
Abstract
Apoptotic cell death is prominent in neurodegenerative disorders, such as Alzheimer's disease and Huntington's disease, and is found in cerebral ischemia. Using a murine model of delayed cell death, we determined that cleavage of zDEVD-amino-4-trifluoromethyl coumarin (zDEVD-afc) in brain homogenate, a measure of caspase activation, increased initially 9 hours after brief (30 minutes) middle cerebral artery occlusion along with caspase-3p20 immunoreactive cleavage product as determined by immunoblotting. zDEVD-afc cleavage activity was blocked by pretreatment or posttreatment with the caspase-inhibitor N-benzyloxycarbonyl-Asp(OMe)-Glu(OMe)-Val-Asp(OMe)-fluoromethyl-ketone (zDEVD-fmk), and ischemic damage was reduced when the drug was injected up to 9 hours after reperfusion. The protection was long lasting (21 days). Hence, the period before caspase activation defined the therapeutic opportunity for this neuroprotective agent after mild ischemic brain injury. Prolonged protection after caspase inhibition plus the extended treatment window may be especially relevant to the treatment of neurodegenerative disorders.
Caspases are mammalian homologues of the death-promoting ced-3 gene product in the nematode Caenorhabditis elegans (Yuan et al., 1993) and have been implicated in triggering and executing programmed cell death (Kumar, 1995). Among 11 known family members (Alnemri et al., 1996), caspase-3 shows the highest amino acid sequence homology to CED-3 (Xue et al., 1996). Caspase-3 is constitutively expressed in adult mouse brain (Namura et al., 1998) as an inactive proform (32 kDa) and is activated by posttranslational cleavage into a large and a small peptide (20 and 12 kDa) plus a short N-terminal prodomain. Caspase activity requires heterodimerization of the large and the small cleavage products and presumably promotes cell death by cleaving nuclear and cytosolic proteins. By caspase cleavage, these substrates are either (1) activated, such as protein kinase C δ, sterol regulatory element binding proteins, caspase-activated DNase, and caspases themselves, (2) inactivated, such as DNA-dependent protein kinase, (3) disassembled, such as nuclear lamins, or (4) modified with unclear functional impact, such as poly(ADP-ribose)polymerase (PARP), U1-ribonuclear protein, and huntingtin (Nicholson and Thornberry, 1997; Villa et al., 1997; Enari et al., 1998).
Cleavage-site directed caspase inhibitors attenuate apoptotic cell death during development (Milligan et al., 1995) and reduce infarct volume 24 hours after middle cerebral artery (MCA) occlusion (2 hours) if administered up to 1 hour after reperfusion (Hara et al., 1997; Ma et al., 1998). Cell damage and caspase activation develop so rapidly after 2 hours of occlusion that mechanisms of protection are difficult to clarify in this model (Namura et al., 1998). Therefore we examined the onset of caspase-like enzyme activity and the treatment window for caspase inhibitors in a murine model of cerebral ischemia in which cell death is delayed and apoptosis unmasked. In this model of focal ischemia, the pattern of cell death (affecting predominantly neurons) resembles a type of delayed cell death that is usually seen in global ischemia. Cells die slowly over days accompanied by the appearance of terminal deoxynucleotidyl transferase-mediated DNA nick-end labeling (TUNEL) plus prominent oligonucleosomal DNA fragmentation (Endres et al., 1998). Neuroprotection was documented even when the caspase-inhibitor N-benzyloxycarbonyl-Asp(OMe)-Glu(OMe)-Val-Asp(OMe)-fluoromethyl-ketone (zDEVD-fmk) was given 6 hours after recirculation in this model and DNA laddering was reduced. Our results now show that the window for neuroprotection is dependent on the time of caspase activation, which is 9 hours after ischemia in this model.
