
Abstract
Effects of prolonged focal ischemia [middle cerebral artery occlusion (MCAO)] of 1, 2, and 4 h followed by 15-h reperfusion on CBF, extracellular amino acids, purine catabolites, evoked potentials, and infarction were studied in core (A: auditory cortex) and border zone (SF: somatosensory cortex) areas of halothane-anesthetized cats. Following MCAO, CBF reduction was severe in A(<15 ml 100 g−1 min−1) and mild to moderate in SF. Prominent elevation of glutamate and abolition of evoked potentials in A contrasted with milder and more variable disturbances in SF. After reperfusion, recovery of CBF, glutamate, and evoked potentials was fast and persistent in the 1- and 2-h groups. In the 4-h group, immediate recovery of CBF, glutamate, and evoked potentials was incomplete, and secondary deterioration of all parameters was obtained at the end of the experiments. Infarction in the 4-h group was significantly larger than in the 1- and 2-h groups. Persistent recovery of extracellular glutamate concentration and electrical function and salvage of neuronal tissue from infarction therefore seem to depend on successful restoration of CBF, which in turn depends on the magnitude and the duration of CBF reduction and of exposure to potentially harmful substances such as glutamate during the ischemic attack.
Ischemic neuronal damage is determined not only by the severity but also by the duration of CBF reduction (Heiss and Rosner, 1983; Heiss and Graf, 1994). Since recirculation occurs in stroke spontaneously as well as intentionally after interventional therapy, transient focal cerebral ischemia is a meaningful model to establish time windows for infarction. However, the duration of ischemia leading to infarction was found to be different in various species: In rats, infarct volumes after 3–4 h of transient occlusion of the middle cerebral artery (MCAO) followed by 20 h of recirculation were not significantly different from those obtained after 24-h permanent occlusion (Kaplan et al., 1991; Memezawa et al., 1992). In larger animals, the time window for successful reperfusion seems to be longer than in rats. In cats, for example, 6 h of transient ischemia was shown to produce smaller infarcts than permanent ischemia (Sundt et al., 1969; Weinstein et al., 1986).
Excitatory amino acids, i.e., glutamate and aspartate, have been considered to play an important role in the evolution of ischemic cerebral infarction. Glutamate in particular is known to cause brain damage (Lucas and Newhouse, 1957; Olney, 1969; Olney et al., 1971). In vitro, dose-dependent glutamate neurotoxicity has been reported (Choi et al., 1987). In experimental stroke, a close correlation between infarct volume and extracellular elevation of excitatory amino acids has been documented (Butcher et al., 1990). Neuronal degeneration after ischemia seems to be proportional to the time of glutamate exposure (Choi et al., 1987). Protective effects of glutamatergic deafferentation prior to ischemia have been demonstrated in rat hippocampus (Benveniste et al., 1989).
Reperfusion after short-term cerebral ischemia (<30 min) results in immediate restoration of CBF and fast return of accumulated glutamate to preischemic concentrations (Benveniste et al., 1984; Hagberg et al., 1985; Baker et al., 1991; Globus et al., 1991a). Delayed secondary postischemic elevations of glutamate and aspartate (Andiné et al., 1991) and of an excitatory index (quotient [glutamate] × [glycine]/[γ-aminobutyrate (GABA)]; Globus et al., 1991b) have been described. In transient ischemia of longer duration, sustained glutamate exposure is to be expected, since in permanent ischemia, glutamate remained elevated throughout a 15-h observation period (Matsumoto et al., 1993). This prolonged exposure may affect progressive neuronal damage during the reperfusion period. Furthermore, it is unknown how transiently elevated extracellular glutamate concentrations develop during reperfusion after longer periods of ischemia. We therefore studied the concentrations of amino acids and purine catabolites in core and border zone areas of an ischemic focus before, during, and after transient ischemia of 1-, 2-, and 4-h duration, respectively, and thus investigated the effects of prolonged transient focal ischemia on the relationship between postischemic CBF recovery and regeneration of elevated levels of amino acids and purine catabolites. A preliminary report of this study has been presented elsewhere (Taguchi et al., 1993).
MATERIALS AND METHODS
Fifteen cats weighing 2.2–3.4 kg were anesthetized with ketamine hydrochloride (25 mg/kg i.m.). After cannulating the left femoral artery and vein, the animals were tracheotomized and immobilized with pancuronium bromide (0.2 mg/kg i.v.). Thereafter, artificial ventilation was initiated; anesthesia was changed to halothane (0.6–1.2% in a 70% nitrous oxide/30% oxygen gas mixture) to avoid protective effects of ketamine against excitotoxic and ischemic damage (Lees, 1989).
Intravenous infusion of 2 ml/kg/h Ringer solution (147.1 mM NaCl/4.0 mM KCl/2.25 mM Ca2+) containing 5 mg/kg/h gallamine triethiodide for muscle relaxation was maintained throughout the experiment. Arterial blood pressure was monitored continuously, and MABP was kept at 80–110 mm Hg. Arterial blood gases were measured intermittently, and Paco2 was kept at 30–40 mm Hg. With use of separate control systems, body and brain surface temperatures were controlled at 37.6 ± 0.4 and 37.2 ± 0.7°C, respectively.
The left MCA was exposed transorbitally; an occluding device was implanted at the proximal portion of the MCA and fixed with dental cement (Graf et al., 1986). Craniectomies were performed in the parietal bone above the auditory cortex (A; middle ectosylvian gyrus) and in the frontal bone above the front limb projection area of the somatosensory cortex (SF; sigmoid gyrus). After removal of the dura mater, microdialysis probes (CMA/12, membrane length 1 mm, diameter 0.5 mm; Carnegie Medicine, Sweden) and platinum electrodes (etched platinum/iridium wire 250 μm in diameter, glass insulated up to 1 mm of the tip) were inserted stereotaxically with a tip-to-tip distance of 1.5 mm into the two regions. The depth of insertion was adjusted to 1.5 mm.
