion. The brains were removed, rapidly frozen in −20°C 2-methyl-butane, and stored at −80°C. They were sectioned with a cryostat into a thickness of 20 µm from the anterior side to the posterior side and stained with cresyl violet.
Gel electrophoresis
Animals were killed 4 and 24 hours after 60 minutes of MCA occlusion. Thirty to 50 mg wet weight of ischemic tissue was taken from the third 2-mm section along with homologous tissue from the contralateral side after the brain was cut coronally. Samples were incubated overnight in 0.6 mL lysis buffer (0.5% sodium dodecyl sulfate, 10 mmol/L Tris-HCl, and 0.1 mol/L EDTA) with 0.6 mg proteinase K (Boehringer Mannheim, Indianapolis, IN, U.S.A.) at 55°C. The DNA was extracted with equal volumes of phenol and phenol-chloroform-isoamyl alcohol (25:24: 1), and precipitated overnight in 0.2 mol/L sodium chloride in 100% ethanol at −80°C. The DNA was washed with 75% ethanol two times, air dried, and resuspended in DNase-free water (Sigma, St. Louis, MO, U.S.A.). The DNA concentration was measured by using To-Pro-1 dye (Molecular Probes, Eugene, OR, U.S.A.). Gel electrophoresis for detecting DNA laddering was performed according to the manufacturer's instructions (Trevigen, Gaithersburg, MD, U.S.A.). Before electrophoresis, 1 µg of DNA was incubated with 50 µg/mL of DNase-free RNase (Boehringer Mannheim) for 30 minutes at 37°C. Then the samples were reacted with Klenow enzyme (Trevigen) and deoxynucleotides (Trevigen) in 1 × Klenow buffer (Trevigen) for 10 minutes at room temperature. Samples were mixed with loading buffer and subjected to electrophoresis on 1.5% agarose gel. Then the gel was washed with 0.25 mol/L HCl, 0.4 mol/L NaOH and 0.8 mol/L NaCl, and 0.5 mol/L Tris buffer (pH 7.5), respectively. DNA was transferred to a nylon membrane overnight in 10 × sodium chloride-sodium citrate. The membrane was first blocked by 5% powdered milk (BioRad, Hercules, CA, U.S.A.) in phosphate-buffered saline for 30 minutes, and incubated with strep-tavidin-horseradish peroxidase conjugate (Trevigen) for 30 minutes. Finally, the bands were visualized by the chemiluminescence method using PeroxyGlow (Trevigen), and the films were exposed to x-ray film.
Immunohistochemistry of APE/Ref-1
Anesthetized animals were perfused with 10 U/mL heparin and subsequently with 4% formaldehyde in 0.1 mol/L phosphate-buffered saline (pH 7.4) after 5 minutes, 30 minutes, 4 hours, and 24 hours of reperfusion after ischemia. Brains were removed, postfixed for 12 hours in 4% formaldehyde, sectioned at 50 µm on a vibratome, and processed for immunohistochemistry. The sections were incubated with blocking solution as described (Fujimura et al., 1998) and reacted with anti-APE/Ref-1 polyclonal antibody (Novus Biologicals, Littleton, CO, U.S.A.) at a dilution of 1:100. Immunohistochemistry was performed using the avidin-biotin technique as described (Fujimura et al., 1998), and then the nuclei were counterstained with methyl green solution for 10 minutes. As a negative control, sections were incubated without primary antibodies. For histologic assessment, alternate slices from each brain section were stained with cresyl violet.
Western blot analysis
Whole cell protein extraction was performed. Approximately 30 mg of both ipsilateral striatum and homologous tissue from the contralateral side were cut into pieces after 4 and 24 hours of reperfusion and put into 10 × volume of Tris-glycine sodium dodecyl sulfate sample buffer (Novex, San Diego, CA, U.S.A.). Samples were then gently homogenized 20 times in a Teflon Dounce homogenizer (Weaton, Millville, NJ, U.S.A.). Equal amounts of the samples (10 µL) were loaded per lane. The primary antibodies were either a 1: 1,000 dilution of polyclonal antibody against APE/Ref-1 (Novus Biologicals) or a 1: 10,000 dilution of anti-β-actin monoclonal antibody (Sigma). For APE/Ref-1 detection, Western blots were performed with horseradish peroxidase-conjugated anti-rabbit immunoglobulin G using the Boehringer Mannheim chemiluminescent system. Recombinant human APE/Ref-1 was a generous gift from Novus Biologicals. As the internal control, Western blot analysis of β-actin was performed with horseradish peroxidase-conjugated anti-mouse immunoglobulin G reagents (Amer-sham International, Buckinghamshire, England).