MATERIALS AND METHODS
Focal cerebral ischemia model
Adult SV129 mice (male, 18 to 22 g; Taconic Farms, Germantown, NY, U.S.A.) were used for this model and monitored for regional cerebral blood flow, mean arterial blood pressure, heart rate, rectal temperature, and blood gases and pH as described (Hara et al., 1996). Rectal temperature was maintained at 37°C ± 1°C and monitored carefully in anesthetized and awake animals. Filament occlusion of the left MCA was performed as described (Hara et al., 1996). Thirty minutes after ischemia, animals were briefly reanesthetized with halothane, and the filament withdrawn. zDEVD-fmk (480 ng; Enzyme Systems, Livermore, CA, U.S.A.) was administered by intracerebroventricular injection (0.9 mm lateral, 0.1 mm posterior, 3.1 mm deep from bregma). Three days after reperfusion, animals were killed and the brains quickly removed and frozen. Five coronal sections (12 μm thick) were taken from the brain each 2 mm caudal from the frontal pole and were stained with hematoxylin and eosin. The ischemic area on each section as reflected by sharply outlined zones of pale staining was measured by an investigator naive to the group identity using an image-analysis system (M4, Imaging Research, St. Catherines, Ontario, Canada) and a Zeiss microscope (×25). The total lesion volume was obtained by integrating from the areas on each section.
Mice were tested for neurologic deficits 30 minutes after ischemia onset at the time of reperfusion and scored as described before (Hara et al., 1996): 0, no observable neurologic deficits; 1, failure to extend right forepaw; 2, circling to the contralateral side; and 3, loss of walking or righting reflex. The rater was naive to the treatment groups. Only animals with a score of 2 or more were included. Animal protocols were approved by the Massachusetts General Hospital's Committee on Research, Subcommittee on Research Animal Care.
Caspase-3-like zDEVD-afc cleavage (DEVDase activity) assay on brain tissue homogenates
Fresh brain hemispheres (after frontal and occipital pole removal) were immediately homogenized in 4 volumes of lysis buffer (HEPES 25 mmol/L, pH 7.5, 4°C) containing 0.1% Triton X-100, 5 mmol/L MgCl2, 2 mmol/L dithiothreitol, 74 μmol/L antipain, 0.15 μmol/L aprotinin, 1.3 mmol/L EDTA, 1 mmol/L ethyleneglycol-bis-β-aminomethylether (EGTA), 15 μmol/L pepstatin, and 20 μmol/L leupeptin and centrifuged at 50,000g at 4°C. Supernatant (100 μL) was added to HEPES buffer (900 μL, 100 mmol/L, pH 7.4) containing 2 mmol/L dithiothreitol at 25°C. After addition of the fluorogenic substrate N-benzyloxycarbonyl-Asp-Glu-Val-Asp-7-amino-4-trifluoromethyl coumarin (zDEVD-afc, 12.5 μmol/L; Enzyme Systems, Livermore, CA, U.S.A.), which mimics the cleavage site of PARP (DEVD216-G217), fluorescent amino-4-trifluoromethyl coumarin production was measured using an Hitachi F-4500 spectrofluorometer (excitation, 400 nm; emission, 505 nm). Increase in fluorescence was linear between 10 and 35 minutes after adding zDEVD-afc. Caspase-3-like activity was calculated from the slope as fluorescence units per milligram of protein per minute and converted to picomoles per milligram per minute based on an amino-4-trifluoromethyl coumarin standard curve. DEVDase activity observed in normal brains from sham-operated animals was considered as background and subtracted from that in ischemic brains. Activity in the contralateral hemisphere was background level, not inhibitable by zDEVD-fmk or N-benzyloxycarbonyl-Val-Ala-Asp (OMe)-fluoromethylketone (zVAD-fmk; 100 μmol/L, respectively; Enzyme Systems) and did not change during ischemia. However, caspase-3-like activity in the ischemic hemisphere was reduced 75% by the in vitro addition of zDEVD-fmk (100 μmol/L; 73% by 10 μmol/L), added 15 minutes before the fluorogenic substrate. One hundred micromolar zDEVD-fmk is 10 times the Km for caspase-3 (9.7 mmol/L; Nicholson et al., 1995) and 10 times the concentration required to completely block caspase-3-like DEVDase activity in staurosporine-exposed HeLa cells (Namura et al., 1998). One hundred micromolar or more was able in vitro to inhibit caspase-3-like activity (Enari et al., 1996; Armstrong et al., 1997; Eldadah et al., 1997). Protein concentration was determined by photometric assay (Bio-Rad Laboratories, Hercules, CA, U.S.A.).