Microdialysis probes were perfused continuously with Ringer solution at a constant flow rate of 2.0 μl/min using a microinfusion pump (CMA/100; Carnegie Medicine). Ten-minute samples of dialysate (20 μl) were divided into two 10-μl aliquots that were analyzed in two different HPLC systems. Amino acids were analyzed with an RF-535 fluorescence detector (excitation wavelength 330 nm, emission wavelength 480 nm; Shimadzu, Japan) after separation by a 5-μm C18-Nucleosil column (60 × 4.0 mm; Knauer, Germany) using a gradient elution profile [buffer A: 0.1 M sodium acetate (pH 6.95)/2.5% tetrahydrofuran/10% methanol; buffer B: 100% methanol] following pre-column derivatization with o-phthaldialdehyde (Westerberg et al., 1988). Purine catabolites were analyzed with a UV detector (Knauer) at a wavelength of 254 nm following separation by a 5-μm C18 column (300 × 4.0 mm; Knauer) using an elution of 10 mM NH4H2PO4/CH3OH (10:1 vol/vol; pH 5.50) with a flow rate of 1.2 ml/min (Harmsen et al., 1981). Amino acids and purine catabolites in the dialysate were identified and quantified by comparing retention times and peak areas with those of external standards.
The platinum electrodes measured hydrogen clearance following inhalation of hydrogen gas that was added (<10%) to the respiratory gas mixture. CBF was calculated from the initial 2 min of the clearance curve using the following formula: CBF (ml 100 g−1 min−1) = 69.3/T1/2. The same electrodes measured auditory evoked potentials (AEPs) in A after click stimulation and somatosensory evoked potentials (SEPs) in SF after electrical stimulation of the front limb. EP amplitudes were evaluated as the primary cortical responses by measuring the difference between the first positive and the first negative peak of the potentials (Graf et al., 1986).
After a 2-h stabilization period, control values for CBF and EPs were determined and control dialysis samples were collected. Thereafter, the MCA was transiently occluded for 1 (n = 5), 2 (n = 5), and 4 (n = 5) h, and the occlusion period was followed in all animals by a 15-h reperfusion period. CBF and EPs were measured every 30 min, and dialysis samples were collected at 10-min intervals during ischemia and 2-h reperfusion period. Then, the perfusion rate in the dialysis probes was decreased to 0.1 μl/min to avoid excessive drainage of substrates during the long reperfusion period. Repeated insertion of the microdialysis probe, which seems necessary in prolonged microdialysis studies in ischemic tissue (Matsumoto et al., 1993), was not used in the reperfused tissue to minimize traumatization. After 14 h of recirculation, the perfusion rate in the dialysate probes was again increased to 2 μl/min for 1 h, and final measurements were obtained from the last 10-min sample just before termination of the experiments. At this time, final measurements of CBF and EPs were also taken.
The experiments were terminated by perfusion fixation (4% paraformaldehyde in 0.01 M phosphate-buffered saline). Brains were removed and embedded in paraffin following an additional immersion fixation period of at least 2 weeks. Thereafter, 10-μm-thick serial coronal sections were cut and stained with hematoxylin–eosin or with a combination of Luxol fast blue and cresyl violet. The areas of ischemic damage were determined on an image analyzer (Gesotec, Germany) under microscopic control. The volume of infarction was calculated by summing up the infarct areas of consecutive sections multiplied by the distance between sections.
All data are expressed as means ± SD. Statistical evaluation was performed using one- or two way analysis of variance (ANOVA) with multiple comparison to test significance (p < 0.05) within and between groups (CSS; StatSoft, U.S.A.).
RESULTS
CBF
In the ischemic core (area A), MCAO resulted in significant CBF reductions (see Fig. 1) in all three transient ischemia groups (<15 ml 100 g−1 min−1; ANOVA, p < 0.05). Between groups, no significant differences were found during occlusion. Upon recirculation, CBF recovered better in the 1- and 2-h groups than in the 4-h group (residual flow −20 ml 100 g−1 min−1) at 1 and 2 h after recirculation. In this group, a secondary decrease of CBF (<10 ml 100 g−1 min−1) was observed at the end of the experiment. In the ischemic border zone (area SF), CBF was mildly reduced during MCAO and recovered immediately upon reperfusion. In the 4-h group, a secondary decrease of CBF (<10 ml 100 g−1 min−1) similar to that seen in A was observed in SF.
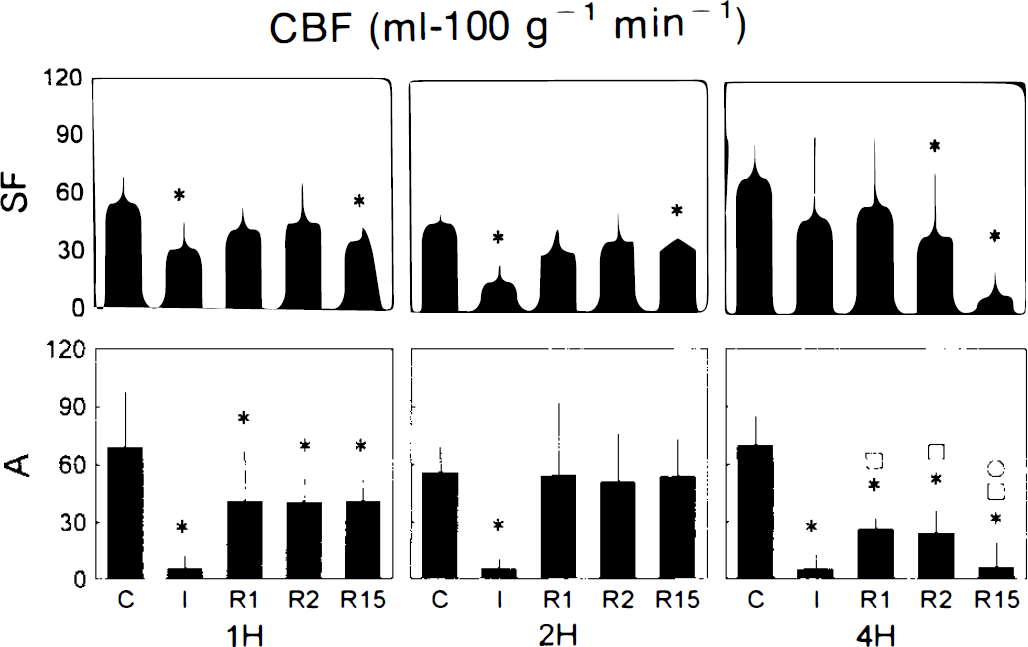
Graph of CBF before, during, and after transient ischemia. 1H, 2H, 4H, groups with 1-, 2-, and 4-h transient ischemia, respectively; A, auditory cortex; SF, somatosensory cortex; C, 1 h before ischemia; I, end of ischemic episode; R1, 1 h after reperfusion; R2, 2 h after reperfusion; R15, 15 h after reperfusion; *, □, O, significantly different from controls (within group), 1-h, and 2-h transient ischemia groups, respectively (analysis of variance, p < 0.05).