RESULTS
Physiologic data and cerebral infarction
Physiologic parameters showed no significant differences in MABP and arterial blood gas analysis between each time point. The preischemic physiologic valuables are as follows: MABP, 71.0 ± 3.8 mm Hg; Pao2, 146.4 ± 9.9 mm Hg; Paco2, 28.9 ± 7.8 mm Hg; pH, 7.35 ± 0.04 (values are mean ± SD, n = 4). There was no deviation from these values during the period of assessment. An ischemic lesion of the core of the caudate putamen is visible as a pale, slightly stained area in the ischemic hemisphere as early as 5 minutes after reperfusion, which extends to the entire MCA territory at 4 hours by cresyl violet staining (data not shown). The time-dependent increase of infarction in mouse brain using intraluminal suture blockade is consistent with previous reports that used the same focal stroke model in mice (Kondo et al., 1997; Murakami et al., 1997).
DNA laddering was detected by genomic DNA gel electrophoresis
To detect the occurrence of apoptosis as characterized by intranucleosomal DNA fragmentation, we analyzed DNA from both the ischemic brain and the homologous sample on the contralateral side. DNA laddering was absent in both the control tissue and ischemic tissue 4 hours after ischemia (Fig. 1, lanes 1 to 3). A significant amount of DNA laddering was detected 24 hours after ischemia (Fig. 1, lane 4).
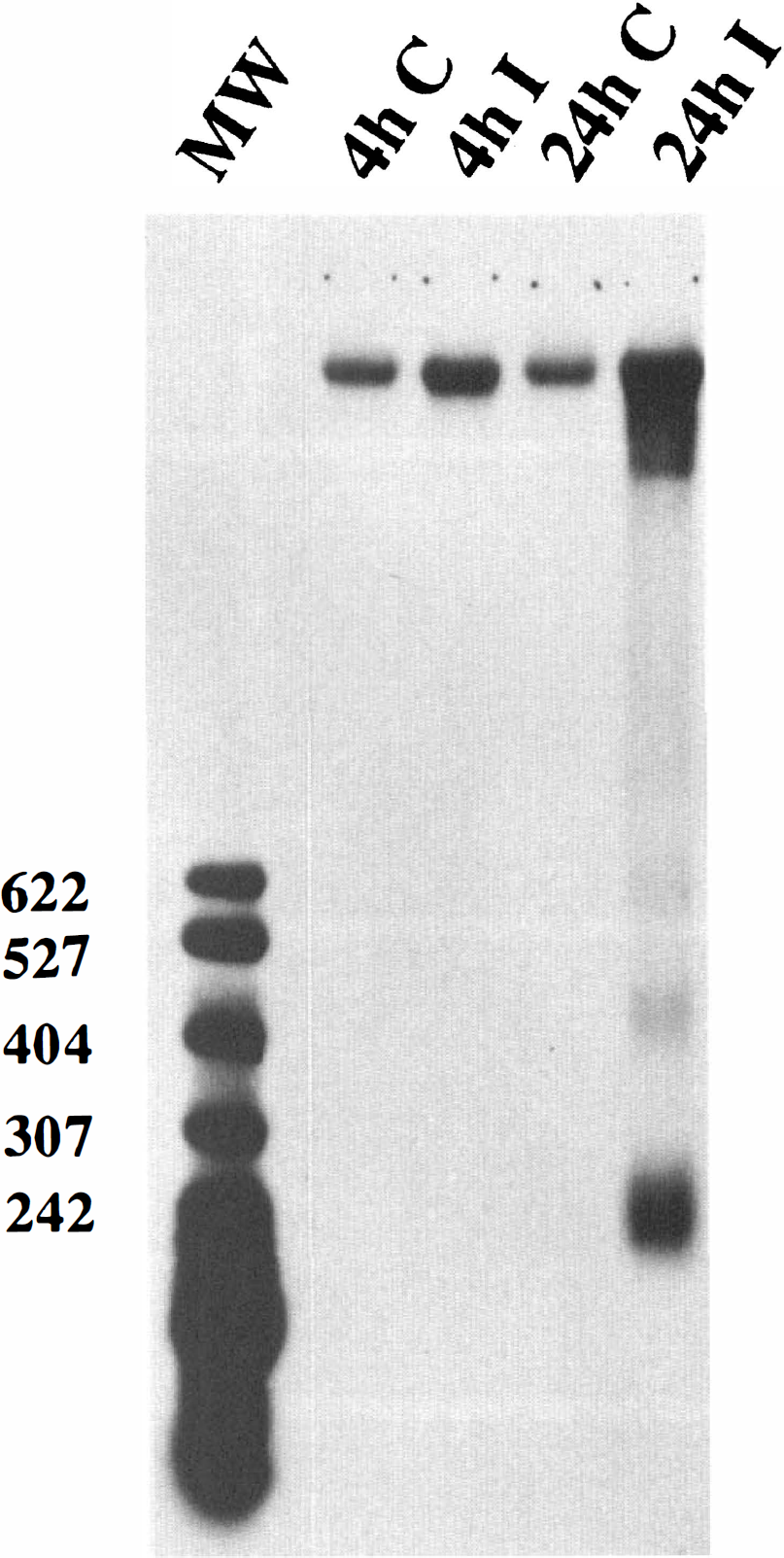
Genomic DNA agarose gel electrophoresis. No DNA laddering was observed in the contralateral brain. In the ischemic brain, DNA laddering was detected at 24 hours but not at 4 hours after 60 minutes of MCA occlusion. DNA was end-labeled with biotinylated deoxynucleotides, underwent electrophoresis on a 1.5% agarose gel, was transferred to a nylon membrane, and was visualized by the chemiluminescent method. The ladders corresponding to 200, 400, and 600 base pairs are shown. The number of each base pair is also shown on the left side. (MW, molecular marker).