CM1 immunoblot on brain tissue lysates
Brain hemispheres minus frontal (2 mm) and occipital (1 mm) poles were frozen in liquid N2, crushed and lysed in 10 mmol/L HEPES buffer (pH 7.6) containing 42 mmol/L KCl, 5 mmol/L MgCl2, 1 mmol/L phenylmethylsulfonylfluoride, 1 mmol/L EDTA, 1 mmol/L EGTA, 1 mmol/L dithiothreitol, 1% sodium lauryl sulfate, 1.5 μmol/L pepstatin, 2 μmol/L leupeptin, and 0.7 μmol/L aprotinin, and centrifuged at 20,800g. Samples were subjected to sodium dodecyl sulfate polyacrylamide gel electrophoresis and transferred to a nitro-cellulose membrane. Blots were probed with two different polyclonal antibodies. Caspase-3 antiserum was raised against a 10-amino acid peptide (C12SIKNFEVKT20 in hamster) located close to the N-terminus of caspase-3 prodomain (amino acids 12 through 20; Wang et al., 1996) and therefore recognized both caspase-3p32 and its p20 cleavage product. A rabbit polyclonal antiserum, CM1, was raised against a 13-amino acid peptide (163CRGTELDCGIETD175) covering the active site cysteine and extending to the C-terminus of the p20 cleavage product of human and mouse caspase-3 (Armstrong et al., 1997; Namura et al., 1998). We previously reported that immunostaining was significantly reduced by preabsorbtion with either peptide immunogen (SIKNFEVKT for caspase-3p32; CRGTELDCGIETD for CM1 (Namura et al., 1998). Blot bands were quantified by measuring optical density (OD; Bio-Rad GS-700). Differences in OD of 20-kDa and 32-kDa bands over time or between ischemic and contralateral hemispheres were always determined based on the comparison with the corresponding α-tubulin bands.
Cell counting
To quantify viable cells in the center of the striatal lesion, cells with a nonpyknotic nucleus were counted in a 0.4-mm2 field in a section (20 μm thick; stained with hematoxylin and eosin) through the anterior commissure. Cells with a rod-shaped nucleus, which were assumed to be glia, were not counted.
Statistics