Amino acids
In A, extracellular glutamate (Glu) was significantly elevated (ANOVA, p < 0.05) in all groups during MCAO (Fig. 2). Upon recirculation, it decreased in the 1- and 2-h groups and remained on these low levels during the reperfusion period. In the 4-h group, Glu decreased after recirculation but remained on an elevated level. In this group, a secondary significant Glu elevation was observed 15 h after MCAO. In SF, Glu was only slightly elevated in the 1- and 2-h groups and returned to control levels immediately after recirculation. In the 4-h group, Glu was significantly elevated during ischemia. In this group, Glu returned to control levels immediately after reperfusion, but a secondary significant Glu increase similar to that in A was observed in SF at the end of the experiment.
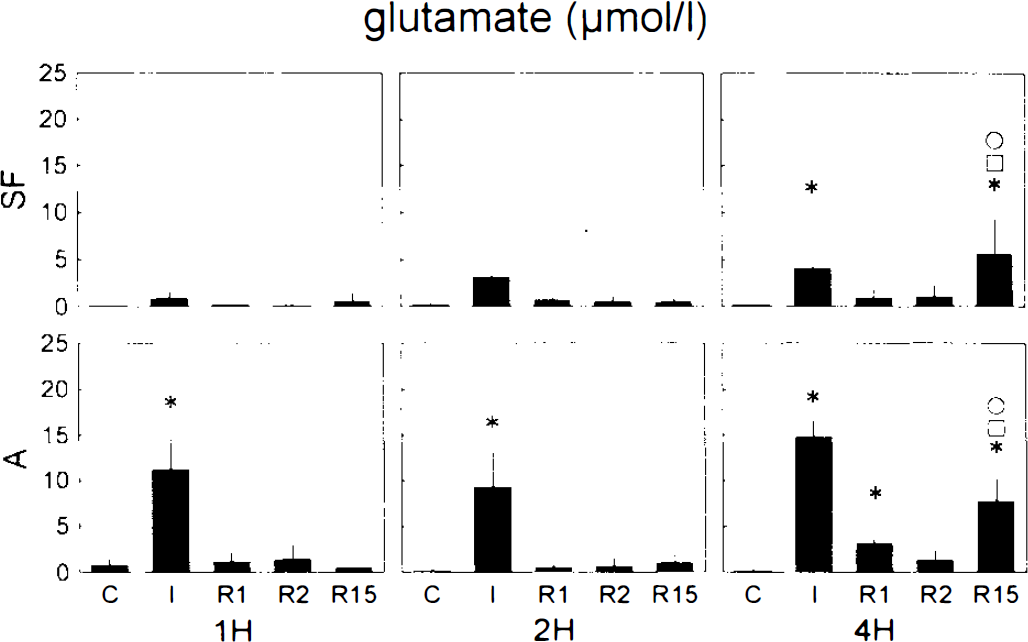
Graph of glutamate concentration in dialysate before, during, and after transient ischemia. 1H, 2H, 4H, groups with 1-, 2-, and 4-h transient ischemia, respectively; A, auditory cortex; SF, somatosensory cortex; C, 1 h before ischemia; I, end of ischemic episode; R1, 1 h after reperfusion; R2, 2 h after reperfusion; R15, 15 h after reperfusion; *, □, O, significantly different from controls (within group), 1-h, and 2-h transient ischemia groups, respectively (analysis of variance, p < 0.05).
Extracellular concentrations of other amino acids, i.e., aspartate (Asp), glycine (Gly), and GABA, showed patterns similar to that of Glu (Table 1). Elevations of these substances during occlusion were predominantly seen in A. At the end of the experiment, glycine was significantly elevated in the 2-h group in A, and all substances were elevated in the 4-h group in both A and SF.