Immunohistochemistry showed the constitutive expression of APE/Ref-1 in normal adult mouse brain, which decreased immediately after reperfusion
The results of APE/Ref-1 immunohistochemistry are summarized in Table 1. The APE/Ref-1 protein was constitutively expressed in the entire region of the normal mouse brain (Fig. 2B-F). It was mainly expressed in the nucleus, which is consistent with previous reports (Ono et al., 1995; Wilson et al., 1996). We observed regional predominance in the hippocampus compared with the cortex and striatum (Fig. 2D, E and F). This regional predominance was confirmed by Western blot analysis of APE/Ref-1 proteins from each region, showing that more expression was observed in the hippocampus and cerebellum compared with the striatum and cortex (data not shown). After 5 minutes of reperfusion, after 60 minutes of ischemia, a moderate reduction of nuclear immunoreactivity of APE/Ref-1 was observed in the lateral caudate putamen (Fig. 2G). Thirty minutes after ischemia, the number of the APE/Ref-1-positive cells was significantly decreased in the lateral caudate putamen (Fig. 2H). Four hours after ischemia, a significant reduction of APE/Ref-1-positive cells was observed in the entire MCA area on the ipsilateral side (Fig. 21 and J), which decreased further at 24 hours after ischemia (Fig. 2K and L). At 24 hours, methyl green counterstaining revealed nuclear fragmentation in several cells, most of which were likely to be lacking APE/Ref-1 immunoreactivity (Fig, 2K). There was no immunoreactivity in the control specimens that were treated without a primary antibody (Fig, 2A).