Caspase-like activity time course and cell counts were analyzed by one-way analysis of variance followed by Dunnett and Scheffé post hoc tests, respectively. Infarct volume, single caspase-like activity data points and optical densities on blots were compared with the corresponding controls by Student's t tests. A P less than 0.05 was considered significant.
RESULTS
Caspase activation after mild focal ischemia
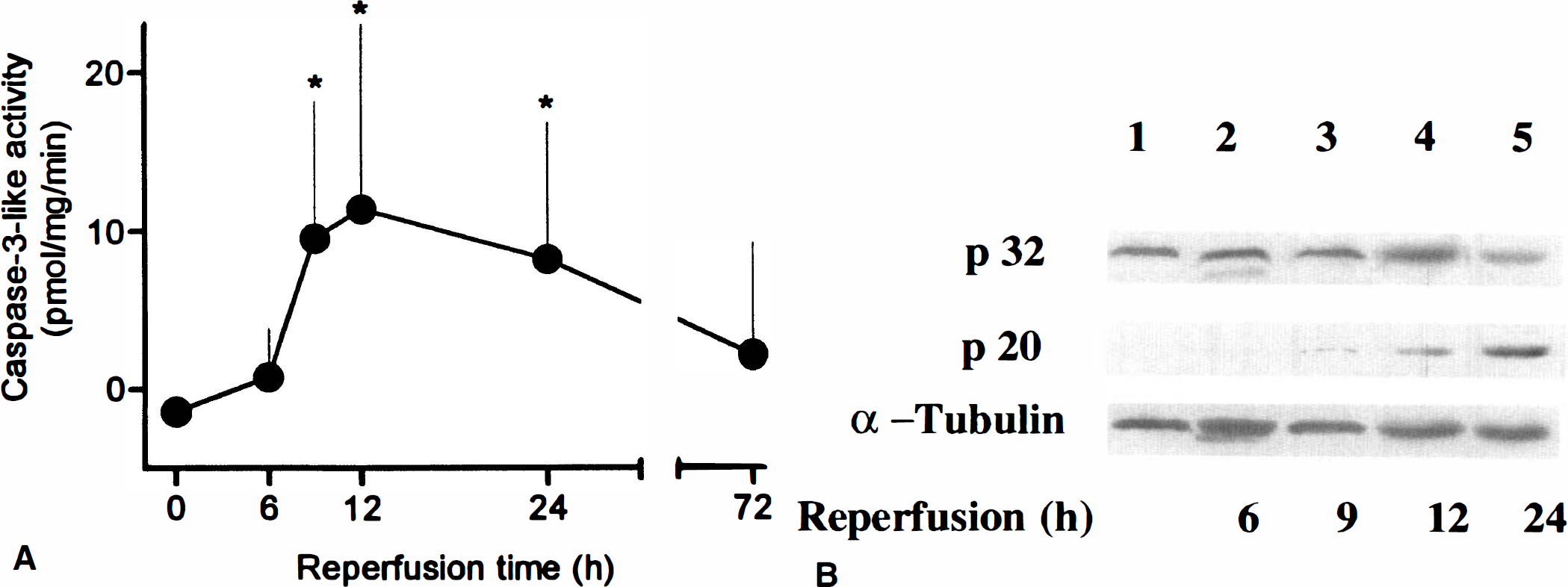
Thirty minutes of reversible MCA occlusion caused the slow development of apoptotic cell death as evidenced by the first appearance of TUNEL-positive cells and DNA laddering at 24 hours, peaking at 72 hours (Endres et al., 1998). When caspase-like activity was measured in brain lysates using the fluorogenic substrate zDEVD-afc, DEVDase activity was detected initially 9 hours after 30 minutes of occlusion, increased at 12 hours, and sustained until 24 hours after reperfusion (Fig. 1A). Using antiserum against the carboxy-terminus of the 20-kDa caspase-3 fragment (p20), we detected a faint band at 9 hours that became more intense at 12 and 24 hours (Fig. 1B, middle panel). A decrease in unprocessed immunoreactive caspase-3 (Fig. 1B, upper panel) was detected at 24 hours (see Fig. 1B legend) and 30 hours (not shown).
zDEVD-fmk administration inhibits caspase activation
When zDEVD-fmk was injected intracerebroventricularly, immediately before MCA occlusion (Fig. 2) caspase-like activity in brain homogenate was almost completely inhibited 12 or 24 hours later. Administration at 6 hours also significantly reduced DEVDase activity when measured during peak activity (12 hours) (18.4 ± 8.5 versus 1.7 ± 2.9 pmol·mg−1·min−1; n = 5 to 6; P < 0.05). Reportedly, caspase-7 and caspase-1 cleave zDEVD-afc (Juan et al., 1997; Thornberry et al., 1997). However, caspase-7 is not expressed in mouse brain (Juan et al., 1997), and zDEVD-fmk treatment did not attenuate elevated interleukin-1β levels in ischemic brain as observed after caspase-1 inhibition (Hara et al., 1997). Therefore, among the known family members expressed in brain, caspase-3 is tentatively identified as the predominant enzyme cleaving the fluorogenic substrate in this study, consistent with a decrease in unprocessed immunoreactive caspase-3 on immunoblots.