Comparison of substrate concentrations in dialysate (μmol/L) obtained in auditory cortex before, during, and after transient ischemia of variable duration
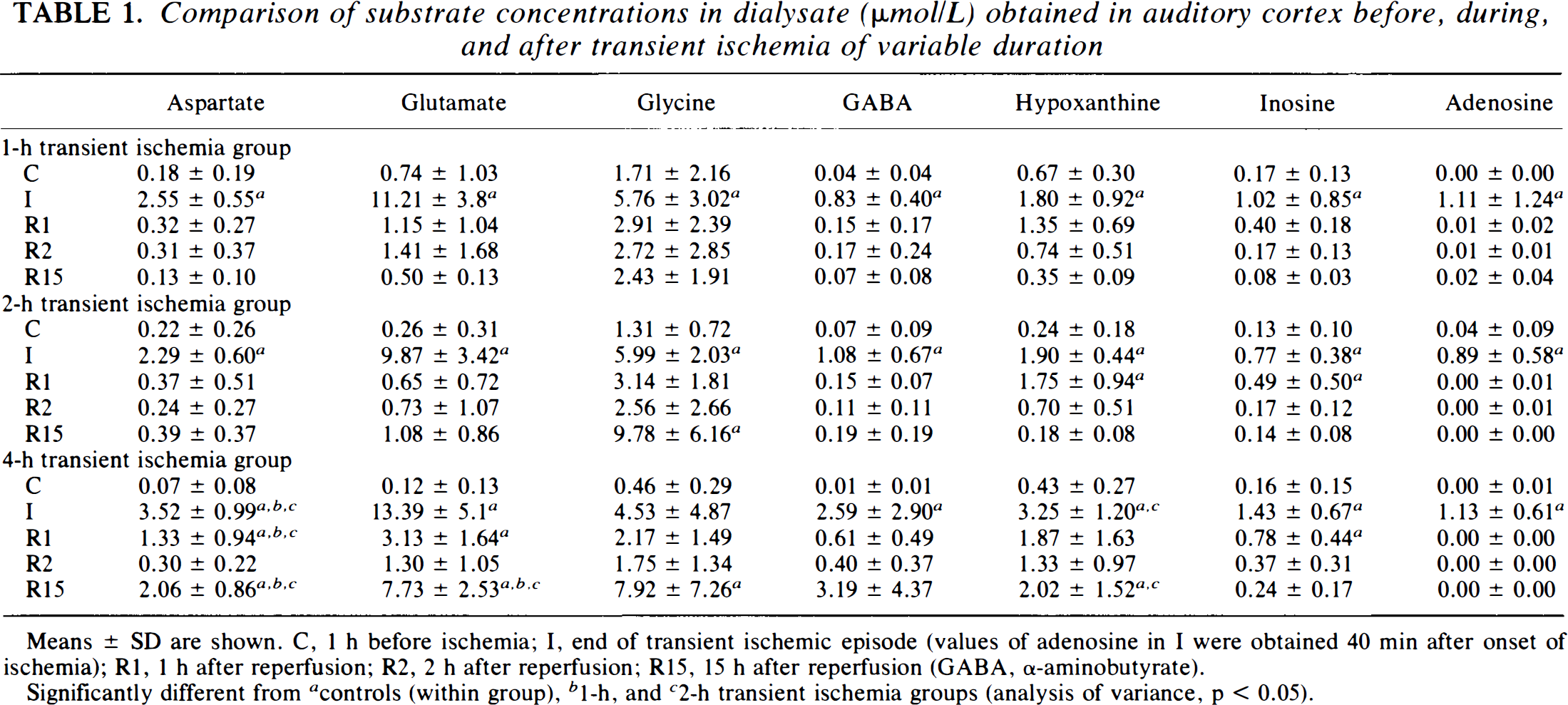
Means ± SD are shown. C, 1 h before ischemia; I, end of transient ischemic episode (values of adenosine in I were obtained 40 min after onset of ischemia); R1, 1 h after reperfusion; R2, 2 h after reperfusion; R15, 15 h after reperfusion (GABA, α-aminobutyrate).
Significantly different from a controls (within group), b 1-h, and c 2-h transient ischemia groups (analysis of variance, p < 0.05).
Purine catabolites
Purine catabolites, i.e., hypoxanthine, inosine, and adenosine, were significantly elevated in A during ischemia (ANOVA, p < 0.05; Table 1). The elevation of hypoxanthine and inosine persisted during ischemia, whereas that of adenosine was only transient, lasting ∼1 h (data not shown). After reperfusion, purine catabolite concentrations returned to control levels, with the exception of the 4-h group, in which the recovery of hypoxanthine and inosine elevation was significantly delayed (Table 1). In SF, elevations were also observed during MCAO, even though significant differences from controls were not consistently obtained. In this location, recovery to control levels after reperfusion was usually fast (Table 2).
Comparison of substrate concentrations in dialysate (μmol/L) obtained in somatosensory cortex before, during, and after transient ischemia of variable duration
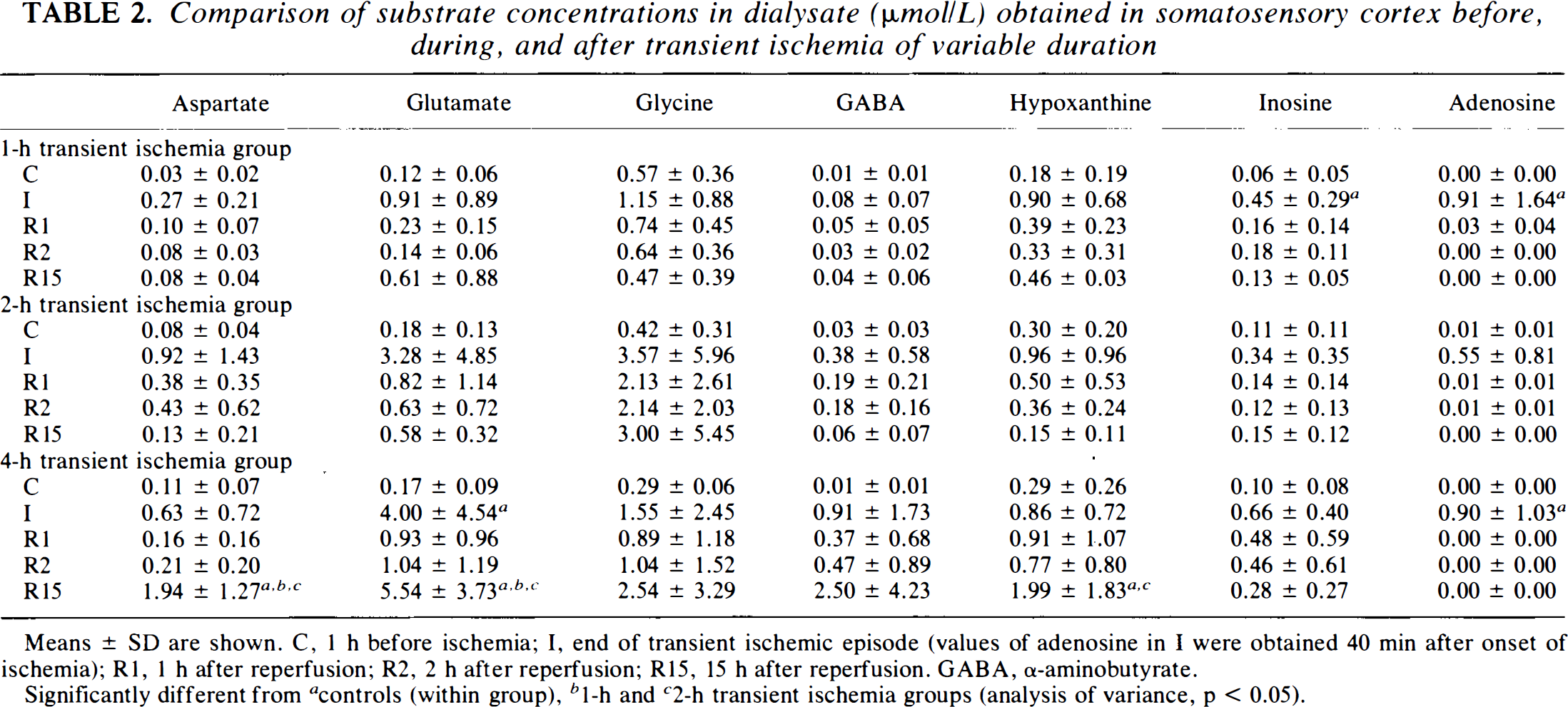
Means ± SD are shown. C, 1 h before ischemia; I, end of transient ischemic episode (values of adenosine in I were obtained 40 min after onset of ischemia); R1, 1 h after reperfusion; R2, 2 h after reperfusion; R15, 15 h after reperfusion. GABA, α-aminobutyrate.