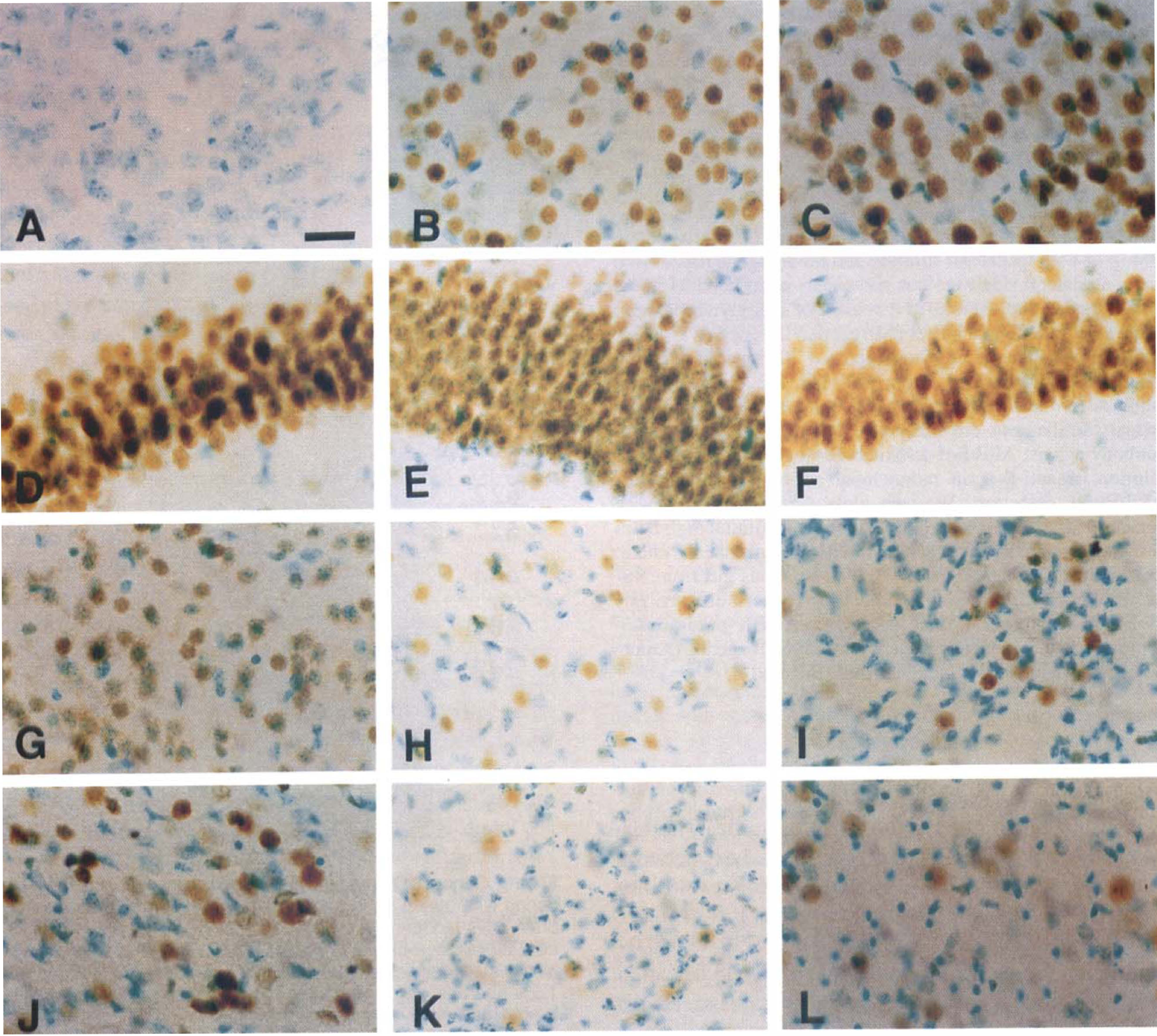
High-power views of immunohistochemical experiments with the APE/Ref-1 protein (counterstained by methyl green). There was no immunoreactivity in the control specimens, which were treated without a primary antibody (
Western blot analysis of APE/Ref-1 protein expression after transient middle cerebral artery occlusion
As shown in Fig, 3, APE/Ref-1 immunoreactivity was evident as a single band of 37-kDa whole cell fraction from the nonischemic hemisphere (lanes 1 and 3 in the upper panel), which was significantly decreased 4 hours after ischemia (lane 2). Twenty-four hours after MCA occlusion, APE/Ref-1 immunoreactivity was absent (lane 4). In contrast, a consistent amount of β-actin immunoreactivity between each lane was shown in the lower panel, suggesting that the amount of the loaded protein was consistent (lower panel). These data not only confirm the specificity of the antibody for APE/Ref-1 used in this study, but also suggest that APE/Ref-1 decreased after transient focal ischemia in a time-dependent manner.

Western blot analysis of the APE/Ref-1 protein and β-actin protein as an internal control. APE/Ref-1 immunoreactivity is evident as a single band of molecular mass 37-kDa whole cell fraction from the nonischemic brain (lanes 1 and 3, upper panel), and was significantly decreased at 4 hours after 60 minutes of MCA occlusion (lane 2). Twenty-four hours after ischemia, immunoreactivity was absent (lane 4). In contrast, a consistent amount of β-actin immunoreactivity is shown in the lower panel.