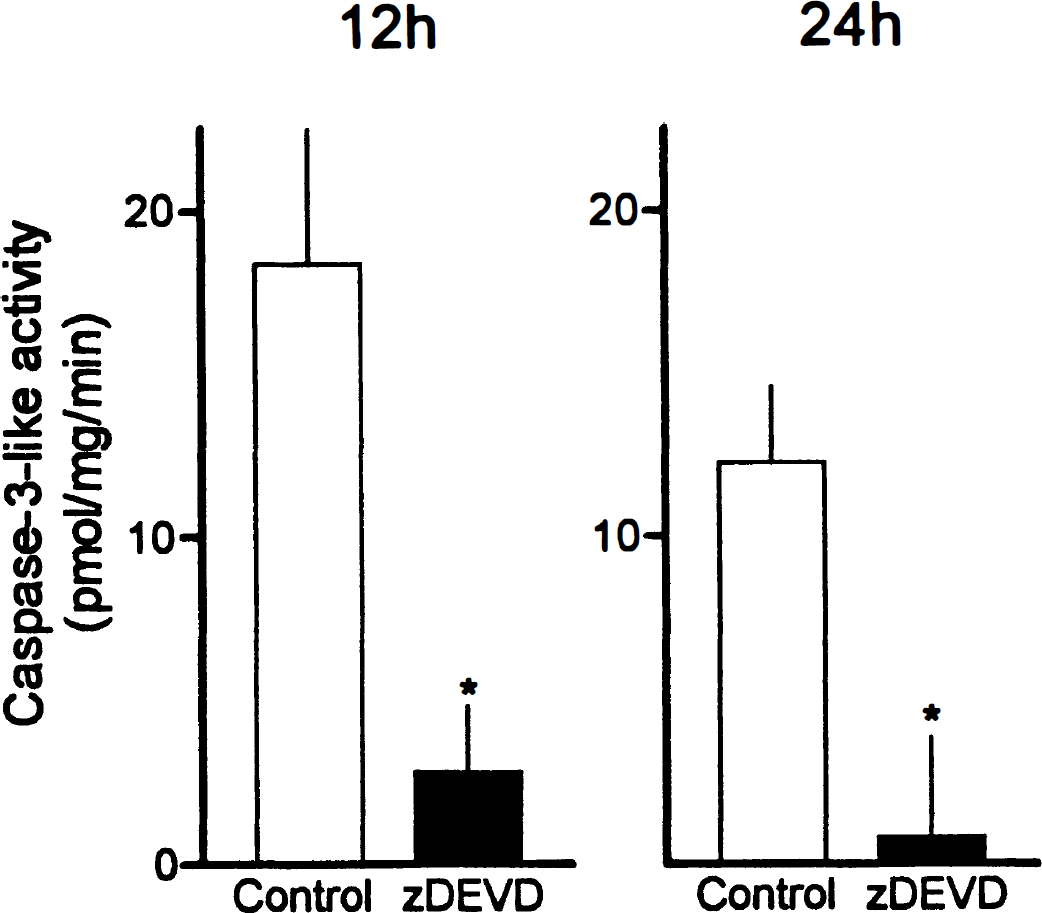
Caspase-3-like DEVDase activity in ischemic brain tissue lysates 12 and 24 hours after reperfusion after 30 minutes occlusion of the left MCA. DEVDase activity was determined by measuring cleavage of the fluorogenic substrate zDEVD-afc. zDEVD-fmk (480 ng) was injected intracerebroventricularly immediately before MCA occlusion. Mean ± SD; * P < 0.01, compared with the corresponding vehicle controls in 4 to 6 experiments.
zDEVD-fmk protects from ischemic cell death
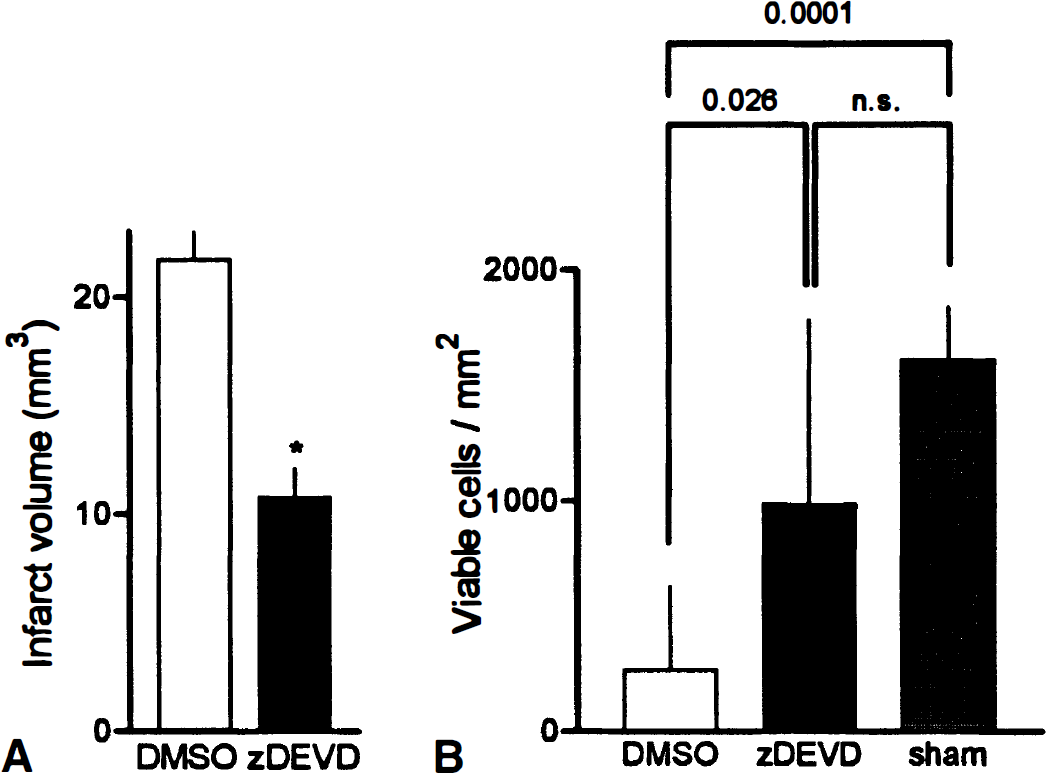
zDEVD-fmk (480 ng) treatment significantly reduced ischemic injury when given immediately before ischemia (55%), or during reperfusion at 6 hours (57%) or 9 hours (45%) (Fig. 3). Neurologic deficits were also improved in the treated groups, suggesting that drug-induced neuronal sparing enhanced sensory-motor performance. Protection sustained when tested at 7 days (Fig. 4A). Even at 21 days, greater numbers of viable