Significantly different from a controls (within group), b 1-h and c 2-h transient ischemia groups (analysis of variance, p < 0.05).
EPs
At the end of the 1- and 2-h ischemic episodes, AEP amplitudes were significantly (ANOVA, p < 0.05) reduced to ∼20% of control, and they were almost completely suppressed in the 4-h group (Fig. 3). Reperfusion resulted in better AEP recovery in the 1- and 2-h groups than in the 4-h group. Additionally, a secondary suppression of AEPs was observed in the 4-h group. SEPs in SF were significantly (ANOVA, p < 0.05) reduced in all groups to ∼20% of control. In this location, EPs recovered soon after recirculation in the 1- and 2-h groups, while in the 4-h group, the reduced EP values remained during the whole reperfusion period.
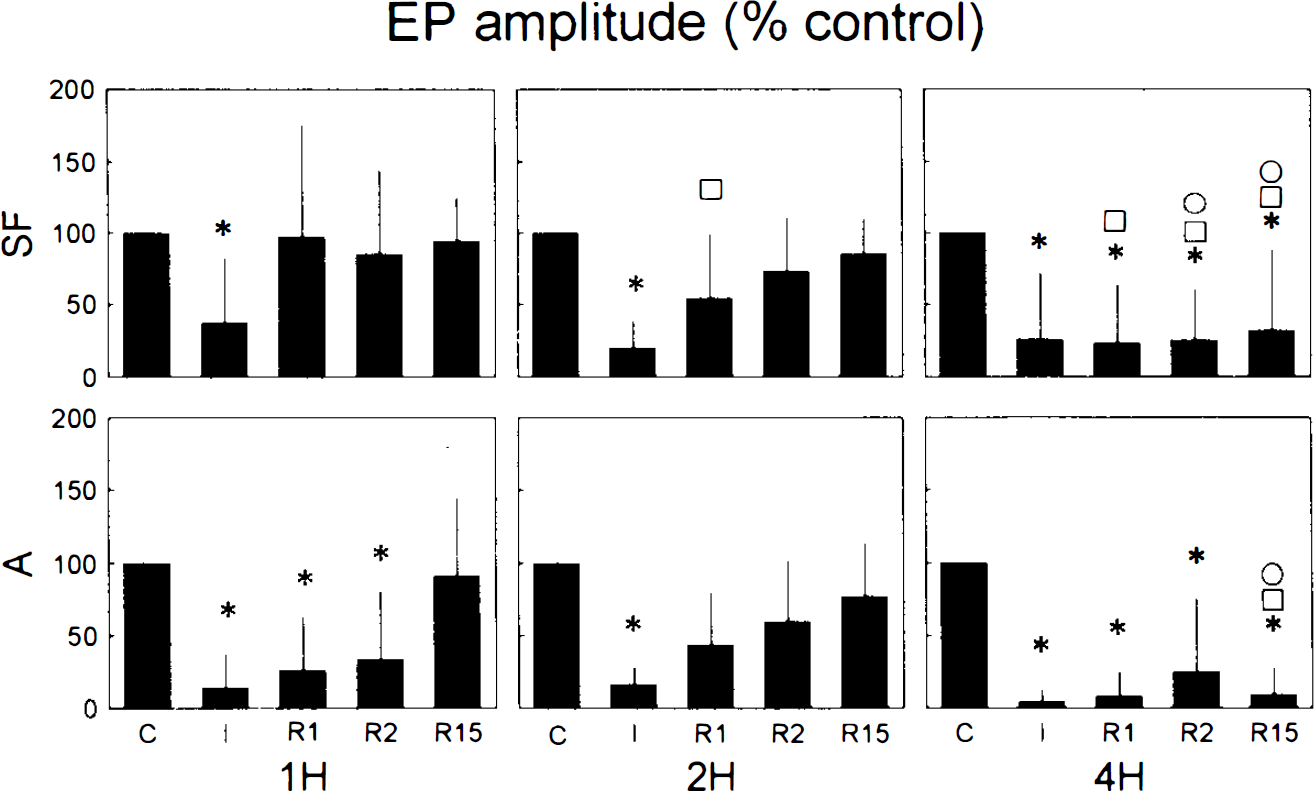
Graph of evoked potential (EP) amplitude (% control) before, during, and after transient ischemia. 1H, 2H, 4H, groups with 1-, 2-, and 4-h transient ischemia, respectively; A, auditory evoked potential recorded in auditory cortex; SF, somatosensory evoked potential recorded in somatosensory cortex; C, 1 h before ischemia; I, end of ischemic episode; R1, 1 h after reperfusion; R2, 2 h after reperfusion; R15, 15 h after reperfusion; *, □, O, significantly different from controls (within group), 1-h, and 2-h transient ischemia groups, respectively (analysis of variance, p < 0.05).