DISCUSSION
The present study provides the first evidence that the DNA repair enzyme APE/Ref-1 rapidly decreases after transient focal cerebral ischemia, and that this reduction precedes the occurrence of DNA fragmentation. Programmed cell death and the DNA repairing mechanism are both assumed to play important roles in cerebral ischemia. However, little is known about the interaction of these process, except the data suggesting a link between apoptosis and the cleavage of the DNA repair enzyme PARP (Nicholson et al., 1995). The interleukin-1β converting enzyme family caspases are the human homologs of the nematode Caenorhabditis elegans Ced-3 (Yuan and Horvitz, 1990) and are considered to play a critical role in programmed cell death (Nicholson et al., 1995). One substrate for caspase is PARP, whose proteolytic cleavage results in a dysfunctional PARP that is unable to contribute to repair or genomic maintenance (Nicholson et al., 1995). Further, the Ca2+/Mg2+–dependent endonuclease that generates internucleosomal DNA cleavage characteristic of apoptosis is negatively regulated by poly(ADP-ribosylation). Therefore, inactivation of PARP could increase DNA cleavage and contribute to programmed cell death.
Neuronal cell death after focal cerebral ischemia has previously been attributed to passive necrotic processes. However, increasing evidence suggests that an active process similar to programmed cell death contributes to the death of neurons (Tominaga et al., 1993; Li et al., 1995; Hara et al., 1997; Namura et al., 1998) and to the expansion of the lesion after focal cerebral ischemia (Du et al., 1996). Fragmented DNA has been shown after focal ischemia by both agarose gel electrophoresis (Tominaga et al., 1993) and by TUNEL staining (Li et al., 1995) after focal ischemia. Our most recent study has shown that cytochrome c, which is assumed to be released from mitochondria and then to activate caspase 3 and downstream apoptotic events, is being released from mitochondria to cytoplasm after transient focal cerebral ischemia (Fujimura et al. 1998). In fact, the activated form of caspase 3 is shown to be detected at the early stage of ischemia-reperfusion injury in the brain (Namura et al., 1998), and the inhibition of the caspase family proteases is reported to reduce infarct volume and the extent of apoptosis after transient ischemia (Hara et al., 1997). In the present study, a significant amount of DNA fragmentation was observed by gel electrophoresis (Fig. 1), suggesting that in addition to ischemic necrosis, apoptosis may play a role in the ischemia-reperfusion model in mice. Because our previous study using the same ischemia model demonstrated the morphologic evidence that apoptotic cells were mostly from neuronal cell population (Kondo et al., 1997), neuronal apoptosis is likely to contribute mainly, if not exclusively, to DNA fragmentation in the current study. However, in light of the increasing evidence that astrocytes and oligodendrocytes could undergo apoptosis after brain injuries, we cannot exclude the possibility that the observed DNA laddering may come from these cells. A double staining of TUNEL staining with various cell markers will provide important information to address this issue.
However, still controversial is whether DNA repairing enzymes, including PARP, play any protective role in ischemic brain damage, including apoptosis. As for PARP, its excessive activation is believed to be deleterious because it might cause energy depletion and ultimately cell death (Zhang et al., 1994). In fact, most recent PARP knockout mice studies implicate the deleterious role of PARP in ischemia (Eliasson et al., 1997; Endres et al., 1997). These studies clearly show the marked reduction of the infarct volume in PARP knockout mice after transient cerebral ischemia (Eliasson et al., 1997; Endres et al., 1997), suggesting that PARP activation plays a detrimental role in neuronal damage after ischemia. However, even from these studies, the relationship of the DNA repairing mechanism to ischemic apoptosis is still unclear because these studies did not exclude the role of PARP in apoptotic cell death. Our data show that the expression of the DNA repair enzyme APE/Ref-1, which is constitutively expressed in the nucleus of the entire region of the normal brain (Fig. 2B-F), decreases as early as 5 minutes after reperfusion at the ischemic core (Fig. 2G). Four hours after ischemia, a significant reduction of APE/Ref-1-positive cells was observed in the entire MCA territory as shown by immunohistochemistry (Fig. 2H) as well as by Western blot analysis (Fig. 3), when the DNA fragmentation was not detected by gel electrophoresis (Fig. 1). APE/Ref-1 was further decreased 24 hours after ischemia (Fig. 2K, L), and had disappeared as shown by Western blot analysis (Fig. 3). At this time point, a significant amount of DNA fragmentation was observed by electrophoresis (Fig. 1). These findings provide the evidence that APE/Ref-1 rapidly decreases after reperfusion, and this reduction precedes the occurrence of DNA fragmentation. Furthermore, it is suggested that the rapid loss of APE/Ref-1 and the failure of the DNA repair mechanism might contribute to DNA fragmentation after transient focal cerebral ischemia in mice. Further study, such as the double staining of APE/Ref-1 and TUNEL, is warranted to elucidate the direct correlation between the reduction of APE/Ref-1 and the occurrence of ischemic apoptosis. The mechanism that causes the early reduction of APE/Ref-1 immunoreactivity after reperfusion is unknown. Because it is well-known that reperfusion increases mitochondrial production of superoxide radicals (Chan, 1996), the reduced expression of APE/Ref-1 may be caused by oxidative damage. This point will be further examined regarding the stability of this enzyme both at transcriptional and translational levels. Furthermore, the activity assays for detecting ischemic DNA lesions, such as apurinic/apyrimidinic sites, would be very useful to provide evidence that DNA base excision repair works after ischemia-reperfusion and could affect APE/Ref-1 expression subsequently. In fact, our preliminary data show the early appearance of ischemic DNA lesion, which is detected as Escherichia coli formamidopyrimidine DNA N–glycosylase-sensitive sites. Alternatively, it will be of interest to study whether the APE/Ref-1 expression is affected by transgenic mice that overexpress the antioxidant enzyme, superoxide dismutase (SOD)-1 (Yang et al., 1994). This transgenic mice study is currently under way in our laboratory.