neurons were present in ischemic striatum (Fig. 4B). Hence, caspase-like inhibition not only postponed apoptotic cell death, but protected cells in a long-lasting way. Because zDEVD-fmk treatment did not modify regional cerebral blood flow, mean arterial blood pressure, heart rate, arterial pH, PaCO2, PaO2, or rectal temperature, we conclude that zDEVD-fmk protects ischemic tissue and preserves neurologic function probably by direct cytoprotective mechanisms involving caspase inhibition.
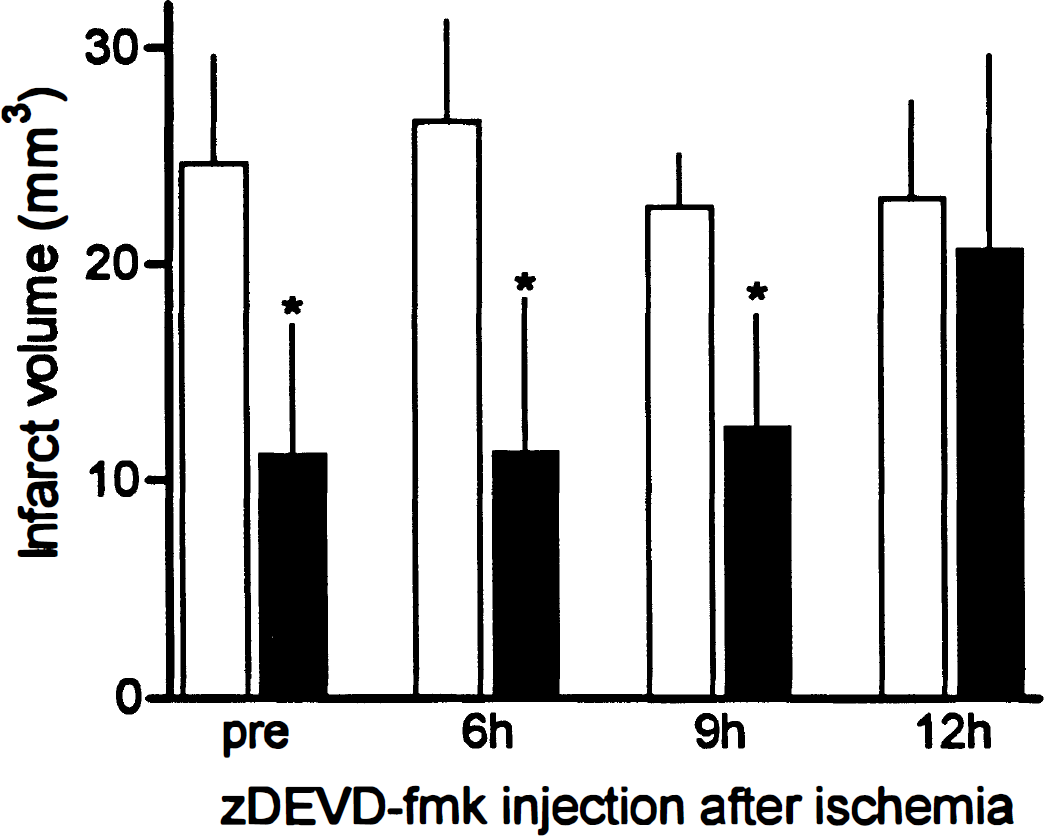
Injury volume after intracerebroventricular injection of zDEVD-fmk (480 ng) before cerebral ischemia or 6 to 12 hours after reperfusion (black bars; n = 7 to 9) or in controls injected with vehicle at the same time points (dimethylsulfoxide, 0.4%; clear bars; n = 6 to 10). Mice were subjected to filament occlusion of the MCA for 30 minutes and reperfused for 72 hours. Mean ± SD; * P < 0.01, compared with the corresponding vehicle controls.