Histopathology
Histopathological analysis of cross-sections stained with hematoxylin–eosin or Luxol fast blue-cresyl violet revealed significantly larger cortical infarct volumes in the 4-h group than in the 1- and 2-h groups (Fig. 4A; ANOVA, p < 0.05). Infarct volumes in the 1- and 2-h groups did not differ significantly. Analysis of infarct areas in consecutive cross sections (Fig. 4B) revealed a comparable result showing that infarcts throughout the cat brain were enlarged as the duration of transient ischemia was prolonged.
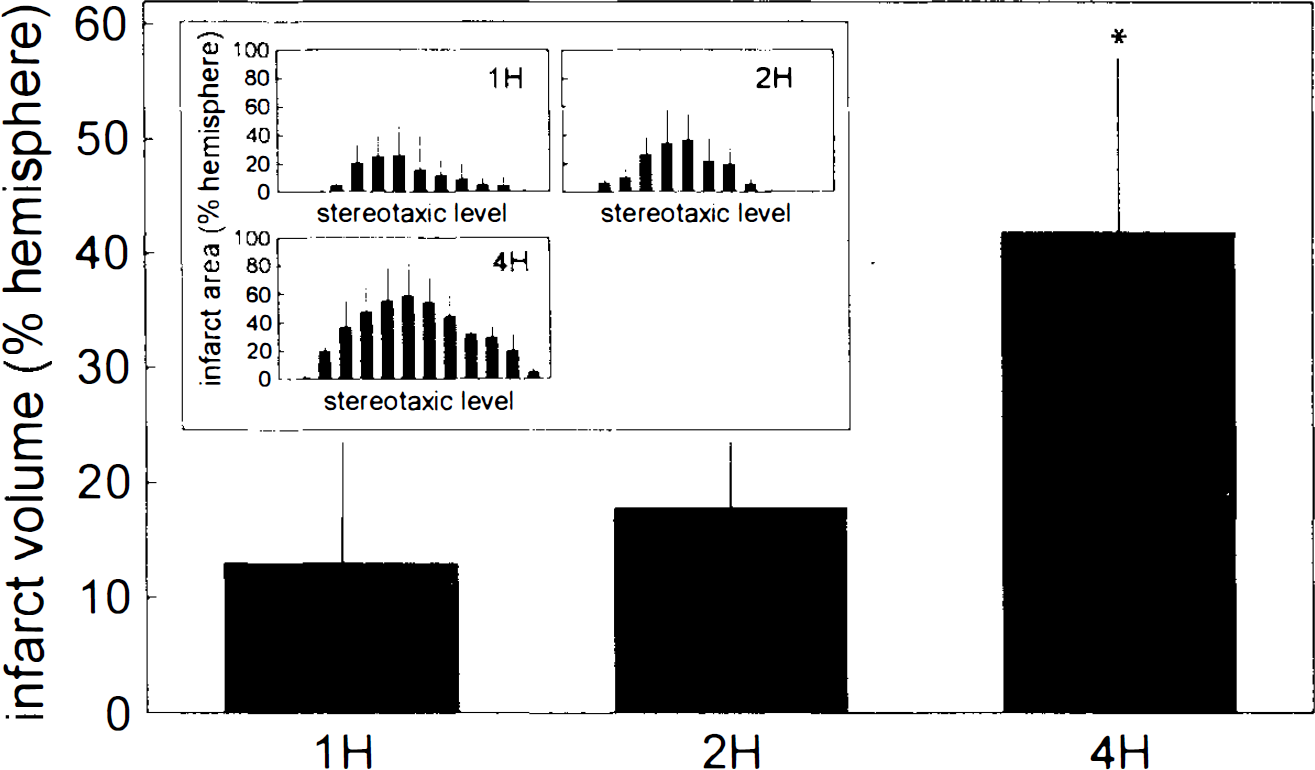
Graph of cortical infarct volume and of cortical infarct areas on different stereotaxic levels
DISCUSSION
In transient ischemia, pathophysiological processes of two different phases contribute to infarct evolution: the initiation of infarction during the ischemia phase and the reversion or maturation of the infarction process during the recirculation phase. Apparently, processes during recirculation depend largely on the initiation, i.e., the magnitude and the duration of blood flow reduction.
Ischemia phase: initiation of infarction
In the investigated ischemic core region, the auditory cortex, the same degree of CBF reduction was observed in all three experimental groups experiencing 1-, 2-, and 4-h transient ischemia. In the border zone region, the somatosensory cortex, smaller CBF reductions were generally found, and among the three groups, residual CBF remained highest in the 4-h group. One-hour transient MCAO was sufficient to initiate infarction, but in the 4-h group, an almost threefold increase of the infarct volume occurred despite the slightly milder CBF disturbance in the ischemic border zone. This result is in good agreement with former reports showing that infarction after transient focal ischemia depends largely on the duration of the ischemic episode. In rats, 4 h of occlusion seemed to mark the upper time limit for infarct completion (Kaplan et al., 1991; Memezawa et al., 1992), whereas in cats, this time limit was at least 6 h (Weinstein et al., 1986). Using positron emission tomography, we have recently shown in cat permanent MCAO that the ischemic penumbra as represented by moderate metabolic impairment propagates for several hours from the center to the periphery of the ischemic territory. This dynamic process may even extend beyond the first day after onset of ischemia (Heiss and Graf, 1994; Heiss et al., 1994).