Antioxidant enzymes and DNA repair proteins are thought to be two major mechanisms by which cells counteract the deleterious effects of reactive oxygen species, and we have reported evidence that antioxidant enzymes such as SOD play a protective role in ischemia-reperfusion injury in the mouse brain (Kinouchi et al., 1991; Chan et al., 1995; Chan, 1996; Kondo et al., 1997). For example, SOD-1 transgenic mice show a significant reduction of infarct volume after transient focal cerebral ischemia (Kinouchi et al., 1991), whereas infarction is increased in SOD-1 knockouts after transient ischemia (Kondo et al., 1997). Also, reactive oxygen species have been assumed to play a detrimental role in the pathogenesis of amyotrophic lateral sclerosis (ALS) since the discovery of a missense mutation in the gene encoding SOD-1 in familial ALS patients (Rosen et al., 1993). Interestingly, recent evidence shows that APE/Ref-1 levels and activity are significantly lower in ALS patients than in control subjects (Kisby et al., 1997), suggesting that the brain tissue of ALS patients is inefficient in repairing oxidative DNA damage. From these observations, it is conceivable that a reduction of APE/Ref-1 after transient ischemia, as shown in the present study, would be extremely detrimental to a neuron in overcoming oxidative stress during ischemia-reperfusion injury. In fact, using the global ischemia model in rats, Gillardon et al. (1997) have also shown that APE/Ref-1 decreased in vulnerable CA1 neurons of the postischemic hippocampus but not in the ischemia-resistant dentate gyrus, suggesting that APE/Ref-1 reduction might contribute to neuronal damage after ischemia-reperfusion injury.
In addition to its important role in the DNA repair mechanism, APE/Ref-1 is drawing particular attention because of its critical role in redox regulation of DNA-binding activity of activator protein-1 family members such as Fos and Jun transcription factors (Xanthoudakis and Curran, 1992; Hirota et al., 1997), which are also considered to be associated with the pathogenesis of cerebral ischemia (An et al., 1993; Hsu et al., 1993). Furthermore, a recent study showed that thioredoxin, an endogenous redox regulating protein with a redox-active site that is closely associated with APE/Ref-1 (Hirota et al., 1997), was downregulated in the ischemic core but was increased in the cortical penumbra after focal ischemia in rats (Takagi et al., 1998), suggesting that thioredoxin could have some neuroprotective function during cerebral ischemia. In our study, APE/Ref-1 rapidly decreased in the ischemic core and spread throughout the entire MCA territory as early as 4 hours after reperfusion. However, there was no significant difference in the outer boundary zones of the cortex compared with the nonischemic area. The relationship of APE/Ref-1 reduction to other redox-regulating proteins, such as thioredoxin, and to transcription factor activity after ischemia should also be determined in a future study.
In conclusion, we have shown that APE/Ref-1 rapidly decreased after transient focal ischemia, and this reduction preceded the occurrence of ischemic apoptosis and infarction. Although further examination is necessary to elucidate the relationship between the APE/Ref-1 decrease and neuronal damage after ischemia-reperfusion, our results suggest the possibility that rapid decrease of APE/Ref-1 and the failure of the DNA repair mechanism may contribute to necrosis or apoptosis after transient focal cerebral ischemia.
Footnotes
Abbreviations used
Acknowledgments
The authors thank K. Murakami, MD, for the valuable suggestion of the surgical procedure and G.W. Kim, MD, PhD, for the advice on DNA extraction procedure. The authors also thank C. Christensen for editorial assistance, and L. Reola and B. Calagui for their technical assistance.