DISCUSSION
The treatment window for zDEVD-fmk corresponds to the onset of caspase activation
zDEVD-fmk (480 ng) protected brain when injected up to 9 hours after restoration of blood flow, but not when administered 12 hours after reperfusion (Fig. 1). The 9-hour treatment window corresponded to the onset of both DEVDase activation and caspase-3 cleavage. Hence, zDEVD-fmk protected against the development of ischemic injury if given before enzyme activation. When injected at time points later than 9 hours, cell death became uncoupled from caspase inhibition presumably because of activation of downstream caspase processing and cleavage of multiple substrates. The long-lasting inhibition of caspase-3-like activity by zDEVD-fmk is caused by irreversible alkylation of the active site cysteine sulfur of caspase-3 by the fluoromethylketone moiety of zDEVD-fmk (Thornberry et al., 1994).
How is caspase-3 activation initiated after mild brain ischemia? Current literature suggests several possible upstream events including caspase-1, caspase-8, and granzyme B (Liu et al., 1997). Cytochrome c release from mitochondria and the dissociation of Bcl-2 from a complex of mammalian CED-4 homologue Apaf-1 (Zou et al., 1997) plus caspase-3 may promote cleavage and caspase-3 processing (Chinnaiyan et al., 1997; Hengartner, 1997). Cycloheximide, which can attenuate mild ischemic damage in this mouse model (Endres et al., 1998) and in the rat (Du et al., 1996), may inhibit new protein synthesis, or alternatively promote bcl-2 upregulation (Furukawa et al., 1997). The importance of free radicals, DNA damage, mitochondrial injury, and p53 upregulation and Fas receptor-mediated formation of death-inducing stimulus complex needs further study (MacManus and Linnik, 1997).
The findings in this study are important for several reasons. DEVDase activity provides the first tissue marker that can be used to assess a therapeutic window for caspase-mediated neuroprotection. The treatment window in this case corresponded to the onset of caspase activation. To our knowledge, this is the first biochemical marker to define a treatment window for any class of therapeutic agents in stroke. Second, the therapeutic window for caspase inhibition after mild ischemia (30 minutes) is considerably longer than for most drugs successful in models of focal cerebral ischemia (generally less than 2 hours after reperfusion) and protection was long lasting (21 days after treatment). Although experience in testing drugs in this model is relatively limited, the therapeutic window for MK-801, an N-methyl-D-aspartate receptor blocker, was not extended in this model (Endres et al., 1998). Because caspase activation may be relevant to slowly evolving cell death after brief cerebral ischemia or in neurodegenerative diseases such as Alzheimer's disease (Yang et al., 1998), Huntington's disease (Goldberg et al., 1996), and amyotrophic lateral sclerosis (Friedlander et al., 1997), our findings emphasize the importance of developing caspase inhibitors, which may be useful for treatment of neurologic diseases in addition to acute focal cerebral ischemia in man.
Footnotes
Acknowledgements
The authors thank Dr. Joseph L. Goldstein, University of Texas Southwestern Medical Center, for providing caspase-3p32 antisera and Drs. Keven J. Tomaselli and Anu Srinivasan, IDUN Pharmaceuticals, Inc., La Jolla, CA, for providing caspase-3p20 antisera.