Among the possible mechanisms involved in the evolution of ischemic infarcts, excitatory amino acid accumulation has been considered as one of the key factors (Rothman and Olney, 1986; Choi et al., 1987). In the present study, the values of extracellular glutamate elevation at the end of the ischemic episodes were significantly higher in the 4-h group than in the 1- and 2-h groups and would therefore support the hypothesis of an involvement of excitatory amino acids in infarct expansion. Further support is provided by the relationship between extracellular glutamate during the ischemic episode and final functional and structural outcome. For this purpose, we calculated cumulative glutamate concentrations (Osuga and Hakin, 1994) in the auditory cortex by adding the results of successive 10-min dialysate samples during the ischemic episodes to have a better estimate of the excitotoxic load as related to the duration of the ischemic episodes. We correlated these values with the final values of AEP amplitudes (Fig. 5A) and with the infarct volumes (Fig. 5B) of the individual experimental animals. Both the negative correlation with EP amplitude and the positive correlation with infarct volume underline the role of glutamate accumulation for neuronal deterioration. Our results obtained in the border zone area are in accordance with this view: In the somatosensory cortex, the largest increase in glutamate was found in the 4-h group, the group exhibiting final suppression of electrical function (EP) and enlarged infarction. Whether or not glutamate toxicity is achieved directly by N-methyl-
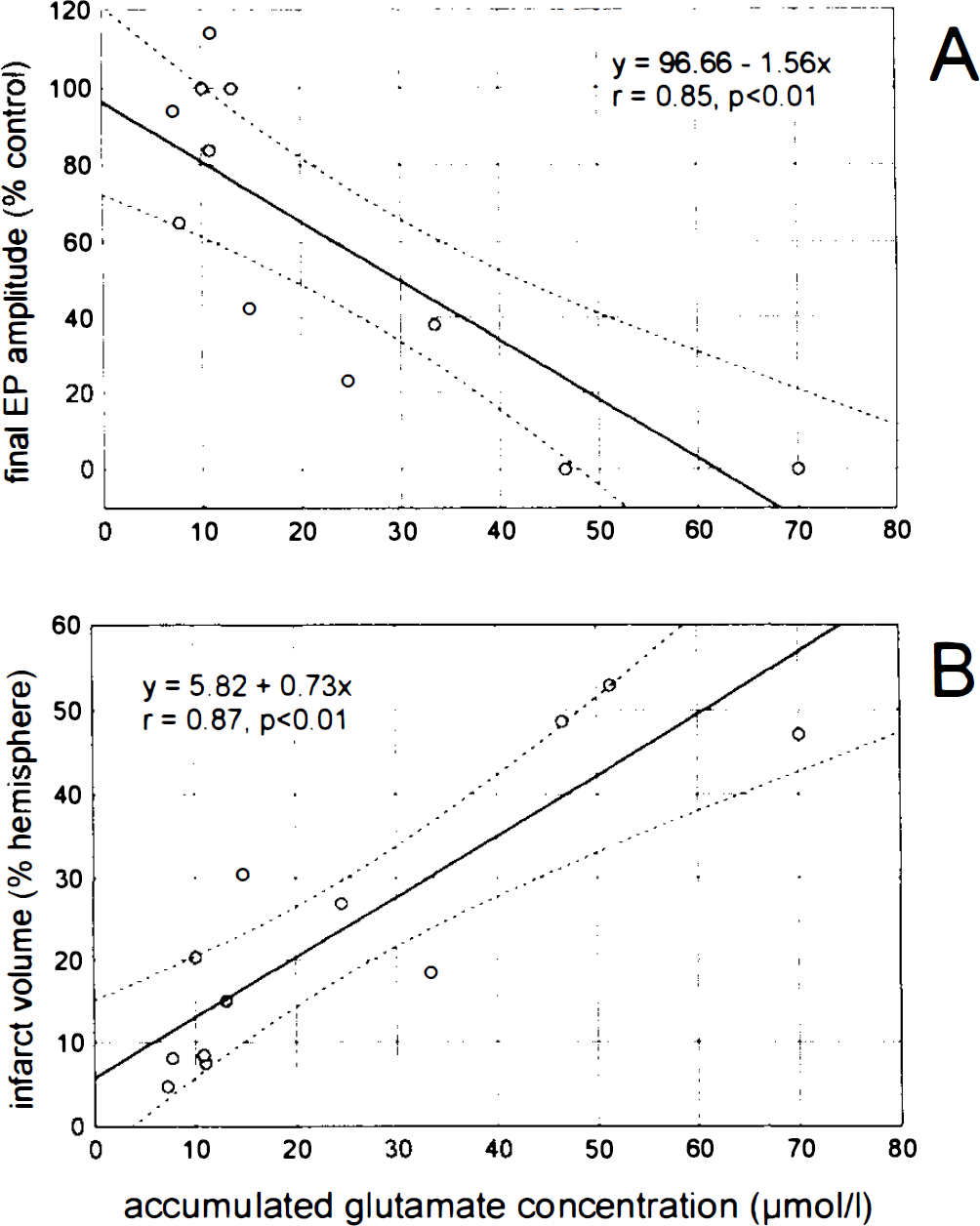
Scatterplot of correlation between accumulated concentration of glutamate in dialysate obtained in the auditory cortex: final auditory evoked potential (EP) amplitude
Progressive deterioration in focal ischemia is determined by a cascade of biochemical and electrical alterations including extracellular elevation of other neuroactive amino acids than glutamate (Matsumoto et al., 1993; Takagi et al., 1993), of purine catabolites such as adenosine (Hagberg et al., 1987; Matsumoto et al., 1992), intracellular elevation of Ca2+ ion activity (Uematsu et al., 1991), or expression of immediate early genes and stress proteins (Welsh et al., 1992; An et al., 1993). Glycine, for example, was shown to increase progressively in the course of permanent focal ischemia in cats (Matsumoto et al., 1993), and it increased in the present study during ischemia. Glycine is an essential factor for augmentation of glutamate-induced Ca2+ entry into cells (Kleckner and Dingledine, 1988), one mechanism by which glutamate toxicity is achieved (Rothman and Olney, 1986). In global ischemia, it increases particularly in selectively vulnerable brain regions (Globus et al., 1990, 1991a), and the glycine blocker HA966 has been shown to protect against ischemic injury (Patel et al., 1989). Inhibitory substances such as GABA, which increased dramatically in the present study, have been suggested to counteract glutamate toxicity (Mattson and Kater, 1989). Some of the pathophysiological processes in early ischemia may be triggered by slow increases of extracellular potassium (Lipton and Wittingham, 1979), however, which may result in a perhaps deleterious chloride influx into cells (Rothman, 1985). Inhibitory transmitters are commonly thought to trigger chloride channel opening. In our view, therefore, the role of increased GABA in ischemic tissue remains obscure. Adenosine, on the other hand, is known to counteract glutamate toxicity (Corradetti et al., 1984). The appearance of adenosine and of other purine catabolites not only in the auditory but also the somatosensory cortex indicates that neurons in these brain areas are exposed to subcritical energy depletion (Matsumoto et al., 1992), since adenosine in the extracellular space is known to derive from ATP breakdown (Hagberg et al., 1987). A potential counteracting role of adenosine against glutamate toxicity, however, is relevant only early in the ischemic episode because this substance virtually disappears from the extracellular compartment by the end of the first hour after MCAO. Consequently, a role of adenosine would have to be expected in our study in the 1- or 2-h groups rather than in the 4-h group (see Tables 1 and 2). Taking our findings together, multiple reasons exist for a worsening of the functional outcome with longer duration of the ischemic episode as suggested by Heiss and Rosner (1983). The enlargement of infarct volumes found in the 4-h transient ischemia group probably depends on these processes.
Recirculation phase: reversal or maturation of infarction
It is known that postischemic neuronal damage is a continuing process, some vulnerable neurons dying only several days after recirculation (Ito et al., 1975; Kirino, 1982). In this study, pathophysiological processes during the recirculation phase clearly differed among the three investigated groups: Only in the 4-h group did severe secondary deterioration occur within the observation period including a late decrease of CBF, accompanied by a secondary increase of glutamate and of other neuroactive substances, a secondary impairment of electric function as determined by the suppression of EPs, and a considerable enlargement of the infarct volume.
Postischemic blood flow disturbances may include a more or less prominent hyperemic episode followed by hypoperfusion (Heiss et al., 1976). In the ischemic core region, hyperperfusion of variable duration occurred immediately after recirculation in the 1- and 2-h but not in the 4-h group (data not shown). Prominent hypoperfusion developed in this study only in the 4-h group. In this group, an additional secondary CBF reduction was observed before termination of the experiments. Postischemic hypoperfusion is deleterious mainly because of deficient oxygen supply (Siesjö, 1981; Hossman, 1982; Klatzo, 1987), being one of the possible reasons for maturation of infarcts and delayed neuronal death. At least partial functional recovery was obtained in all groups of the present study. This result contrasts with former findings in the same model showing permanent EP suppression after 2 h of ischemia (Kataoka et al., 1987). The better outcome may, for example, result from protective effects of halothane anesthesia used in the present study as compared to α-chloralose in the former study. A mechanistic link to this hypothesis is provided by the finding that the generation of spreading depression, which is thought to play a major role in the pathogenesis of ischemic border zones, is inhibited in cats by halothane (Saito et al., 1993). The cause of secondary CBF reduction and subsequent EP suppression seen in the 4-h group was not clear. Since in the early reperfusion period, CBF recovered at least partially in A and almost completely in SF, intravascular factors are probably not responsible. Continued metabolic disturbances are evident for this group since purine catabolites reflecting intracellular ATP degradation remain elevated for longer periods than in the groups with shorter duration of ischemia (see Tables 1 and 2). The main cause for secondary CBF deterioration, however, may be capillary compression resulting from edema formation in the neuronal or endothelial tissue. It is known that in the cat focal ischemia model, transient ischemia of 2 h results in a significant increase in tissue water content (Kataoka et al., 1987).
In the 1- and 2-h groups, elevations of Glu and of other neuroactive substances returned almost to control levels immediately upon recirculation. Among the possible mechanisms responsible for this fast recovery, clearance by restored blood flow and by still effective neuronal and glial uptake systems seem to be most important. Similar or even faster reestablishment of preischemic neurotransmitter levels has been obtained by others after shorter ischemic episodes (Benveniste et al., 1984; Hagberg et al., 1985; Globus et al., 1988). In the 4-h group, in contrast, elevated levels of neuroactive substances are not completely reestablished after recirculation. This may partially result from continued CBF impairment, even though blood flow returns also in this group to levels above 20 ml 100 g− min−1, which is considered to be the critical level for ischemic Glu elevation (Shimada et al., 1989). Impairment of uptake mechanisms may contribute to sustained substrate elevation, even though better recovery of GABA than Glu levels (see Tables 1 and 2) does not favor this idea since uptake mechanisms for GABA have to be assigned to neuronal and those for Glu to glial membranes (Schousboe et al., 1977, 1983). One would suspect neuronal elements to be more affected than glial elements. Similar to Glu and GABA, glycine remained elevated during reperfusion in the 4-h group. Glycine potentiates the NMD A response (Johnson and Ascher, 1987), and elevated levels during reperfusion have led to the suggestion that it might be involved in the evolution of delayed neuronal death (Baker et al., 1991). It should be considered, however, that the glycine site at the NMDA receptor may be saturated at normal extracellular glycine levels. It should be noted that even in the 1- and 2-h groups, slight (nonsignificant) substrate elevations remained in the early reperfusion period. Surprisingly, this was also true for the investigated border zone area SF (see Tables 1 and 2). It remains unclear whether a noncritical state is really (and permanently) reestablished by reperfusion in the groups with shorter duration of ischemia. Longer observation periods after reperfusion than those used in the present study may be necessary to rule out the possibility of delayed secondary deterioration after 1 or 2 h of transient focal ischemia.
In the 4-h group, functional and biochemical recovery after reperfusion is clearly transient in nature. Secondary elevation of Glu and of other neuroactive substances seen in the late reperfusion phase may be caused by the secondary decrease of blood flow discussed, which in turn may result from edema formation. A progressing imbalance of neuroactive substances and of extracellular ions during reperfusion will result in progressive water imbalance and edema formation and thus in secondary blood flow deterioration. A hint for the importance of sustained extracellular Glu elevation is provided by findings showing an attenuation of ischemic cell damage after postischemic administration of NMDA receptor antagonists (Gill et al., 1988; Park et al., 1988). Postischemic hyperthermia has been introduced as another mechanism involved in reperfusion injury and delayed neuronal death, and the application of postischemic hypothermia has been shown to effectively reduce ischemic neuronal injury (Busto et al., 1989; Kuroiwa et al., 1990; Coimbra and Wieloch, 1992). In our study, brain temperature elevations were not observed during the 15-h reperfusion (data not shown). Time course studies throughout longer reperfusion periods looking in more detail into the interaction between CBF, brain temperature, and substrate alterations are needed to shed more light on causal mechanisms responsible for secondary deterioration after prolonged transient ischemia.